Chapter 20
Alterations of Neurologic Function in Children
Lynne M. Kerr and Sue E. Huether

Structure and Function of the Nervous System in Children
Both genetics and environment shape nervous system development. The proper proportions of essential nutrients are necessary for growth and proliferation of the nervous system tissue (see Nutrition & Disease: Iron and Cognitive Function). Maternal lifestyle, nutrition, exposure to potential toxins, and state of health also have a crucial effect on nervous system development at critical periods of fetal maturation.
The central nervous system (CNS) develops from a dorsal thickening of the ectoderm known as the neural plate. This plate appears around the middle of the third gestational week and unfolds to form a neural groove and neural folds. During the fourth gestational week the neural groove deepens, its folds develop laterally, and it closes dorsally to form the neural tube, epithelial tissue that ultimately becomes the CNS. The neural tube closes first in the cervical region and then “zippers” in two directions—cranially and caudally (Figure 20-1).
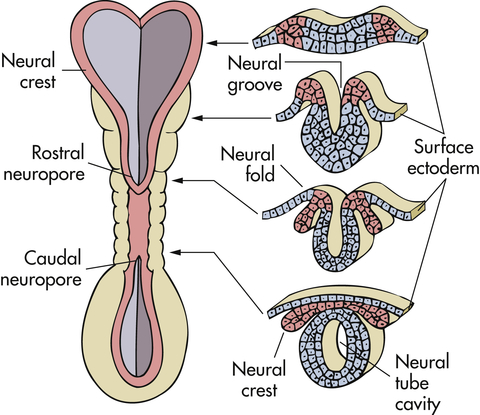
Neural folds have begun to fuse at the cervical level of the future spinal cord. Right, Cross sections of the neural tube at four different levels; at any given level the embryonic central nervous system (CNS) goes through a series of stages resembling these four cross sections. Total length of neural tube at this time is about 2.5 mm.
some peripheral sensory nerve endings, and peripheral nerve coverings.
The cranial end of the neural tube forms the brain, and the remainder develops into the spinal cord. The lumen of the neural tube becomes the ventricles of the brain and the central canal of the spinal cord (Figure 20-2). On either side of the neural tube’s inner surface is a longitudinal groove (sulcus limitans). Anterior to this region (basal plate) the gray matter differentiates into the nuclei of the lower motor neurons. The region posterior to the sulcus (alar plate) differentiates into the sensory nuclei of the spinal cord.
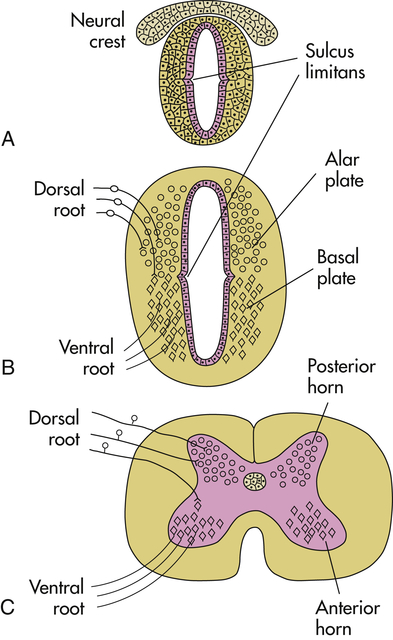
A, Neural tube during the fourth week of gestation. B, Embryonic spinal cord during the sixth week of gestation; dorsal root ganglion cells, derived from the neural crest, send their central processes into the spinal cord to terminate mainly in alar plate cells; basal plate cells become motor neurons, whose axons exit in the ventral roots. C, Adult spinal cord.
Embryonic development of the nervous system occurs in six stages: (1) dorsal (posterior) induction, (2) ventral (anterior) induction, (3) proliferation, (4) migration, (5) organization, and (6) myelination. Figure 20-3 summarizes the embryonic development of the nervous system and identifies disorders associated with interference in any of these stages. Many different events happen simultaneously, and critical periods must pass uninterrupted if the vulnerable fetus is to develop normally.
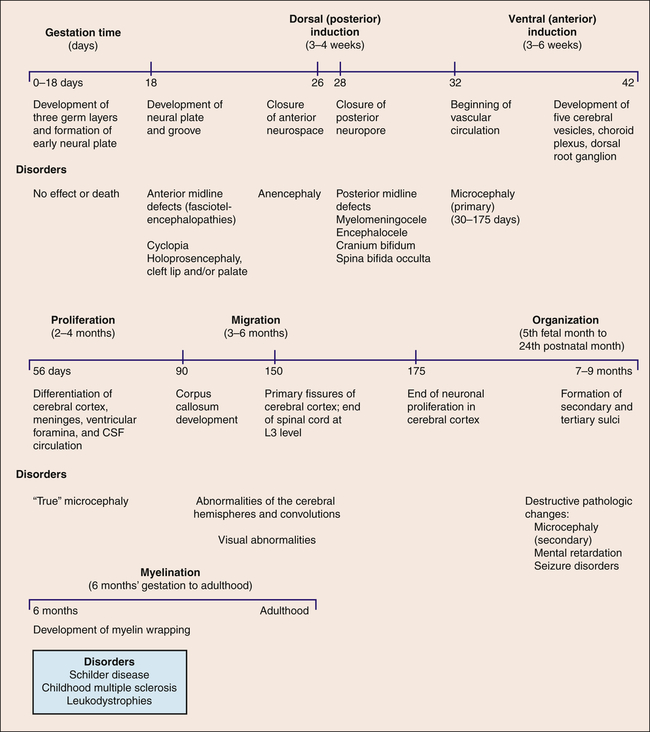
CSF, Cerebrospinal fluid.
