24
Antiseizure Drugs
CASE STUDY
A 23-year-old woman presents to the office for consultation regarding her antiseizure medications. Seven years ago, this otherwise healthy young woman had a generalized tonic-clonic seizure (GTCS) at home. She was rushed to the emergency department, at which time she was alert but complained of headache. A consulting neurologist placed her on levetiracetam, 500 mg bid. Four days later, EEG showed rare right temporal sharp waves. MRI was normal. One year after this episode, a repeat EEG was unchanged, and levetiracetam was gradually increased to 1000 mg bid. The patient had no significant adverse effects from this dosage. At age 21, she had a second GTCS while in college; further discussion with her roommate at that time revealed a history of two recent episodes of 1–2 minutes of altered consciousness with lip smacking (complex partial seizures). A repeat EEG showed occasional right temporal spikes. What is one possible strategy for controlling her present symptoms?
Approximately 1% of the world’s population has epilepsy, the third most common neurologic disorder after dementia and stroke. Although standard therapy permits control of seizures in 80% of these patients, millions (500,000 people in the USA alone) have uncontrolled epilepsy. Epilepsy is a heterogeneous symptom complex—a chronic disorder characterized by recurrent seizures. Seizures are finite episodes of brain dysfunction resulting from abnormal discharge of cerebral neurons. The causes of seizures are many and include the full range of neurologic diseases—from infection to neoplasm and head injury. In some subgroups, heredity has proved to be a predominant factor. Single gene defects, usually of an autosomal dominant nature involving genes coding voltage-gated ion channels or GABAA receptors, have been shown to account for a small number of familial generalized epilepsies. Commonly, one family shows multiple epilepsy syndromes including, for example, febrile seizures, absence attacks, and juvenile myoclonic epilepsy.
The antiseizure drugs described in this chapter are also used in patients with febrile seizures or with seizures occurring as part of an acute illness such as meningitis. The term “epilepsy” is not usually applied to such patients unless chronic seizures develop later. Seizures are occasionally caused by an acute underlying toxic or metabolic disorder, in which case appropriate therapy should be directed toward the specific abnormality, eg, hypocalcemia. In most cases of epilepsy, however, the choice of medication depends on the empiric seizure classification.
DRUG DEVELOPMENT FOR EPILEPSY
For a long time it was assumed that a single antiepileptic drug (AED) could be developed for the treatment of all forms of epilepsy. However, the causes of epilepsy are extremely diverse, encompassing genetic and developmental defects and infective, traumatic, neoplastic, and degenerative disease processes. Drug therapy to date shows little evidence of etiologic specificity. There is some specificity according to seizure type (Table 24–1), which is most clearly seen with generalized seizures of the absence type. These are typically seen with 2–3 Hz spike-and-wave discharges on the electroencephalogram, which respond to ethosuximide and valproate but can be exacerbated by phenytoin and carbamazepine. Drugs acting selectively on absence seizures can be identified by animal screens, using either threshold pentylenetetrazol clonic seizures in mice or rats or mutant mice showing absence-like episodes (so-called lethargic, star-gazer, or tottering mutants). In contrast, the maximal electroshock (MES) test, with suppression of the tonic extensor phase, identifies drugs such as phenytoin, carbamazepine, and lamotrigine, which are active against generalized tonic-clonic seizures and complex partial seizures. The maximal electroshock test as the major initial screen for new drugs led predominantly to the early identification of drugs with a mechanism of action involving prolonged inactivation of the voltage-gated Na+ channel (see Chapter 14). Limbic seizures induced in rats by the process of electrical kindling (involving repeated episodes of focal electrical stimulation) probably provide a better screen for predicting efficacy in complex partial seizures.
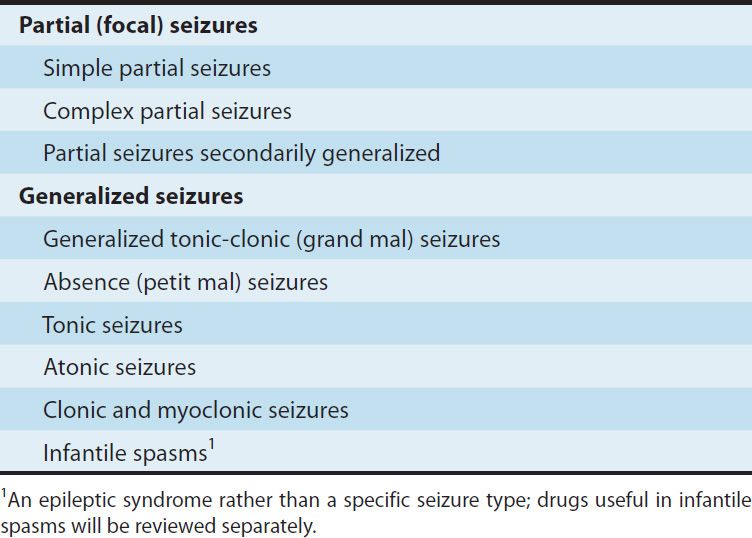
TABLE 24–1 Classification of seizure types.
