47
Antimycobacterial Drugs
CASE STUDY
A 45-year-old homeless man presents to the emergency department complaining of a 2-month history of fatigue, weight loss (10 kg), fevers, night sweats, and a productive cough. He is currently living on the street and has spent time in homeless shelters and prison in the last several years. He reports drinking 2–3 pints of hard alcohol per day for the last 15 years and also reports a history of intravenous drug use. In the emergency department, a chest X-ray shows a right apical infiltrate. Given the high suspicion for pulmonary tuberculosis, the patient is placed in respiratory isolation. His first sputum smear shows many acid-fast bacilli, and a rapid HIV antibody test returns with a positive result. What drugs should be started for treatment of presumptive pulmonary tuberculosis? Does the patient have a heightened risk of developing medication toxicity? If so, which medication(s) would be likely to cause toxicity?
Mycobacteria are intrinsically resistant to most antibiotics. Because they grow more slowly than other bacteria, antibiotics that are most active against rapidly growing cells are relatively ineffective. Mycobacterial cells can also be dormant and thus completely resistant to many drugs or killed only very slowly. The lipid-rich mycobacterial cell wall is impermeable to many agents. Mycobacterial species are intracellular pathogens, and organisms residing within macrophages are inaccessible to drugs that penetrate these cells poorly. Finally, mycobacteria are notorious for their ability to develop resistance. Combinations of two or more drugs are required to overcome these obstacles and to prevent emergence of resistance during the course of therapy. The response of mycobacterial infections to chemotherapy is slow, and treatment must be administered for months to years, depending on which drugs are used. The drugs used to treat tuberculosis, atypical mycobacterial infections, and leprosy are described in this chapter.
 DRUGS USED IN TUBERCULOSIS
DRUGS USED IN TUBERCULOSIS
Isoniazid (INH), rifampin (or other rifamycin), pyrazinamide, ethambutol, and streptomycin are the traditional five first-line agents for treatment of tuberculosis (Table 47–1). Streptomycin is no longer recommended as first-line therapy in most settings. Isoniazid and rifampin are the most active drugs. An isoniazid-rifampin combination administered for 9 months will cure 95–98% of cases of tuberculosis caused by susceptible strains. The addition of pyrazinamide to an isoniazid-rifampin combination for the first 2 months allows the total duration of therapy to be reduced to 6 months without loss of efficacy (Table 47–2). In practice, therapy is usually initiated with a four-drug regimen of isoniazid, rifampin, pyrazinamide, and ethambutol until susceptibility of the clinical isolate has been determined. Neither ethambutol nor other drugs such as streptomycin adds substantially to the overall activity of the regimen (ie, the duration of treatment cannot be further reduced if another drug is used), but the fourth drug provides additional coverage if the isolate proves to be resistant to isoniazid, rifampin, or both. The prevalence of isoniazid resistance among clinical isolates in the United States is approximately 10%. Prevalence of resistance to both isoniazid and rifampin (which is termed multidrug resistance) is about 3%. Resistance to rifampin alone is rare.
TABLE 47–1 Antimicrobials used in the treatment of tuberculosis.
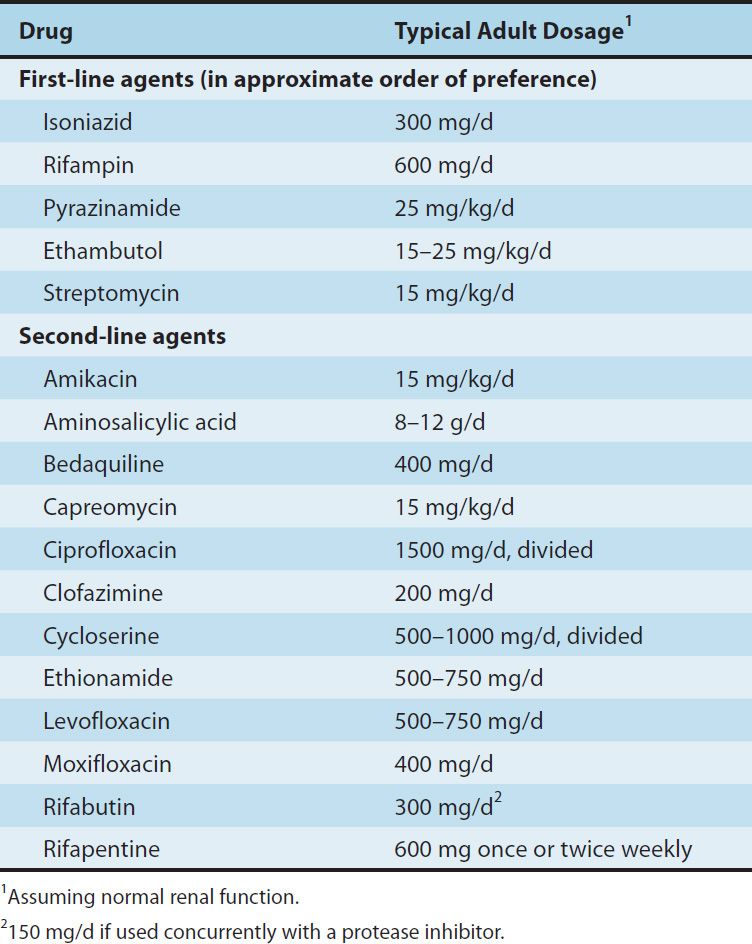
TABLE 47–2 Recommended duration of therapy for tuberculosis.