Brain Development
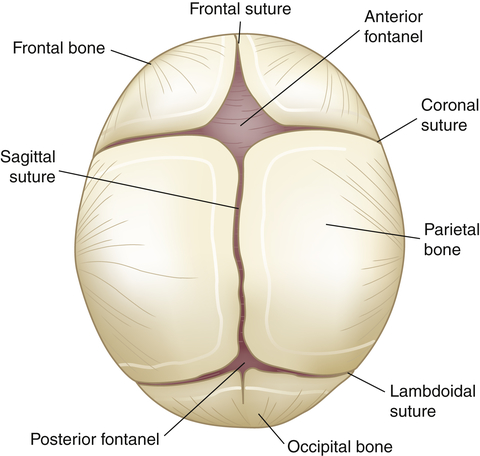
When an infant is born, the bones of the skull are separated at the suture lines, thus forming two fontanels or “soft spots”: one diamond-shaped anterior fontanel and one triangular-shaped posterior fontanel. The posterior fontanel may be open until 2 to 3 months of age; the anterior fontanel normally closes by 18 months (Figure 20-4). The fact that the sutures are not closed allows for increases in head circumference as part of normal growth for 5 to 8 years after birth; in fact, the head is the fastest growing body part during infancy. Although basically all of the neurons that an individual will ever have are present at birth, the development of skills, such as walking, talking, and thinking, depends on these cells making correct connections with other cells and on myelination of the axons making those connections. This leads to brain and then to skull growth. Increased intracranial pressure may result in an increased head circumference in excess of that expected with normal growth. Healthcare providers carefully monitor head growth during the first 5 years of life by measuring head circumference and comparing the results with a standardized growth chart. In contrast, the adult cranium is a closed cavity with sutures firmly holding the cranial bones together. An adult’s head size will not expand regardless of intracranial events, e.g., tumor growth, bleeding, or increased production of cerebrospinal fluid (CSF).
Fibrous union of suture lines and interlocking of serrated edges (occurs by 6 months; solid union requires approximately 12 years).
Most neurons are formed during fetal life but the connections between brain circuits, myelination of those connections, and dendritic development are most significant after birth. Human neurologic functioning is primarily at a subcortical level at birth with many reflex patterns mediated by brainstem and spinal cord mechanisms that disappear at predictable times during infancy while brain axons are myelinated and connecting pathways develop. Myelination is a process by which lipid-protein material made by specialized connective tissue cells (Schwann cells in the peripheral nervous system and oligodendrocytes in the central nervous system) covers the axons of neurons, allowing electrical signals to travel more quickly (see Chapter 16). The development of myelin in the brain follows predictable patterns over time and is most extensive after birth and throughout childhood. The primitive reflex pathways require myelination and by 6 months of age they are integrated and inhibited for progression to voluntary movement. Table 20-1 summarizes the ages at which primitive reflexes appear and disappear. Testing for the presence or absence of these reflexes at various times enables the examiner to evaluate the function and development of the nervous system.
TABLE 20-1
| REFLEX | AGE AT APPEARANCE OF REFLEX | AGE AT WHICH REFLEX SHOULD NO LONGER BE OBTAINABLE |
| Stepping | Birth | 6 weeks |
| Moro | Birth | 3 months |
| Sucking | Birth | 4 months awake |
| 7 months asleep | ||
| Rooting | Birth | 4 months awake |
| 7 months asleep | ||
| Palmar grasp | Birth | 6 months |
| Plantar grasp | Birth | 10 months |
| Tonic neck | 2 months | 5 months |
| Neck righting | 4-6 months | 24 months |
| Landau | 3 months | 24 months |
| Parachute reaction | 9 months | Persists for life |
Structural Malformations
Defects of Neural Tube Closure
Neural tube defects (NTDs) are caused by an arrest of the normal development of the brain and spinal cord during the first month of embryonic development and occur in about 3000 pregnancies in the United States each year.1 There is a strong association of fetal death with NTDs, reducing the actual prevalence of neural defects at birth. Maternal folate deficiency is associated with NTDs. The specific mechanism that relates to how folate supplements prevent these anomalies is unknown but genes involving folate metabolism and environmental factors are involved.2 In 1996 the United States mandated folate fortification in many foods and since that time neural tube defects have decreased by 20% to 30%3 (see What’s New? Reduction of Risks for Neural Tube Defects [NTDs]).
Anencephaly
Anencephaly is an anomaly in which the soft, bony component of the skull and much of the brain are missing. This is a relatively common disorder, with an incidence rate of approximately 1 per 4859 live births in the United States each year.4 These infants are stillborn or die within a few days after birth. The pathologic mechanism is unknown. Diagnosis is often made prenatally using ultrasound or evaluating maternal serum alpha fetoprotein (AFP).5
Encephalocele
Encephalocele refers to a herniation or protrusion of various amounts of brain and meninges through a defect in the skull, resulting in a saclike structure. The incidence rate is approximately 1 in 10,000 live births in the United States each year.6
Clinical Manifestations
Encephalocele usually is seen at birth as a midline skull defect through which a large mass protrudes (Figure 20-5). If the defect is located in the nasopharynx, no external anomaly is visible, but the child may experience nasal airway obstruction. On examination with a nasal speculum, a smooth, round mass will be visible in the nasal passages. A frontal encephalocele may extend into the orbit of the eye and produce proptosis on the affected side.