Existing antiseizure drugs provide adequate seizure control in about two thirds of patients. So-called “drug resistance” may be observed from the onset of attempted therapy or may develop after a period of relatively successful therapy. Explanations are being sought in terms of impaired access of the drugs to target sites or insensitivity of target molecules to them. In children, some severe seizure syndromes associated with progressive brain damage are very difficult to treat. In adults, some focal seizures are refractory to medications. Some, particularly in the temporal lobe, are amenable to surgical resection. Some of the drug-resistant population may respond to vagus nerve stimulation (VNS), a nonpharmacologic treatment for epilepsy now widely approved for treatment of patients with partial seizures. Another device approved in the USA for the treatment of medically refractory partial epilepsy is the responsive neurostimulator (RNS) system. The RNS neurostimulator is designed to detect abnormal electrical activity in the brain and deliver electrical brain stimulation to normalize activity before the patient experiences seizures. Other devices, using various paradigms of electrical stimulation, are in clinical development. One of these, a deep brain stimulation device, has been approved in Canada and in Europe, but not in the USA.
New antiseizure drugs are being sought not only by the screening tests noted above but also by more focused approaches. Compounds are sought that act by one of three mechanisms: (1) enhancement of GABAergic (inhibitory) transmission, (2) diminution of excitatory (usually glutamatergic) transmission, or (3) modification of ionic conductances. Presynaptic effects on transmitter release appear particularly important, and some molecular targets are known, eg, SV2A (see Figure 24–2).
Although it is widely recognized that current antiseizure drugs are palliative rather than curative, successful strategies for identifying drugs that are either disease modifying or that prevent epileptogenesis have proved elusive. Neuronal targets for current and potential antiseizure drugs include both excitatory and inhibitory synapses. Figure 24–1 represents a glutamatergic (excitatory) synapse, and Figure 24–2 indicates targets in a GABAergic (inhibitory) synapse.
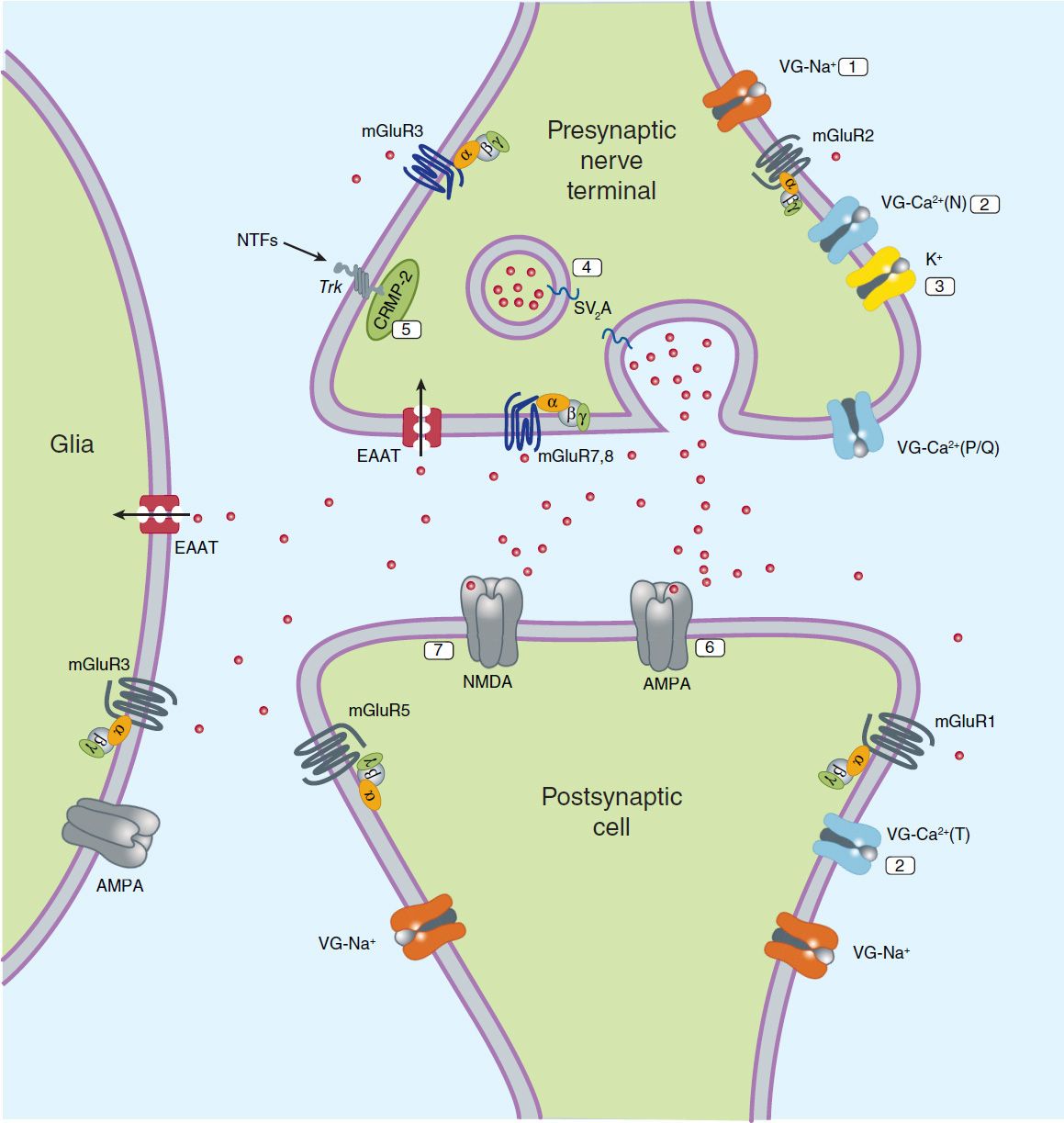
FIGURE 24–1 Molecular targets for antiseizure drugs at the excitatory, glutamatergic synapse. Presynaptic targets diminishing glutamate release include 1, voltage-gated (VG) Na+ channels (phenytoin, carbamazepine, lamotrigine, and lacosamide); 2, VG-Ca2+ channels (ethosuximide, lamotrigine, gabapentin, and pregabalin); 3, K+ channels (retigabine); synaptic vesicle proteins, 4, SV2A (levetiracetam); and 5, CRMP-2, collapsin-response mediator protein-2. Postsynaptic targets include 6, AMPA receptors (blocked by phenobarbital, topiramate, lamotrigine, and perampanel) and 7, NMDA receptors (blocked by felbamate). EAAT, excitatory amino acid transporter; NTFs, neurotrophic factors; SV2A, synaptic vesicular proteins. Red dots represent glutamate.