ISONIAZID
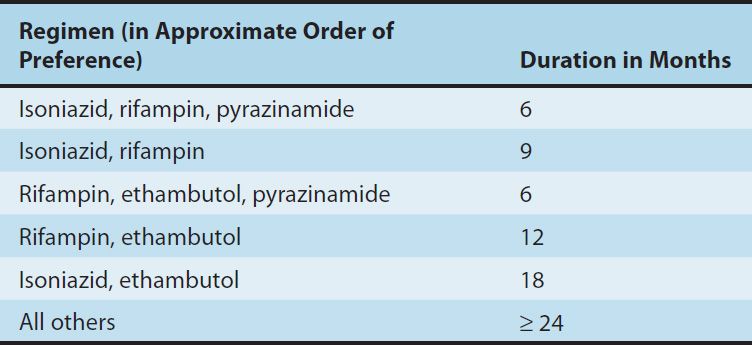
Isoniazid is the most active drug for the treatment of tuberculosis caused by susceptible strains. It is a small molecule (MW 137) that is freely soluble in water. The structural similarity to pyridoxine is shown below.
In vitro, isoniazid inhibits most tubercle bacilli at a concentration of 0.2 mcg/mL or less and is bactericidal for actively growing tubercle bacilli. It is less effective against atypical mycobacterial species. Isoniazid penetrates into macrophages and is active against both extracellular and intracellular organisms.
Mechanism of Action & Basis of Resistance
Isoniazid inhibits synthesis of mycolic acids, which are essential components of mycobacterial cell walls. Isoniazid is a prodrug that is activated by KatG, the mycobacterial catalase-peroxidase. The activated form of isoniazid forms a covalent complex with an acyl carrier protein (AcpM) and KasA, a beta-ketoacyl carrier protein synthetase, which blocks mycolic acid synthesis. Resistance to isoniazid is associated with mutations resulting in overexpression of inhA, which encodes an NADH-dependent acyl carrier protein reductase; mutation or deletion of the katG gene; promoter mutations resulting in overexpression of ahpC, a gene involved in protection of the cell from oxidative stress; and mutations in kasA. Overproducers of inhA express low-level isoniazid resistance and cross-resistance to ethionamide. KatG mutants express high-level isoniazid resistance and often are not cross-resistant to ethionamide.
Drug-resistant mutants are normally present in susceptible mycobacterial populations at about 1 bacillus in 106. Since tuberculous lesions often contain more than 108 tubercle bacilli, resistant mutants are readily selected if isoniazid or any other drug is given as a single agent. The use of two independently acting drugs in combination is much more effective. The probability that a bacillus is initially resistant to both drugs is approximately 1 in 106 × 106, or 1 in 1012, several orders of magnitude greater than the number of infecting organisms. Thus, at least two (or more in certain cases) active agents should always be used to treat active tuberculosis to prevent emergence of resistance during therapy.
Pharmacokinetics
Isoniazid is readily absorbed from the gastrointestinal tract. A 300 mg oral dose (5 mg/kg in children) achieves peak plasma concentrations of 3–5 mcg/mL within 1–2 hours. Isoniazid diffuses readily into all body fluids and tissues. The concentration in the central nervous system and cerebrospinal fluid ranges between 20% and 100% of simultaneous serum concentrations.
Metabolism of isoniazid, especially acetylation by liver N-acetyltransferase, is genetically determined (see Chapter 4). The average plasma concentration of isoniazid in rapid acetylators is about one third to one half of that in slow acetylators, and average half-lives are less than 1 hour and 3 hours, respectively. More rapid clearance of isoniazid by rapid acetylators is usually of no therapeutic consequence when appropriate doses are administered daily, but subtherapeutic concentrations may occur if drug is administered as a once-weekly dose or if there is malabsorption.
Isoniazid metabolites and a small amount of unchanged drug are excreted mainly in the urine. The dosage need not be adjusted in renal failure. Dose adjustment is not well defined in patients with severe preexisting hepatic insufficiency and should be guided by serum concentrations if a reduction in dose is contemplated.
Clinical Uses
The typical dosage of isoniazid is 5 mg/kg/d; a typical adult dose is 300 mg given once daily. Up to 10 mg/kg/d may be used for serious infections or if malabsorption is a problem. A 15 mg/kg dose, or 900 mg, may be used in a twice-weekly dosing regimen in combination with a second antituberculous agent (eg, rifampin, 600 mg). Pyridoxine, 25–50 mg/d, is recommended for those with conditions predisposing to neuropathy, an adverse effect of isoniazid. Isoniazid is usually given by mouth but can be given parenterally in the same dosage.
Isoniazid as a single agent is also indicated for treatment of latent tuberculosis. The dosage is 300 mg/d (5 mg/kg/d) or 900 mg twice weekly, and the duration is usually 9 months.