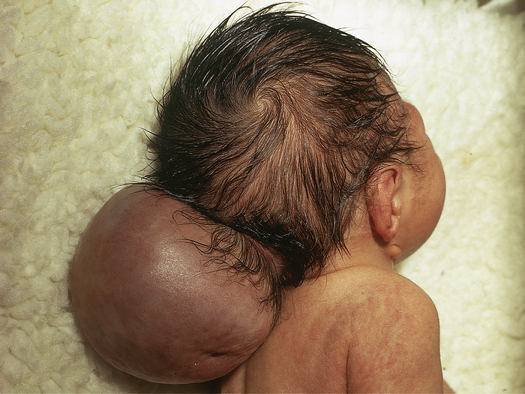
Meningocele and Myelomeningocele
Meningocele and myelomeningocele are different types of spina bifida that occur because of an incompletely formed or absent posterior vertebral arch in the spinal column, allowing protrusion of either a saclike cyst of meninges filled with spinal fluid (a meningocele) or a similar saclike cyst that also has neural tissue, spinal cord, or nerves in it, in which case it is a myelomeningocele. Both develop during the first 4 weeks of pregnancy when the neural tube fails to close completely (Figure 20-6). Meningoceles occur with equal frequency in the cervical, thoracic, and lumbar spine areas, whereas 80% of myelomeningoceles are located in the lumbar and lumbosacral regions, the last regions of the neural tube to close. Myelomeningocele is much more common than meningocele and has an incidence rate ranging from 0.5 to 1 per 1000 pregnancies in the United States.7 It is thus is one of the most common developmental anomalies of the nervous system.
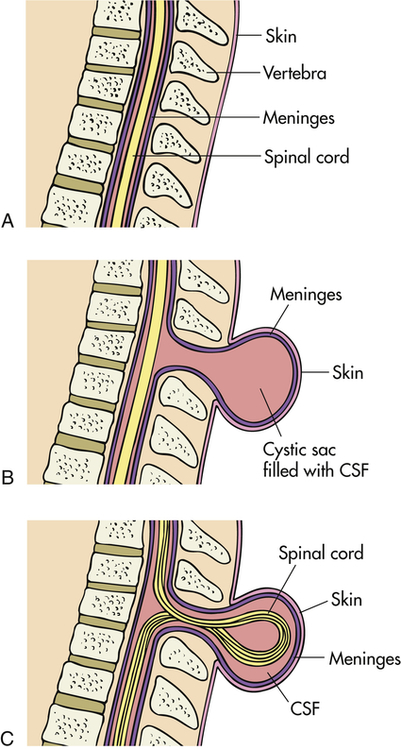
Diagram depicts section through normal spine (A), spine with meningocele (B), and spine with myelomeningocele (C).
Pathophysiology
Because of changes in cerebrospinal fluid (CSF) flow, myelomeningoceles also are likely to be associated with a Chiari II malformation, which is a downward displacement of the cerebellum, brainstem, and fourth ventricle (Figure 20-7). The Chiari II malformation compresses and essentially stretches the posterior region of the cerebellum and brainstem downward through the foramen magnum. The brainstem houses cranial nerve nuclei and pressure on this region may result in altered function of these nerves or actual palsies. The Chiari II malformation is associated with hydrocephalus, an increase in intracranial pressure attributable to obstruction of the flow of CSF; syringomyelia, an abnormality causing cysts at multiple levels within the spinal cord; and cognitive and motor deficits.8
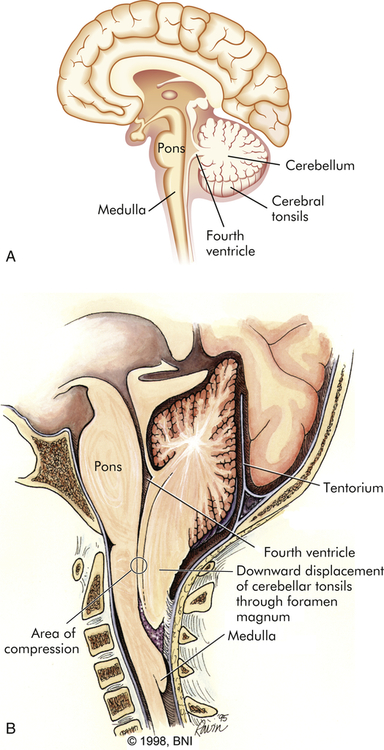
A, Diagram of normal brain. B, Diagram of Chiari II malformation with downward displacement of cerebellar tonsils and medulla through foramen magnum causing compression and obstruction to flow of CSF. (B modified from Barrow Neurological Institute of St. Joseph’s Hospital and Medical Center, Phoenix, Arizona. Reprinted with permission.)
Other forms of Chiari malformation include type I, which is usually asymptomatic; type III, in which the brainstem or cerebellum extends into a high cervical myelomeningocele; and type IV, which is characterized by lack of cerebellar development.9
Seizures are present in about 30% of children with myelomeningocele,10 and visual problems (perceptual and astigmatism) and intellectual deficits (ranging from learning problems to intellectual disability) also are common.11
Clinical Manifestations
Myeloceles and myelomeningoceles are evident at birth as a pronounced skin defect on the infant’s back (see Figure 20-6). The severity of the clinical presentation will depend on the amount of tissue involved (generally the more neural tissue that is included in the sac, the worse the clinical presentation; some children with meningocele will not have any functional problems) and the level of the defect (thoracic vs. sacral; a sacral defect will cause fewer problems than a defect higher in the neural axis). The bony prominences of the unfused neural arches can be palpated at the lateral border of the defect. The defect is usually covered by a transparent membrane that may have neural tissue attached to its inner surface. This membrane may be intact at birth or may leak CSF, thereby increasing the risks of infection and neuronal damage. Until the defect is closed surgically, CSF may accumulate and cause increased dilation and enlargement of the sac, which may further endanger the nervous system.