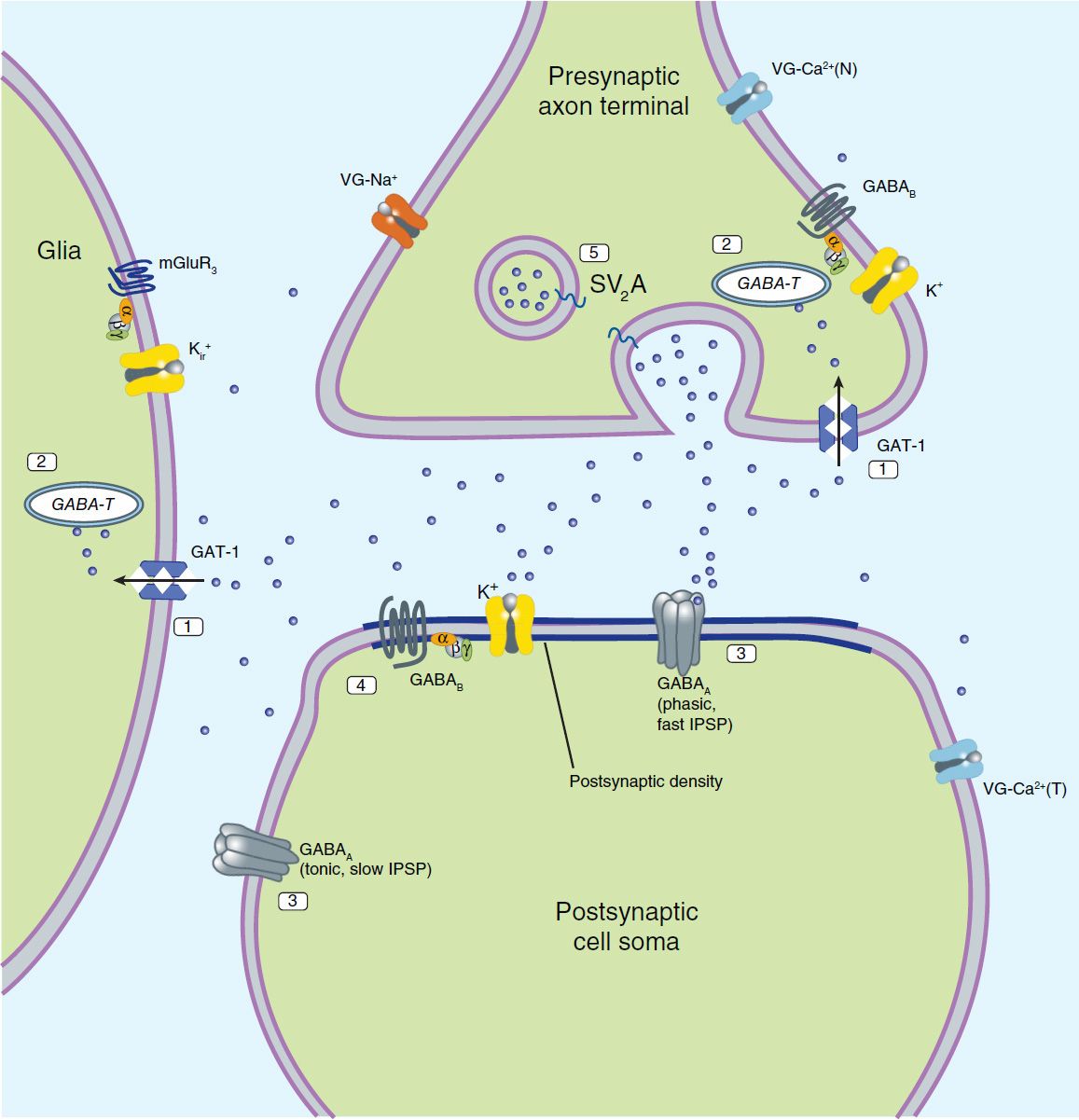
FIGURE 24–2 Molecular targets for antiseizure drugs at the inhibitory, GABAergic synapse. These include “specific” targets: 1, GABA transporters (especially GAT-1, tiagabine); 2, GABA-transaminase (GABA-T, vigabatrin); 3, GABAA receptors (benzodiazepines); potentially, 4, GABAB receptors; and 5, synaptic vesicular proteins (SV2A). Effects may also be mediated by “nonspecific” targets such as by voltage-gated (VG) ion channels and synaptic proteins. IPSP, inhibitory postsynaptic potential. Blue dots represent GABA.