Adverse Reactions
The incidence and severity of untoward reactions to isoniazid are related to dosage and duration of administration.
A. Immunologic Reactions
Fever and skin rashes are occasionally seen. Drug-induced systemic lupus erythematosus has been reported.
B. Direct Toxicity
Isoniazid-induced hepatitis is the most common major toxic effect. This is distinct from the minor increases in liver aminotransferases (up to three or four times normal), which do not require cessation of the drug and which are seen in 10–20% of patients, who usually are asymptomatic. Clinical hepatitis with loss of appetite, nausea, vomiting, jaundice, and right upper quadrant pain occurs in 1% of isoniazid recipients and can be fatal, particularly if the drug is not discontinued promptly. There is histologic evidence of hepatocellular damage and necrosis. The risk of hepatitis depends on age. It occurs rarely under age 20, in 0.3% of those aged 21–35, 1.2% of those aged 36–50, and 2.3% for those aged 50 and above. The risk of hepatitis is greater in individuals with alcohol dependence and possibly during pregnancy and the postpartum period. Development of isoniazid hepatitis contraindicates further use of the drug.
Peripheral neuropathy is observed in 10–20% of patients given dosages greater than 5 mg/kg/d, but it is infrequently seen with the standard 300 mg adult dose. Peripheral neuropathy is more likely to occur in slow acetylators and patients with predisposing conditions such as malnutrition, alcoholism, diabetes, AIDS, and uremia. Neuropathy is due to a relative pyridoxine deficiency. Isoniazid promotes excretion of pyridoxine, and this toxicity is readily reversed by administration of pyridoxine in a dosage as low as 10 mg/d. Central nervous system toxicity, which is less common, includes memory loss, psychosis, and seizures. These effects may also respond to pyridoxine.
Miscellaneous other reactions include hematologic abnormalities, provocation of pyridoxine deficiency anemia, tinnitus, and gastrointestinal discomfort. Isoniazid can reduce the metabolism of phenytoin, increasing its blood level and toxicity.
RIFAMPIN
Rifampin is a semisynthetic derivative of rifamycin, an antibiotic produced by Streptomyces mediterranei. It is active in vitro against gram-positive and gram-negative cocci, some enteric bacteria, mycobacteria, and chlamydiae. Susceptible organisms are inhibited by less than 1 mcg/mL. Resistant mutants are present in all microbial populations at approximately 1 in 106 organisms and are rapidly selected out if rifampin is used as a single drug, especially in a patient with active infection. There is no cross-resistance to other classes of antimicrobial drugs, but there is cross-resistance to other rifamycin derivatives, eg, rifabutin and rifapentine.
Mechanism of Action, Resistance, & Pharmacokinetics
Rifampin binds to the β subunit of bacterial DNA-dependent RNA polymerase and thereby inhibits RNA synthesis. Resistance results from any one of several possible point mutations in rpoB, the gene for the β subunit of RNA polymerase. These mutations result in reduced binding of rifampin to RNA polymerase. Human RNA polymerase does not bind rifampin and is not inhibited by it. Rifampin is bactericidal for mycobacteria. It readily penetrates most tissues and penetrates into phagocytic cells. It can kill organisms that are poorly accessible to many other drugs, such as intracellular organisms and those sequestered in abscesses and lung cavities.
Rifampin is well absorbed after oral administration and excreted mainly through the liver into bile. It then undergoes enterohepatic recirculation, with the bulk excreted as a deacylated metabolite in feces and a small amount excreted in the urine. Dosage adjustment for renal or hepatic insufficiency is not necessary. Usual doses result in serum levels of 5–7 mcg/mL. Rifampin is distributed widely in body fluids and tissues. The drug is relatively highly protein-bound, and adequate cerebrospinal fluid concentrations are achieved only in the presence of meningeal inflammation.
Clinical Uses
A. Mycobacterial Infections
Rifampin, usually 600 mg/d (10 mg/kg/d) orally, must be administered with isoniazid or other antituberculous drugs to patients with active tuberculosis to prevent emergence of drug-resistant mycobacteria. In some short-course therapies, 600 mg of rifampin is given twice weekly. Rifampin, 600 mg daily or twice weekly for 6 months, also is effective in combination with other agents in some atypical mycobacterial infections and in leprosy. Rifampin, 600 mg daily for 4 months as a single drug, is an alternative to isoniazid for patients with latent tuberculosis who are unable to take isoniazid or who have had exposure to a case of active tuberculosis caused by an isoniazid-resistant, rifampin-susceptible strain.
B. Other Indications
Rifampin has other uses in bacterial infections. An oral dosage of 600 mg twice daily for 2 days can eliminate meningococcal carriage. Rifampin, 20 mg/kg/d for 4 days, is used as prophylaxis in contacts of children with Haemophilus influenzae type b disease. Rifampin combined with a second agent is used to eradicate staphylococcal carriage. Rifampin combination therapy is also indicated for treatment of serious staphylococcal infections such as osteomyelitis and prosthetic valve endocarditis.
Adverse Reactions
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