Tethered cord syndrome may develop in children with myelomeningocele.12 The cord becomes abnormally attached or tethered as a result of scar tissue that develops as the cord transcends the vertebral canal with growth. Traction decreases blood flow and impairs oxidative metabolism. Symptoms are related to excessive tension on the lumbosacral cord and can include scoliosis, altered gait pattern, changes in muscle strength at or below the lesion, disturbance in urinary and bowel patterns, and back pain. The cord can be untethered surgically.12
It is possible for a defect to occur without any visible exposure of meninges or neural tissue. There is a cleft in the spine because the posterior vertebral laminae have failed to fuse and the overlying skin is intact. Because the defect is not apparent to the unaided eye (i.e., it is “occult” or hidden), the term spina bifida occulta is used.13 This extremely common defect occurs to some degree in 10% to 25% of infants. Approximately 80% of these vertebral defects are located in the lumbosacral regions, most commonly in the fifth lumbar vertebra and the first sacral vertebra. Spina bifida occulta usually causes no serious neurologic dysfunctions because the spinal cord and spinal nerves are generally anatomically and functionally normal, but the spinal defect can cause hydrocephalus.14
Evaluation and Treatment
Diagnosis is based on clinical manifestations and examination of the meningeal sac. Prenatal diagnosis of myelomeningocele is possible through ultrasonography. In the United States, most pregnant women receive triple or quad screening between 15 and 20 weeks of a pregnancy. The presence of a neural tube defect may result in an elevated amniotic fluid AFP level and subsequent maternal serum AFP levels. Additional testing is then performed if screening tests are positive, including a detailed ultrasound, which may allow for prenatal diagnosis of spina bifida. Prenatal diagnosis offers the parents the option to terminate the pregnancy or become a candidate for fetal intrauterine repair.15 Cesarean delivery may be recommended to minimize trauma to the open myelomeningocele.16
In an effort to preserve neuronal function and minimize damage that may occur from infection and manipulation of the fragile sac, surgical closure is optimal prenatally or during the first 72 hours of life, as recommended by the management of Myelomeningocele Study (MOMS).17 Functional implications depend on the level and severity of the defect (Table 20-2) and the presence of associated conditions, such as abnormal renal structure or function, or both, and the presence of a Chiari II malformation (see p. 665). These conditions are screened at birth in any child born with myelomeningocele or meningocele.
TABLE 20-2
FUNCTIONAL ALTERATIONS IN MYELODYSPLASIA RELATED TO LEVEL OF LESION
| LEVEL OF LESION | FUNCTIONAL IMPLICATIONS |
| Thoracic | Flaccid paralysis of lower extremities; variable weakness in abdominal trunk musculature; high thoracic level may mean respiratory compromise; absence of bowel and bladder control |
| High lumbar | Voluntary hip flexion and adduction; flaccid paralysis of knees, ankles, and feet; may walk with extensive braces and crutches; absence of bowel and bladder control |
| Midlumbar | Strong hip flexion and adduction; fair knee extension; flaccid paralysis of ankles and feet; absence of bowel and bladder control |
| Low lumbar | Strong hip flexion, extension, and adduction and knee extension; weak ankle and toe mobility; may have limited bowel and bladder function |
| Sacral | Normal function of lower extremities; normal bowel and bladder function |
Data from Farley JA, Dunleavy MJ: Myelodysplasia. In Allen PJ, Vessey JA, editors: Primary care of the child with a chronic condition, ed 4, St Louis, 2004, Mosby.
Craniosynostosis
Craniosynostosis (craniostenosis) is the premature closure of one or more of the cranial sutures during the first 18 to 20 months of an infant’s life. The incidence of craniosynostosis is 1 in 1800 to 2200 live births.18 Males are affected twice as often as females. Craniosynostosis can occur as part of a syndrome or, more commonly, as an isolated defect.19 Nonsyndromic craniosynostosis is classified as simple craniosynostosis when only one suture is involved and compound or complex craniosynostosis when two or more sutures are involved. Craniosynostosis prevents normal skull expansion and causes asymmetric skull growth. Premature closure of a suture causes failure of the growth of the bone located at a right angle to the involved suture. Compensatory growth occurs in regions where the sutures are patent, and this causes the various cosmetic deformities of head shape. Where sutures are fused, cerebral growth may exceed the space present. Brain growth may be restricted, and compression may cause neurologic symptoms (Figure 20-8). Clinically, however, most children who present with small heads and developmental delay have microcephaly (see below) caused by defects in brain growth, not compression by prematurely closed sutures.