 BASIC PHARMACOLOGY OF ANTISEIZURE DRUGS
BASIC PHARMACOLOGY OF ANTISEIZURE DRUGS
CHEMISTRY
Until 1990, approximately 16 antiseizure drugs were available, and 13 of them can be classified into five very similar chemical groups: barbiturates, hydantoins, oxazolidinediones, succinimides, and acetylureas. These groups have in common a similar heterocyclic ring structure with a variety of substituents (Figure 24–3). For drugs with this basic structure, the substituents on the heterocyclic ring determine the pharmacologic class, either anti-MES or antipentylenetetrazol. Very small changes in structure can dramatically alter the mechanism of action and clinical properties of the compound. The remaining drugs in this older group—carbamazepine, valproic acid, and the benzodiazepines—are structurally dissimilar, as are the newer compounds marketed since 1990, ie, eslicarbazepine, felbamate, gabapentin, lacosamide, lamotrigine, levetiracetam, oxcarbazepine, perampanel, pregabalin, retigabine, rufinamide, stiripentol, tiagabine, topiramate, vigabatrin, and zonisamide.
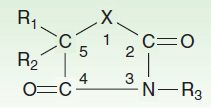
FIGURE 24–3 Antiseizure heterocyclic ring structure. The X varies as follows: hydantoin derivatives, –N–; barbiturates, –C–N–; oxazolidinediones, –O–; succinimides, –C–; acetylureas, –NH2 (N connected to C2). R1, R2, and R3 vary within each subgroup.
PHARMACOKINETICS
The antiseizure drugs exhibit many similar pharmacokinetic properties—even those whose structural and chemical properties are quite diverse—because most have been selected for oral activity and all must enter the central nervous system. Although many of these compounds are only slightly soluble, absorption is usually good, with 80–100% of the dose reaching the circulation. Most antiseizure drugs (other than phenytoin, tiagabine, and valproic acid) are not highly bound to plasma proteins.
Antiseizure drugs are cleared chiefly by hepatic mechanisms, although they have low extraction ratios (see Chapter 3). Many are converted to active metabolites that are also cleared by the liver. These drugs are predominantly distributed into total body water. Plasma clearance is relatively slow; many antiseizure drugs are therefore considered to be medium to long acting. Some have half-lives longer than 12 hours. Many of the older antiseizure drugs are potent inducers of hepatic microsomal enzyme activity. Compliance is better with less frequent administration; thus extended-release formulations permitting once- or twice-daily administration may offer an advantage.
DRUGS USED IN PARTIAL SEIZURES & GENERALIZED TONIC-CLONIC SEIZURES
The classic major drugs for partial and generalized tonic-clonic seizures are phenytoin (and congeners), carbamazepine, valproate, and the barbiturates. However, the availability of newer drugs—eslicarbazepine, lamotrigine, levetiracetam, gabapentin, oxcarbazepine, pregabalin, retigabine, topiramate, vigabatrin, lacosamide, and zonisamide—is altering clinical practice in countries where these compounds are available. The next section of the chapter is a description of major drugs from a historical and structural perspective. Factors involved in the clinical choice of drugs are described in the last section of the chapter.
PHENYTOIN
Phenytoin is the oldest nonsedative antiseizure drug, introduced in 1938 after a systematic evaluation of compounds such as phenobarbital that altered electrically induced seizures in laboratory animals. It was known for decades as diphenylhydantoin.
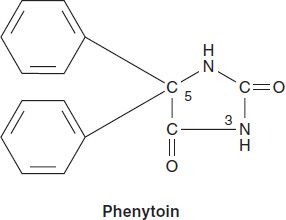
Chemistry
Phenytoin is a diphenyl-substituted hydantoin with the structure shown. It has much lower sedative properties than compounds with alkyl substituents at the 5 position. A more soluble prodrug of phenytoin, fosphenytoin, is available for parenteral use; this phosphate ester compound is rapidly converted to phenytoin in the plasma.
Mechanism of Action
Phenytoin has major effects on several physiologic systems. It alters Na+, K+, and Ca2+ conductance, membrane potentials, and the concentrations of amino acids and the neurotransmitters norepinephrine, acetylcholine, and γ-aminobutyric acid (GABA). Studies with neurons in cell culture show that phenytoin blocks sustained high-frequency repetitive firing of action potentials (Figure 24–4). This effect is seen at therapeutically relevant concentrations. It is a use-dependent effect (see Chapter 14) on Na+ conductance, arising from preferential binding to—and prolongation of—the inactivated state of the Na+ channel. This effect is also seen with therapeutically relevant concentrations of carbamazepine, lamotrigine, and valproate and probably contributes to their antiseizure action in the electroshock model and in partial seizures. Phenytoin also blocks the persistent Na+ current, as do several other AEDs including valproate, topiramate, and ethosuximide.
