Chapter 29
Alterations of Leukocyte, Lymphoid, and Hemostatic Function
Anna Schwartz and Neal S. Rote

Alterations of Leukocyte Function
Qualitative leukocyte disorders consist of disruptions of leukocyte function. Phagocytic cells (granulocytes, monocytes, macrophages) may lose their phagocytic capacity to function. Lymphocytes may lose their capacity to respond to antigens. (Qualitative disruptions of inflammatory and immune processes caused by leukocyte disorders are described in Chapter 9.) Other leukocyte alterations include infectious mononucleosis and cancers of the blood—leukemia and multiple myeloma.
Quantitative Alterations of Leukocytes
Leukocytosis occurs as a normal protective response to physiologic stressors, such as infection, strenuous exercise, emotional changes, temperature changes, anesthesia, surgery, pregnancy, and some drugs, hormones, and toxins. It is also caused by pathologic conditions, such as malignancies and hematologic disorders. Leukopenia is never normal and is defined as an absolute blood cell count less than 4000 cells/mm3. Leukopenia is associated with a decrease in neutrophils, which increases risk for infection. The absolute neutrophil count (ANC) is calculated by multiplying the white blood cell count by the percent of band and segmented neutrophils. The ANC is classified as mild (1000 to 1500 cells/mm3), moderate (500 to 1000 cells/mm3), or severe (<500 cells/mm3). When the ANC is less than 500/mm3, the possibility for life-threatening infections is high. Leukopenia can be caused by radiation, anaphylactic shock, autoimmune disease (e.g., systemic lupus erythematosus), immune deficiencies (see Chapter 9), and exposure to certain drugs and chemotherapeutic agents.
Granulocytes and Monocytes
Granulocytosis—an increase in the number of granulocytes (neutrophils, eosinophils, basophils)—begins with the release of stored leukocytes from the venous sinuses of the marrow. Neutrophilia is another term that may be used to describe granulocytosis because neutrophils are the most numerous of the granulocytes (Table 29-1). Neutrophilia occurs in the early stages of infection or inflammation and is established when the absolute neutrophil count exceeds 7500/μL. Stored neutrophils are approximately 20 to 40 times greater in number than circulating neutrophils. On rare occasions when the neutrophil count increases greatly—more than 100,000/μL (usually seen only in those with myelocytic leukemia)—the blood viscosity may increase greatly so that thrombosis or occlusion of blood vessels occurs. Release and depletion of stored neutrophils from the venous sinuses stimulate granulopoiesis to replenish neutrophil reserves. Specific conditions associated with neutrophilia are identified in Table 29-1.
TABLE 29-1
OTHER CONDITIONS ASSOCIATED WITH NEUTROPHILS, EOSINOPHILS, BASOPHILS, MONOCYTES, AND LYMPHOCYTES
| CONDITION | CAUSE | EXAMPLE |
| Neutrophil | ||
| Neutrophilia (granulocytosis) | Inflammation or tissue necrosis | Surgery, burns, MI, pneumonitis, rheumatic fever, rheumatoid arthritis |
| Infection | Bacterial: gram-positive (staphylococci, streptococci, pneumococci), gram-negative (Escherichia coli, Pseudomonas species) | |
| Physiologic | Exercise, extreme heat or cold, third-trimester pregnancy, emotional distress | |
| Hematologic | Acute hemorrhage, hemolysis, myeloproliferative disorder, chronic granulocytic leukemia | |
| Drugs or chemicals | Epinephrine, steroids, heparin, histamine, endotoxin | |
| Metabolic | Diabetes (acidosis), eclampsia, gout, thyroid storm | |
| Neoplasm | Liver, GI tract, bone marrow | |
| Neutropenia | Decreased marrow production | Radiation, chemotherapy, leukemia, aplastic anemia, abnormal granulopoiesis |
| Increased destruction | Splenomegaly, hemodialysis, autoimmune disease | |
| Infection | Gram-negative (typhoid), viral (influenza, hepatitis B, measles, mumps, rubella), severe infections, protozoal infections (malaria) | |
| Eosinophil | ||
| Eosinophilia | Allergy | Asthma, hay fever, drug sensitivity |
| Infection | Parasites (trichinosis, hookworm), chronic (fungal, leprosy, TB) | |
| Malignancy | CML, lung, stomach, ovary, Hodgkin disease | |
| Dermatosis | Pemphigus, exfoliative dermatitis (drug-induced) | |
| Drugs | Digitalis, heparin, streptomycin, tryptophan (eosinophilia-myalgia syndrome), penicillins, propranolol | |
| Eosinopenia | Stress response | Trauma, shock, burns, surgery, mental distress |
| Drugs | Steroids (Cushing syndrome) | |
| Basophil | ||
| Basophilia | Inflammation | Infection (measles, chickenpox), hypersensitivity reaction (immediate) |
| Hematologic | Myeloproliferative disorders (CML, polycythemia vera, Hodgkin lymphoma, hemolytic anemia) | |
| Endocrine | Myxedema, antithyroid therapy | |
| Basopenia | Physiologic | Pregnancy, ovulation, stress |
| Endocrine | Graves disease | |
| Monocyte | ||
| Monocytosis | Infection | Bacterial (subacute bacterial endocarditis, TB), recovery phase of infection |
| Hematologic | Myeloproliferative disorders, Hodgkin disease, agranulocytosis | |
| Physiologic | Normal newborn | |
| Monocytopenia | Rare | |
| Lymphocyte | ||
| Lymphocytosis | Physiologic | 4 months to 4 years |
| Acute infection | Infectious mononucleosis, CMV infection, pertussis, hepatitis, mycoplasma pneumonia, typhoid | |
| Chronic infection | Congenital syphilis, tertiary syphilis | |
| Endocrine | Thyrotoxicosis, adrenal insufficiency | |
| Malignancy | ALL, CLL, lymphosarcoma cell leukemia | |
| Lymphocytopenia | Immunodeficiency syndrome | AIDS, agammaglobulinemia |
| Lymphocyte destruction | Steroids (Cushing syndrome), radiation, chemotherapy, Hodgkin lymphoma, CHF, renal failure, TB, SLE, aplastic anemia | |
When the demand for circulating mature neutrophils exceeds the supply, the marrow begins to release immature neutrophils (and other leukocytes) into the blood. Premature release of the immature white cells is responsible for the phenomenon known as a shift-to-the-left or leukemoid reaction. This refers to the microscopic detection of disproportionate numbers of immature leukocytes in peripheral blood smears. Many diagrams present cellular differentiation and maturation progressing from left to right within the drawing, instead of vertically as shown in Figure 27-10. When immature leukocytes are released prematurely they cause a shift in the distribution of cells in the blood toward the left side, or immaturity side, of the diagram. This phenomenon is also seen in the blood smear of individuals with leukemia as well, hence the term leukemoid reaction. As infection or inflammation diminishes and as granulopoiesis replenishes circulating granulocytes, a shift back to normal occurs.