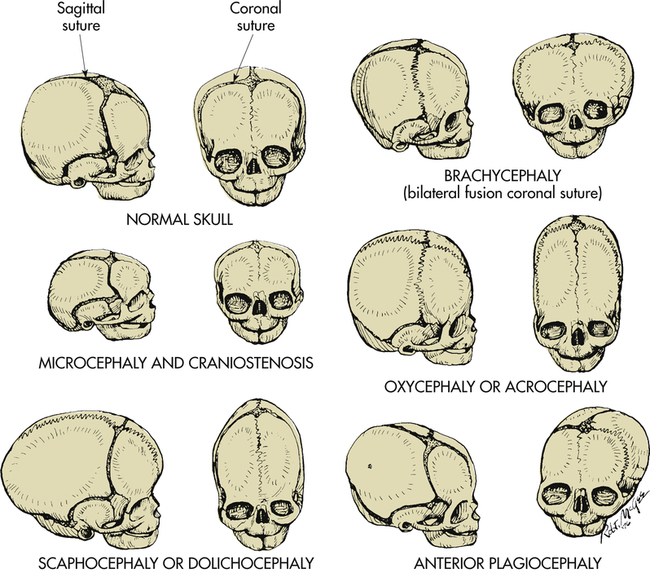
Abnormal head configuration resulting from premature closing of cranial sutures. Normal skull: Bones separated by membranous seams until sutures gradually close. Microcephaly and craniostenosis: Microcephaly is head circumference more than 2 standard deviations below the mean for age, gender, race, and gestation and reflects a small brain; craniosynostosis is premature closure of sutures. Scaphocephaly or dolichocephaly (frequency 56%): Premature closure of sagittal suture, resulting in restricted lateral growth. Brachycephaly: Premature closure of coronal suture, resulting in excessive lateral growth. Oxycephaly or acrocephaly (frequency 5.8% to 12%): Premature closure of all coronal and sagittal sutures, resulting in accelerated upward growth and small head circumference. Anterior plagiocephaly (frequency 13%): Unilateral premature closure of coronal suture, resulting in asymmetric growth. (From Hockenberry JH et al: Wong’s nursing care of infants and children, ed 9, St Louis, 2011, Mosby.)
Clinical Manifestations
Craniosynostosis is classified according to head contour or suture involvement. Final skull contour is determined by the sutures that close, the duration and order of closure, and the ability of other sutures to compensate by expansion (see Figures 20-8 and 20-9).
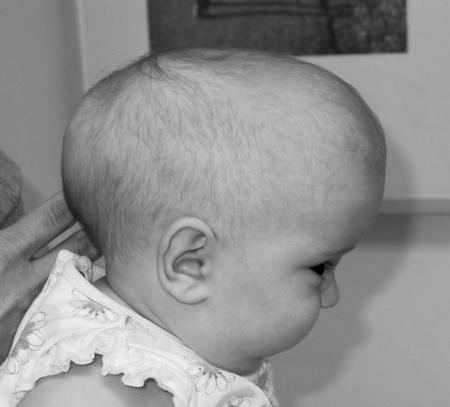
Note the frontal bossing, elongation along the anteroposterior (AP) axis, and prominent occiput, all of which are characteristic of this condition. (From Coran A et al: Pediatric surgery, ed 7, St Louis, 2012, Mosby.)
Evaluation and Treatment
Diagnosis of craniosynostosis is made on the basis of physical examination, head circumference measurements, and radiologic examination. Surgical treatment is often indicated for craniosynostosis and is performed when closure of multiple sutures causes chronic increased intracranial pressure. Surgery then limits the extent of brain damage. In children with craniosynostosis of one suture, surgery often is performed for cosmetic purposes.21
Malformations of Cortical Development
Structural malformations of cortical development are a diverse, heterogeneous group of disorders caused by abnormal neuronal cell migration, abnormal neuronal and glial proliferation, and malformations secondary to abnormal postmigrational neuronal development. The processes of malformation can occur at any time during fetal development and after birth. There is a specific genetic defect for some of these disorders; others are multifactorial or acquired (i.e., intrauterine trauma or infection). Most increase the risk for seizures that are difficult to control, developmental delay, and motor dysfunction. Diagnosis is made by magnetic resonance imaging (MRI), family history, and clinical information. Genetic testing is done to assess risk in other family members and to guide prospective therapy.22–25
Microcephaly
Reduced proliferation or accelerated apoptosis causes congenital microcephaly (microencephaly—small brain), whereas increased proliferation causes megalencephaly (abnormally large brain). Microcephaly is a rare defect in brain growth as a whole (see Figure 20-8). The word microcephaly is derived from the Greek (mikro, “small”; kephale, “head”). Cranial size is significantly below average for the infant’s age, sex, race, and gestation. The small size of the skull reflects a small brain (microencephaly) that is caused by reduced proliferation or accelerated apoptosis. The condition is not treatable.
True (primary) microcephaly is present at birth and can be caused by genetic alterations, including autosomal dominant, autosomal recessive, or X-linked genetic abnormalities, or various chromosomal abnormalities. Environmental causes include toxin or radiation exposure or intrauterine infection during the period of induction and neural cell migration (Box 20-1). Secondary microcephaly develops postnatally (the baby’s head size is normal at birth but then fails to grow as expected) and is associated with a variety of causes including infection, trauma, metabolic disorders, maternal anorexia experienced during the third trimester of pregnancy, and the presence of some genetic syndromes (e.g., Rett syndrome).26
Cortical Dysplasias
Cortical dysplasias are a heterogeneous group of disorders caused by defects in neuronal cell migration and subsequent abnormalities in connections between cells. These disorders may range from a small area of abnormal tissue (e.g., heterotopia, which are pieces of gray matter that did not migrate to their normal position in the cortex of the brain; or focal cortical dysplasias, where brain organization in one small area is abnormal) to an entire brain that is smooth without the normal configuration of gyri and sulci of a developed brain (lissencephaly) (Figure 20-10).