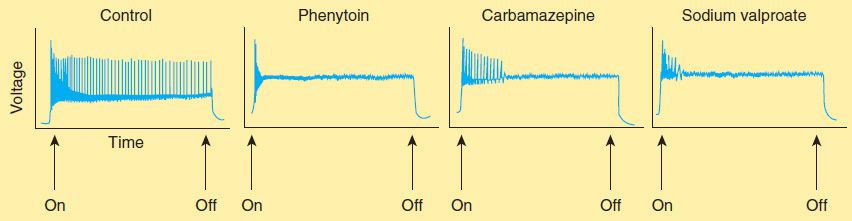
FIGURE 24–4 Effects of three antiseizure drugs on sustained high-frequency firing of action potentials by cultured neurons. Intracellular recordings were made from neurons while depolarizing current pulses, approximately 0.75 s in duration, were applied (on-off step changes indicated by arrows). In the absence of drug, a series of high-frequency repetitive action potentials filled the entire duration of the current pulse. Phenytoin, carbamazepine, and sodium valproate all markedly reduced the number of action potentials elicited by the current pulses. (Adapted, with permission, from Macdonald RL, Meldrum BS: Principles of anti-epileptic drug action. In: Levy RH et al [editors]: Antiepileptic Drugs, 4th ed. Raven Press, 1995.)
In addition, phenytoin paradoxically causes excitation in some cerebral neurons. A reduction of calcium permeability, with inhibition of calcium influx across the cell membrane, may explain the ability of phenytoin to inhibit a variety of calcium-induced secretory processes, including release of hormones and neurotransmitters. Recording of excitatory and inhibitory postsynaptic potentials show that phenytoin decreases the synaptic release of glutamate and enhances the release of GABA. The mechanism of phenytoin’s action probably involves a combination of actions at several levels. At therapeutic concentrations, the major action of phenytoin is to block Na+ channels and inhibit the generation of rapidly repetitive action potentials. Presynaptic actions on glutamate and GABA release probably arise from actions other than those on voltage-gated Na+ channels.
Clinical Uses
Phenytoin is effective against partial seizures and generalized tonic-clonic seizures. In the latter, it appears to be effective against attacks that are either primary or secondary to another seizure type.
Pharmacokinetics
Absorption of phenytoin is highly dependent on the formulation of the dosage form. Particle size and pharmaceutical additives affect both the rate and the extent of absorption. Absorption of phenytoin sodium from the gastrointestinal tract is nearly complete in most patients, although the time to peak may range from 3 to 12 hours. Absorption after intramuscular injection is unpredictable, and some drug precipitation in the muscle occurs; this route of administration is not recommended for phenytoin. In contrast, fosphenytoin, a more soluble phosphate prodrug of phenytoin, is well absorbed after intramuscular administration.
Phenytoin is highly bound to plasma proteins. The total plasma level decreases when the percentage that is bound decreases, as in uremia or hypoalbuminemia, but correlation of free levels with clinical states remains uncertain. Drug concentration in cerebrospinal fluid is proportionate to the free plasma level. Phenytoin accumulates in brain, liver, muscle, and fat.
Phenytoin is metabolized to inactive metabolites that are excreted in the urine. Only a very small proportion of the dose is excreted unchanged.
The elimination of phenytoin is dose-dependent. At very low blood levels, phenytoin metabolism follows first-order kinetics. However, as blood levels rise within the therapeutic range, the maximum capacity of the liver to metabolize phenytoin is approached. Further increases in dosage, though relatively small, may produce very large changes in phenytoin concentrations (Figure 24–5). In such cases, the half-life of the drug increases markedly, steady state is not achieved in routine fashion (since the plasma level continues to rise), and patients quickly develop symptoms of toxicity.
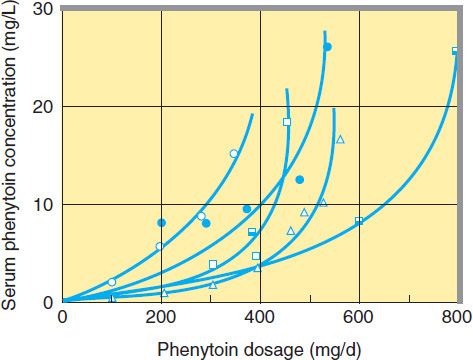
FIGURE 24–5 Nonlinear relationship of phenytoin dosage and plasma concentrations. Five patients (identified by different symbols) received increasing dosages of phenytoin by mouth, and the steady-state serum concentration was measured at each dosage. The curves are not linear since, as the dosage increases, the metabolism is saturable. Note also the marked variation among patients in the serum levels achieved at any dosage. (Adapted, with permission, from Jusko WJ: Bioavailability and disposition kinetics of phenytoin in man. In: Kellaway P, Petersen I [editors]: Quantitative Analytic Studies in Epilepsy. Raven Press, 1977.)
The half-life of phenytoin varies from 12 to 36 hours, with an average of 24 hours for most patients in the low to mid therapeutic range. Much longer half-lives are observed at higher concentrations. At low blood levels, it takes 5–7 days to reach steady-state blood levels after every dosage change; at higher levels, it may be 4–6 weeks before blood levels are stable.
Therapeutic Levels & Dosage
The therapeutic plasma level of phenytoin for most patients is between 10 and 20 mcg/mL. A loading dose can be given either orally or intravenously; the latter, using fosphenytoin, is the method of choice for convulsive status epilepticus (discussed later). When oral therapy is started, it is common to begin adults at a dosage of 300 mg/d, regardless of body weight. This may be acceptable in some patients, but it frequently yields steady-state blood levels below 10 mcg/mL, which is the minimum therapeutic level for most patients. If seizures continue, higher doses are usually necessary to achieve plasma levels in the upper therapeutic range. Because of its dose-dependent kinetics, some toxicity may occur with only small increments in dosage. The phenytoin dosage should be increased each time by only 25–30 mg in adults, and ample time should be allowed for the new steady state to be achieved before further increasing the dosage. A common clinical error is to increase the dosage directly from 300 mg/d to 400 mg/d; toxicity frequently occurs at a variable time thereafter. In children, a dosage of 5 mg/kg/d should be followed by readjustment after steady-state plasma levels are obtained.
Two types of oral phenytoin sodium are currently available in the USA, differing in their respective rates of dissolution; one is absorbed rapidly and one more slowly. Only the slow-release extended-action formulation can be given in a single daily dosage, and care must be used when changing brands (see Preparations Available). Although a few patients being given phenytoin on a long-term basis have been proved to have low blood levels from poor absorption or rapid metabolism, the most common cause of low levels is poor compliance. Fosphenytoin sodium is available for intravenous or intramuscular use and replaces intravenous phenytoin sodium, a much less soluble form of the drug.
Drug Interactions & Interference with Laboratory Tests
Drug interactions involving phenytoin are primarily related to protein binding or to metabolism. Since phenytoin is 90% bound to plasma proteins, other highly bound drugs, such as phenylbutazone and sulfonamides, can displace phenytoin from its binding site. In theory, such displacement may cause a transient increase in free drug. A decrease in protein binding—eg, from hypoalbuminemia—results in a decrease in the total plasma concentration of drug but not the free concentration. Intoxication may occur if efforts are made to maintain total drug levels in the therapeutic range by increasing the dose. The protein binding of phenytoin is decreased in the presence of renal disease. The drug has an affinity for thyroid-binding globulin, which confuses some tests of thyroid function; the most reliable screening test of thyroid function in patients taking phenytoin appears to be measurement of thyroid-stimulating hormone (TSH).
Phenytoin has been shown to induce microsomal enzymes responsible for the metabolism of a number of drugs. Autostimulation of its own metabolism, however, appears to be insignificant.
Toxicity
Dose-related adverse effects caused by phenytoin are often similar to those caused by other antiseizure drugs in this group, making differentiation difficult in patients receiving multiple drugs. Nystagmus occurs early, as does loss of smooth extraocular pursuit movements, but neither is an indication for decreasing the dose. Diplopia and ataxia are the most common dose-related adverse effects requiring dosage adjustment; sedation usually occurs only at considerably higher levels. Gingival hyperplasia and hirsutism occur to some degree in most patients; the latter can be especially unpleasant in women. Long-term use is associated in some patients with coarsening of facial features and with mild peripheral neuropathy, usually manifested by diminished deep tendon reflexes in the lower extremities. Long-term use may also result in abnormalities of vitamin D metabolism, leading to osteomalacia. Low folate levels and megaloblastic anemia have been reported, but the clinical importance of these observations is unknown.
Idiosyncratic reactions to phenytoin are relatively rare. A skin rash may indicate hypersensitivity of the patient to the drug. Fever may also occur, and in rare cases the skin lesions may be severe and exfoliative. Lymphadenopathy may be difficult to distinguish from malignant lymphoma, and although some studies suggest a causal relationship between phenytoin and Hodgkin’s disease, the data are far from conclusive. Hematologic complications are exceedingly rare, although agranulocytosis has been reported in combination with fever and rash.
MEPHENYTOIN, ETHOTOIN, & PHENACEMIDE
Many congeners of phenytoin have been synthesized, but only three have been marketed in the USA, and one of these (phenacemide) has been withdrawn. The other two congeners, mephenytoin and ethotoin, like phenytoin, appear to be most effective against generalized tonic-clonic seizures and partial seizures. No well-controlled clinical trials have documented their effectiveness. The incidence of severe reactions such as dermatitis, agranulocytosis, or hepatitis is higher for mephenytoin than for phenytoin.
Ethotoin may be recommended for patients who are hypersensitive to phenytoin, but larger doses are required. The adverse effects and toxicity are generally less severe than those associated with phenytoin, but the drug appears to be less effective.
Both ethotoin and mephenytoin share with phenytoin the property of saturable metabolism within the therapeutic dosage range. Careful monitoring of the patient during dosage alterations with either drug is essential. Mephenytoin is metabolized to 5,5-ethylphenylhydantoin via demethylation. This metabolite, nirvanol, contributes most of the antiseizure activity of mephenytoin. Both mephenytoin and nirvanol are hydroxylated and undergo subsequent conjugation and excretion. Therapeutic levels for mephenytoin range from 5 to 16 mcg/mL, and levels above 20 mcg/mL are considered toxic.
Therapeutic blood levels of nirvanol are between 25 and 40 mcg/mL. A therapeutic range for ethotoin has not been established.