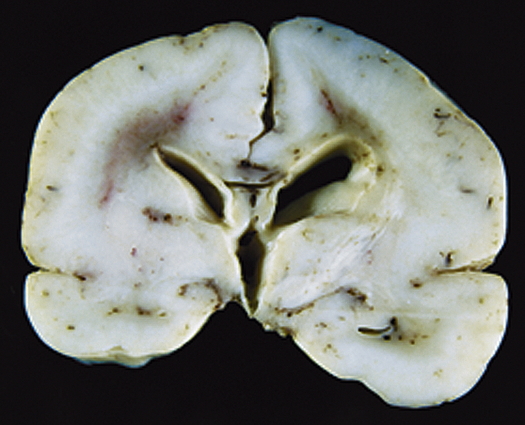
Congenital Hydrocephalus
Congenital hydrocephalus is present at birth and characterized by increased intracranial pressure (ICP). This increase may be caused by a blockage within the ventricular system in which the CSF flows, an imbalance in the production of CSF, or a reduced reabsorption of CSF that results in ventricular enlargement and increased ICP. The pressure within the ventricular system dilates the ventricles and pushes and compresses the brain tissue against the skull. When hydrocephalus develops before fusion of the cranial sutures, the skull has the capacity to increase its size to accommodate this additional space-occupying volume and to preserve neuronal function. The overall incidence of hydrocephalus is approximately 1 to 3 per 1000 live births.27 The incidence of hydrocephalus, excluding the hydrocephalus associated with myelomeningocele, is approximately 0.5 to 1 per 1000 live births, with aqueductal stenosis as the cause for approximately one third of these cases.28 (Types of hydrocephalus are discussed in Chapter 17.)
Clinical Manifestations
Congenital hydrocephalus is present at birth, whereas acquired hydrocephalus usually becomes apparent in the first weeks to months of life. Symptoms of this condition depend directly on the cause and rate of hydrocephalus development. Typically, the fontanels enlarge and become full and bulging (Figures 20-11 and 20-12). The eyes may assume a staring expression, with sclera visible above the iris, a finding called sunsetting eyes.
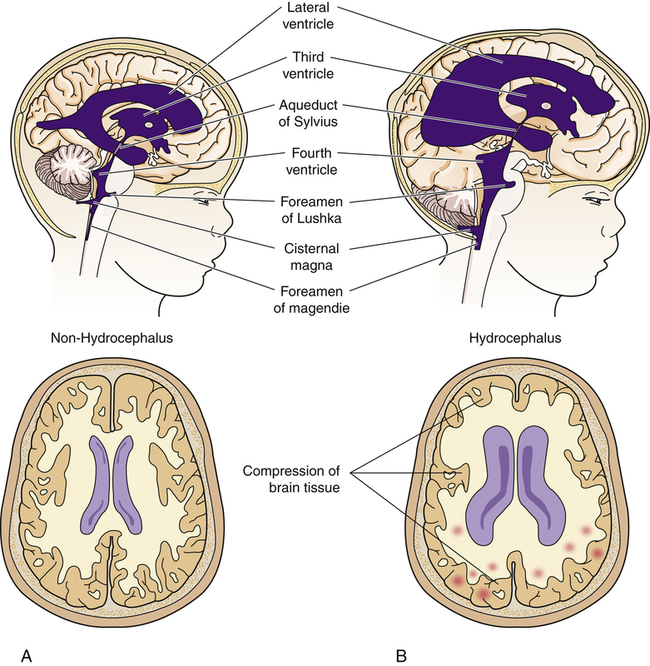
A block in the flow of cerebrospinal fluid (CSF). A, Patent cerebrospinal fluid circulation. B, Enlarged lateral and third ventricles caused by obstruction of circulation—stenosis of aqueduct of Sylvius.

The infant may have difficulty holding the head upright. The scalp skin is thin and shiny, and scalp veins may become prominent. The large cranial vault and the face are disproportionate, and frontal bossing may be present. The infant’s cry becomes high pitched as ICP rises; irritability, lethargy, vomiting, and other signs of increased ICP may develop. Dramatic head growth and enlargement, compression of the optic nerves, and optic chiasm occur in chronic, untreated hydrocephalus. However, because of early surgical intervention, these signs of hydrocephalus rarely are seen in developed countries. When hydrocephalus develops late in childhood, the head may not have the capacity to enlarge, and symptoms of increased ICP occur quickly (see Chapter 17).
The relationship between hydrocephalus and intellectual disability has been heavily debated. Correlation between the degree of hydrocephalus and impaired cognitive function often is a result of additional complications, such as severe congenital malformations, acute or chronic infections, or progressive brain tumors. Approximately two thirds of children with uncomplicated congenital hydrocephalus treated successfully with shunting may have normal to borderline intelligence.29
Evaluation and Treatment
Both cranial ultrasound and computed tomography (CT) are used to evaluate hydrocephalus. Magnetic resonance imaging (MRI) may add information about the specific cause of the hydrocephalus. In infancy, head circumference measurements are obtained and monitored, and if the head is enlarging too quickly the child is referred for imaging first and then, if needed, to neurosurgery for further evaluation. The treatment is surgical placement of a shunt to divert the excess CSF from the ventricular cavity to other areas of the body. Several types of shunts are available. The main objective of any shunt system is to decrease ICP and preserve neuronal function. With neurosurgical intervention and follow-up, the 5-year survival usually is greater than 80%. Most deaths that occur within this category result from severe congenital malformations and/or progressive disorders such as brain tumors. Children with hydrocephalus depend on the internal shunt system to maintain safe ICPs and therefore are at risk for sudden failure of this system, which leads to acute increased ICP and death. Endoscopic ventriculostomy with choroid plexus cauterization are two newer procedures performed together and offer hope to replace shunts, and avoiding shunt blocks, malfunction, and infection.29a
Alterations in Function: Encephalopathies