CARBAMAZEPINE
Closely related to imipramine and other antidepressants, carbamazepine is a tricyclic compound effective in treatment of bipolar depression. It was initially marketed for the treatment of trigeminal neuralgia but has proved useful for epilepsy as well.
Chemistry
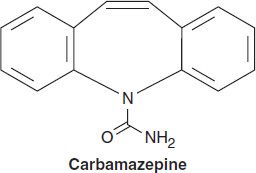
Although not obvious from a two-dimensional representation of its structure, carbamazepine has many similarities to phenytoin. The ureide moiety (–N–CO–NH2) in the heterocyclic ring of most antiseizure drugs is also present in carbamazepine. Three-dimensional structural studies indicate that its spatial conformation is similar to that of phenytoin.
Mechanism of Action
The mechanism of action of carbamazepine appears to be similar to that of phenytoin. Like phenytoin, carbamazepine shows activity against maximal electroshock seizures. Carbamazepine, like phenytoin, blocks Na+ channels at therapeutic concentrations and inhibits high-frequency repetitive firing in neurons in culture (Figure 24–4). It also acts presynaptically to decrease synaptic transmission. Potentiation of a voltage-gated K+ current has also been described. These effects probably account for the anticonvulsant action of carbamazepine. Binding studies show that carbamazepine interacts with adenosine receptors, but the functional significance of this observation is not known.
Clinical Uses
Although carbamazepine has long been considered a drug of choice for both partial seizures and generalized tonic-clonic seizures, some of the newer antiseizure drugs are beginning to displace it from this role. Carbamazepine is not sedative in its usual therapeutic range. The drug is also very effective in some patients with trigeminal neuralgia, although older patients may tolerate higher doses poorly, with ataxia and unsteadiness. Carbamazepine is also useful for controlling mania in some patients with bipolar disorder.
Pharmacokinetics
The rate of absorption of carbamazepine varies widely among patients, although almost complete absorption apparently occurs in all. Peak levels are usually achieved 6–8 hours after administration. Slowing absorption by giving the drug after meals helps the patient tolerate larger total daily doses.
Distribution is slow, and the volume of distribution is roughly 1 L/kg. The drug is approximately 70% bound to plasma proteins; no displacement of other drugs from protein binding sites has been observed.
Carbamazepine has a very low systemic clearance of approximately 1 L/kg/d at the start of therapy. The drug has a notable ability to induce microsomal enzymes. Typically, the half-life of 36 hours observed in subjects after an initial single dose decreases to as little as 8–12 hours in subjects receiving continuous therapy. Considerable dosage adjustments are thus to be expected during the first weeks of therapy. Carbamazepine also alters the clearance of other drugs (see below).
Carbamazepine is completely metabolized in humans to several derivatives. One of these, carbamazepine-10,11-epoxide, has been shown to have anticonvulsant activity. The contribution of this and other metabolites to the clinical activity of carbamazepine is unknown.
Therapeutic Levels & Dosage
Carbamazepine is available only in oral form. The drug is effective in children, in whom a dosage of 15–25 mg/kg/d is appropriate. In adults, daily doses of 1 g or even 2 g are tolerated. Higher dosage is achieved by giving multiple divided doses daily. Extended-release preparations permit twice-daily dosing for most patients. In patients in whom the blood is drawn just before the morning dose (trough level), the therapeutic level is usually 4–8 mcg/mL. Although many patients complain of diplopia at drug levels above 7 mcg/mL, others can tolerate levels above 10 mcg/mL, especially with monotherapy. Extended-release formulations that overcome some of these issues are now available.
Drug Interactions
Drug interactions involving carbamazepine are almost exclusively related to the drug’s enzyme-inducing properties. As noted previously, the increased metabolic capacity of the hepatic enzymes may cause a reduction in steady-state carbamazepine concentrations and an increased rate of metabolism of other drugs, eg, primidone, phenytoin, ethosuximide, valproic acid, and clonazepam. Other drugs such as valproic acid may inhibit carbamazepine clearance and increase steady-state carbamazepine blood levels. Other anticonvulsants, however, such as phenytoin and phenobarbital, may decrease steady-state concentrations of carbamazepine through enzyme induction. No clinically significant protein-binding interactions have been reported.
Toxicity
The most common dose-related adverse effects of carbamazepine are diplopia and ataxia. The diplopia often occurs first and may last less than an hour during a particular time of day. Rearrangement of the divided daily dose can often remedy this complaint. Other dose-related complaints include mild gastrointestinal upsets, unsteadiness, and, at much higher doses, drowsiness. Hyponatremia and water intoxication have occasionally occurred and may be dose related.
Considerable concern exists regarding the occurrence of idiosyncratic blood dyscrasias with carbamazepine, including fatal cases of aplastic anemia and agranulocytosis. Most of these have been in elderly patients with trigeminal neuralgia, and most have occurred within the first 4 months of treatment. The mild and persistent leukopenia seen in some patients is not necessarily an indication to stop treatment but requires careful monitoring. The most common idiosyncratic reaction is an erythematous skin rash; other responses such as hepatic dysfunction are unusual.
OXCARBAZEPINE
Oxcarbazepine is closely related to carbamazepine and is useful in the same seizure types, but it may have an improved toxicity profile. Oxcarbazepine has a half-life of only 1–2 hours. Its activity, therefore, resides almost exclusively in the 10-hydroxy metabolite (especially the S(+) enantiomer, eslicarbazepine), to which it is rapidly converted and which has a half-life similar to that of carbamazepine, ie, 8–12 hours. The drug is mostly excreted as the glucuronide of the 10-hydroxy metabolite.
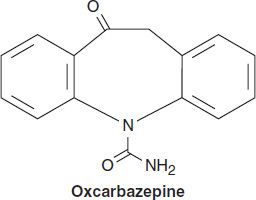
Oxcarbazepine is less potent than carbamazepine, both in animal models of epilepsy and in epileptic patients; clinical doses of oxcarbazepine may need to be 50% higher than those of carbamazepine to obtain equivalent seizure control. Some studies report fewer hypersensitivity reactions to oxcarbazepine, and cross-reactivity with carbamazepine does not always occur. Furthermore, the drug appears to induce hepatic enzymes to a lesser extent than carbamazepine, minimizing drug interactions. Although hyponatremia may occur more commonly with oxcarbazepine than with carbamazepine, most adverse effects that occur with oxcarbazepine are similar in character to reactions reported with carbamazepine.
ESLICARBAZINE
Eslicarbazepine acetate (ESL) is a prodrug that has been approved as adjunctive therapy in adults with partial-onset seizures, with or without secondary generalization. ESL is more rapidly converted to S(+)-licarbazine (eslicarbazine) than is oxcarbazepine; clearly both prodrugs have the same metabolite as active product. The mechanism of action of carbamazepine, oxcarbazepine, and ESL appears to be the same, ie, blocking of voltage-gated Na+ channels. The R(−) enantiomer has some activity, but much less than its counterpart.
Clinically, the drug is similar to carbamazepine and oxcarbazepine in its spectrum of action, but it is less well studied in other possible indications. A possible advantage of ESL is its once-daily dosing regimen. The measured half-life of the S(+) enantiomer is 9–11 hours. The drug is administered at a dosage of 400–1200 mg/d; titration is typically required for the higher doses.
Minimal drug level effects are observed with co-administration of carbamazepine, levetiracetam, lamotrigine, topiramate, and valproate. Oral contraceptives may be less effective with concomitant ESL administration.
PHENOBARBITAL
Aside from the bromides, phenobarbital is the oldest of the currently available antiseizure drugs. Although it has long been considered one of the safest of the antiseizure agents, the use of other medications with lesser sedative effects has been urged. Many consider the barbiturates the drugs of choice for seizures only in infants.
Chemistry
The four derivatives of barbituric acid clinically useful as antiseizure drugs are phenobarbital, mephobarbital, metharbital, and primidone. The first three are so similar that they are considered together. Metharbital is methylated barbital, and mephobarbital is methylated phenobarbital; both are demethylated in vivo. The pKas of these three weak acid compounds range from 7.3 to 7.9. Slight changes in the normal acid-base balance, therefore, can cause significant fluctuation in the ratio of the ionized to the nonionized species. This is particularly important for phenobarbital, the most commonly used barbiturate, whose pKa is similar to the plasma pH of 7.4.
The three-dimensional conformations of the phenobarbital and N-methylphenobarbital molecules are similar to that of phenytoin. Both compounds possess a phenyl ring and are active against partial seizures.
Mechanism of Action
The exact mechanism of action of phenobarbital is unknown, but enhancement of inhibitory processes and diminution of excitatory transmission probably contribute significantly. Recent data indicate that phenobarbital may selectively suppress abnormal neurons. Like phenytoin, phenobarbital suppresses high-frequency repetitive firing in neurons in culture through an action on Na+ conductance, but only at high concentrations. Also at high concentrations, barbiturates block some Ca2+ currents (L-type and N-type). Phenobarbital binds to an allosteric regulatory site on the GABAA receptor, and it enhances the GABA receptor-mediated current by prolonging the openings of the Cl– channels (see Chapter 22). Phenobarbital can also decrease excitatory responses. An effect on glutamate release is probably more significant than blockade of AMPA responses (see Chapter 21). Both the enhancement of GABA-mediated inhibition and the reduction of glutamate-mediated excitation are seen with therapeutically relevant concentrations of phenobarbital.
Clinical Uses
Phenobarbital is useful in the treatment of partial seizures and generalized tonic-clonic seizures, although the drug is often tried for virtually every seizure type, especially when attacks are difficult to control. There is little evidence for its effectiveness in generalized seizures such as absence, atonic attacks, and infantile spasms; it may worsen certain patients with these seizure types.
Some physicians prefer either metharbital (no longer readily available) or mephobarbital (especially the latter) to phenobarbital because of supposed decreased adverse effects. Only anecdotal data are available to support such comparisons.
Pharmacokinetics, Therapeutic Levels, & Dosage
For pharmacokinetics, drug interactions, and toxicity of phenobarbital, see Chapter 22.
The therapeutic levels of phenobarbital in most patients range from 10 mcg/mL to 40 mcg/mL. Documentation of effectiveness is best in febrile seizures, and levels below 15 mcg/mL appear ineffective for prevention of febrile seizure recurrence. The upper end of the therapeutic range is more difficult to define because many patients appear to tolerate chronic levels above 40 mcg/mL.
PRIMIDONE
Primidone, or 2-desoxyphenobarbital (Figure 24–6
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