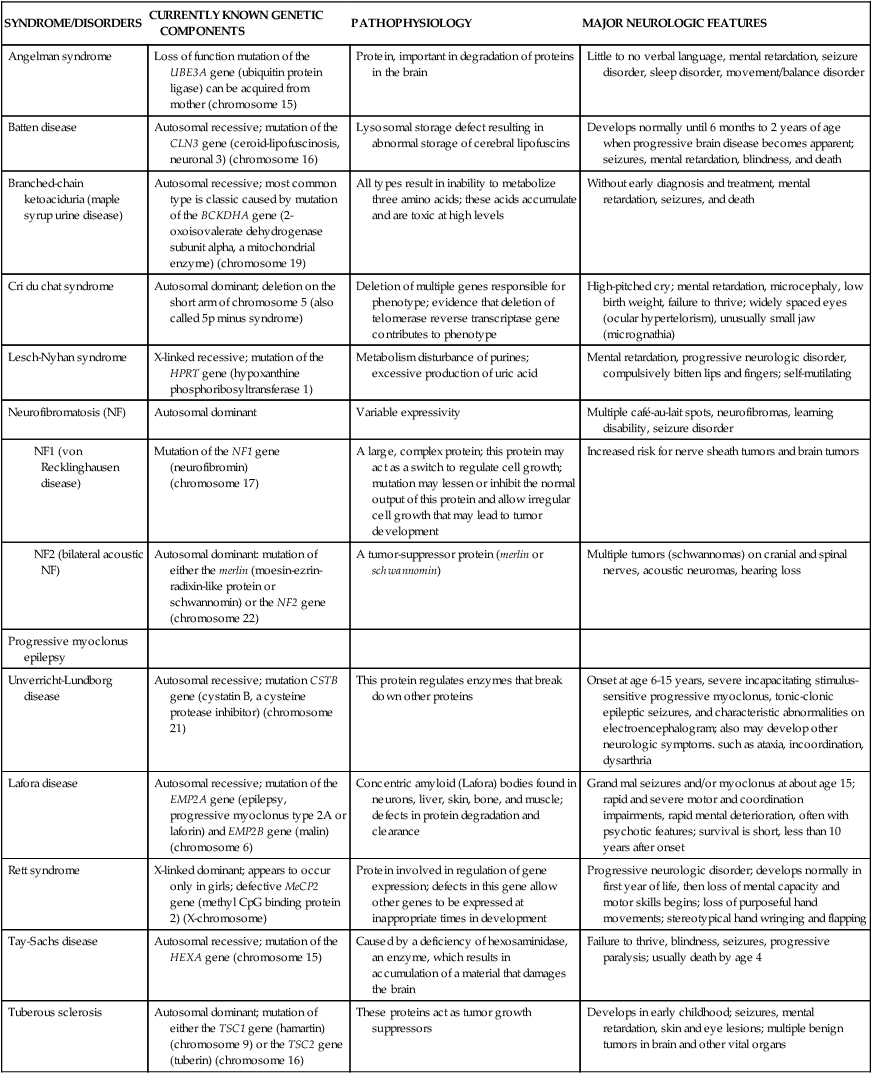
Encephalopathy, a pathology involving the brain, is a general category of syndromes and diseases that include infections, ischemia, metabolic and toxic causes, and others. Encephalopathies in children are associated with a great variety of known and suspected causes. They can be divided into non-progressive and progressive subtypes. Progressive subtypes can be either acute or slowly progressive. Rare neurologic disorders with a genetic basis are summarized in Table 20-3.
TABLE 20-3
SELECTED NEUROLOGIC DISORDERS WITH A GENETIC BASIS
| SYNDROME/DISORDERS | CURRENTLY KNOWN GENETIC COMPONENTS | PATHOPHYSIOLOGY | MAJOR NEUROLOGIC FEATURES |
| Angelman syndrome | Loss of function mutation of the UBE3A gene (ubiquitin protein ligase) can be acquired from mother (chromosome 15) | Protein, important in degradation of proteins in the brain | Little to no verbal language, mental retardation, seizure disorder, sleep disorder, movement/balance disorder |
| Batten disease | Autosomal recessive; mutation of the CLN3 gene (ceroid-lipofuscinosis, neuronal 3) (chromosome 16) | Lysosomal storage defect resulting in abnormal storage of cerebral lipofuscins | Develops normally until 6 months to 2 years of age when progressive brain disease becomes apparent; seizures, mental retardation, blindness, and death |
| Branched-chain ketoaciduria (maple syrup urine disease) | Autosomal recessive; most common type is classic caused by mutation of the BCKDHA gene (2-oxoisovalerate dehydrogenase subunit alpha, a mitochondrial enzyme) (chromosome 19) | All types result in inability to metabolize three amino acids; these acids accumulate and are toxic at high levels | Without early diagnosis and treatment, mental retardation, seizures, and death |
| Cri du chat syndrome | Autosomal dominant; deletion on the short arm of chromosome 5 (also called 5p minus syndrome) | Deletion of multiple genes responsible for phenotype; evidence that deletion of telomerase reverse transcriptase gene contributes to phenotype | High-pitched cry; mental retardation, microcephaly, low birth weight, failure to thrive; widely spaced eyes (ocular hypertelorism), unusually small jaw (micrognathia) |
| Lesch-Nyhan syndrome | X-linked recessive; mutation of the HPRT gene (hypoxanthine phosphoribosyltransferase 1) | Metabolism disturbance of purines; excessive production of uric acid | Mental retardation, progressive neurologic disorder, compulsively bitten lips and fingers; self-mutilating |
| Neurofibromatosis (NF) | Autosomal dominant | Variable expressivity | Multiple café-au-lait spots, neurofibromas, learning disability, seizure disorder |
| Mutation of the NF1 gene (neurofibromin) (chromosome 17) | A large, complex protein; this protein may act as a switch to regulate cell growth; mutation may lessen or inhibit the normal output of this protein and allow irregular cell growth that may lead to tumor development | Increased risk for nerve sheath tumors and brain tumors | |
| Autosomal dominant: mutation of either the merlin (moesin-ezrin-radixin-like protein or schwannomin) or the NF2 gene (chromosome 22) | A tumor-suppressor protein (merlin or schwannomin) | Multiple tumors (schwannomas) on cranial and spinal nerves, acoustic neuromas, hearing loss | |
| Progressive myoclonus epilepsy | |||
| Unverricht-Lundborg disease | Autosomal recessive; mutation CSTB gene (cystatin B, a cysteine protease inhibitor) (chromosome 21) | This protein regulates enzymes that break down other proteins | Onset at age 6-15 years, severe incapacitating stimulus-sensitive progressive myoclonus, tonic-clonic epileptic seizures, and characteristic abnormalities on electroencephalogram; also may develop other neurologic symptoms. such as ataxia, incoordination, dysarthria |
| Lafora disease | Autosomal recessive; mutation of the EMP2A gene (epilepsy, progressive myoclonus type 2A or laforin) and EMP2B gene (malin) (chromosome 6) | Concentric amyloid (Lafora) bodies found in neurons, liver, skin, bone, and muscle; defects in protein degradation and clearance | Grand mal seizures and/or myoclonus at about age 15; rapid and severe motor and coordination impairments, rapid mental deterioration, often with psychotic features; survival is short, less than 10 years after onset |
| Rett syndrome | X-linked dominant; appears to occur only in girls; defective MeCP2 gene (methyl CpG binding protein 2) (X-chromosome) | Protein involved in regulation of gene expression; defects in this gene allow other genes to be expressed at inappropriate times in development | Progressive neurologic disorder; develops normally in first year of life, then loss of mental capacity and motor skills begins; loss of purposeful hand movements; stereotypical hand wringing and flapping |
| Tay-Sachs disease | Autosomal recessive; mutation of the HEXA gene (chromosome 15) | Caused by a deficiency of hexosaminidase, an enzyme, which results in accumulation of a material that damages the brain | Failure to thrive, blindness, seizures, progressive paralysis; usually death by age 4 |
| Tuberous sclerosis | Autosomal dominant; mutation of either the TSC1 gene (hamartin) (chromosome 9) or the TSC2 gene (tuberin) (chromosome 16) | These proteins act as tumor growth suppressors | Develops in early childhood; seizures, mental retardation, skin and eye lesions; multiple benign tumors in brain and other vital organs |
Static Encephalopathies
Cerebral Palsy
Cerebral palsy (CP) is a disorder of movement, muscle tone, or posture that is caused by injury or abnormal development in the immature brain, before, during, or after birth up to 1 year of age. CP is one of the most common crippling disorders of childhood, affecting nearly 500,000 children in the United States alone. Although the exact incidence is unknown, studies suggest that the prevalence is approximately 3.3 per 1000 live births in the United States.30 Although the term cerebral palsy refers specifically to the motor deficits, associated intellectual disability, seizures, and other problems also are common in children with this disorder.
Pathophysiology
Several factors, alone or in combination, can produce brain damage that leads to CP (Table 20-4). Prenatal factors that affect the developing nervous system may be endogenous or exogenous. The fetus may be affected by impaired embryo implantation, chromosomal abnormalities, infection, trauma, radiation exposure, and toxic substances. Maternal toxemia, diabetes mellitus, and maternal nutritional deficiencies can produce neurologic damage in the fetus. Anoxia, trauma, and infections are the most common factors that cause injury to the nervous system in the perinatal period. Low birth weight and birth asphyxia are commonly identified risk factors for cerebral palsy. Vascular abnormalities, arterial or venous stasis, and thrombosis can occur as a result of tissue hypoxia or as unrelated structural alterations. These anomalies may result in direct brain trauma that leads to infarction, intraventricular hemorrhage, and subarachnoid hemorrhage. Physical trauma to the central nervous system can occur during the birthing process. Genetic, teratogenic, and early pregnancy influences on the development of cerebral palsy are multifactorial and not yet fully understood.31,32 The severity of the damage depends on the gestational age at the time of the injury and the type and degree of injury sustained. Magnesium sulfate in preterm newborns and head cooling during or after resuscitation at birth may reduce brain damage and risk of CP.33,34
TABLE 20-4
CEREBRAL PALSY: PREDISPOSING FACTORS AND KNOWN CAUSES
| RISK FACTORS | ASSOCIATED CAUSES |
| Prenatal | |
| Maternal | Metabolic diseases |
| Nutritional deficiencies (e.g., anemia) | |
| Twin or multiple births | |
| Bleeding | |
| Toxemia | |
| Blood incompatibilities | |
| Exposure to radiation | |
| Infection (e.g., rubella, toxoplasmosis, cytomegalic inclusion disease) | |
| Premature labor | |
| Prematurity | Asphyxia leading to cerebral hemorrhage |
| Genetic factors | Absence of corpus callosum, aqueductal stenosis, cerebellar hypoplasia |
| Congenital anomalies of the brain | Unknown causes not evident on clinical examination |
| Perinatal | Anesthesia or analgesia during labor and delivery |
| Mechanical trauma during delivery | |
| Immaturity at birth | |
| Metabolic disorders (e.g., hyperbilirubinemia, hypoglycemia, amino acid disorders, hyperosmolality) | |
| Electrolyte disturbances (e.g., hypernatremia, hypoglycemia) | |
| Postnatal | Head trauma |
| Infections (e.g., meningitis, encephalitis) | |
| Cerebrovascular accidents | |
| Toxicosis | |
| Environmental toxins (e.g., lead ingestion, methyl mercury ingestion from contaminated fish) | |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