Neutropenia is a condition associated with a reduction in the number of circulating neutrophils. Clinically, neutropenia exists when the neutrophil count is less than 2000/μL.1 The absolute neutrophil count reflects not only the degree of neutropenia but also the risk for infection (see preceding discussion of leukocytosis). A reduction in the number of neutrophils can occur in severe, prolonged infections when production of granulocytes cannot keep up with demand.
Eosinophilia is an absolute increase (more than 450/μL) in the total numbers of circulating eosinophils. Allergic disorders (type I hypersensitivity) associated with asthma, hay fever, and drug reactions, as well as parasitic infections (particularly with metazoal parasites) are often cited as causes. Hypersensitivity reactions and the normal defense against parasites trigger the release of eosinophil chemotactic factor of anaphylaxis (ECF-A) from mast cells, attracting eosinophils to the area. (These processes are described and illustrated in Chapters 8 and 9.) Tissues with abundant mast cells, such as the respiratory and gastrointestinal tracts, are particularly common sites for eosinophil invasion. Mast cells also release interleukin-5 (IL-5), which stimulates the bone marrow to produce and release more eosinophils into the blood. Eosinophilia may also be associated with dermatologic disorders, such as atopic dermatitis, eczema, and pemphigus. Various types of eosinophilic scleroderma-like diseases also have been reported to occur in association with hemato-oncogenic disorders (i.e., eosinophilic cellulitis [Wells syndrome] and eosinophilic fasciitis [Shulman syndrome]). Increased numbers of eosinophils have been observed in individuals with eosinophilia-myalgia syndrome (EMS), which is associated with ingestion of the supplement l-tryptophan. EMS may develop in individuals with fibromyalgia syndrome as an allergic reaction to l-tryptophan.2
Eosinopenia, a decrease in circulating numbers of eosinophils, generally is caused by migration of eosinophils into inflammatory sites. It also may be seen in Cushing syndrome and as a result of stress caused by surgery, shock, trauma, burns, or mental distress. Other conditions causing eosinopenia are detailed in Table 29-1.
Basophilia, an increase in circulating numbers of basophils, is rare and generally is a response to inflammation and immediate hypersensitivity reactions. Basophils contain histamine that is released during an allergic reaction. An increase in the levels of basophils is seen also in myeloproliferative disorders, such as chronic myeloid leukemia and myeloid metaplasia. Other conditions associated with basophilia are listed in Table 29-1.
Basopenia (also known as basophilic leukopenia), a decrease in circulating numbers of basophils, is seen in hyperthyroidism, acute infection, and long-term therapy with steroids. A decrease in the number of basophils may be seen during ovulation and pregnancy. Other conditions associated with basopenia are listed in Table 29-1.
Monocytosis is an increase (generally greater than 800/μL) in numbers of circulating monocytes. The condition is often transient and not related to a dysfunction of monocyte production. When present, it most commonly occurs with neutropenia associated with bacterial infections, particularly in the late stages or recovery stage, when monocytes are needed to phagocytize surviving microorganisms and debris. Monocytosis often is seen in chronic infections, usually with intracellular bacteria, such as tuberculosis (TB), brucellosis, listeriosis, and subacute bacterial endocarditis (SBE). Peripheral monocytosis has been found to correlate with the extent of myocardial damage following myocardial infarction. Increased numbers of monocytes also may indicate marrow recovery from agranulocytosis. Other conditions associated with monocytosis are identified in Table 29-1.
Lymphocytes
Quantitative alteration of lymphocytes occurs when lymphocytes are activated by antigenic stimuli, usually microorganisms (see Chapter 8). Lymphocytosis is rare in acute bacterial infections and occurs most commonly in acute viral infections, particularly those caused by the Epstein-Barr virus (EBV), a causative agent in infectious mononucleosis. Other specific disorders associated with lymphocytosis are listed in Table 29-1.
Lymphocytopenia may be attributable to (1) abnormalities of lymphocyte production associated with neoplasias and immune deficiencies, and (2) destruction by drugs, viruses, or radiation. It also can occur in individuals for no apparent reason. Other conditions associated with lymphocytopenia are identified in Table 29-1. The lymphocytopenia associated with heart failure and other acute illnesses may be caused by elevated levels of cortisol. Lymphocytopenia is a major problem in acquired immunodeficiency syndrome (AIDS) in which the human immunodeficiency virus (HIV) is cytopathic for T helper lymphocytes. (For a more detailed discussion of AIDS, see Chapter 10.)
Infectious Mononucleosis
Evaluation and Treatment
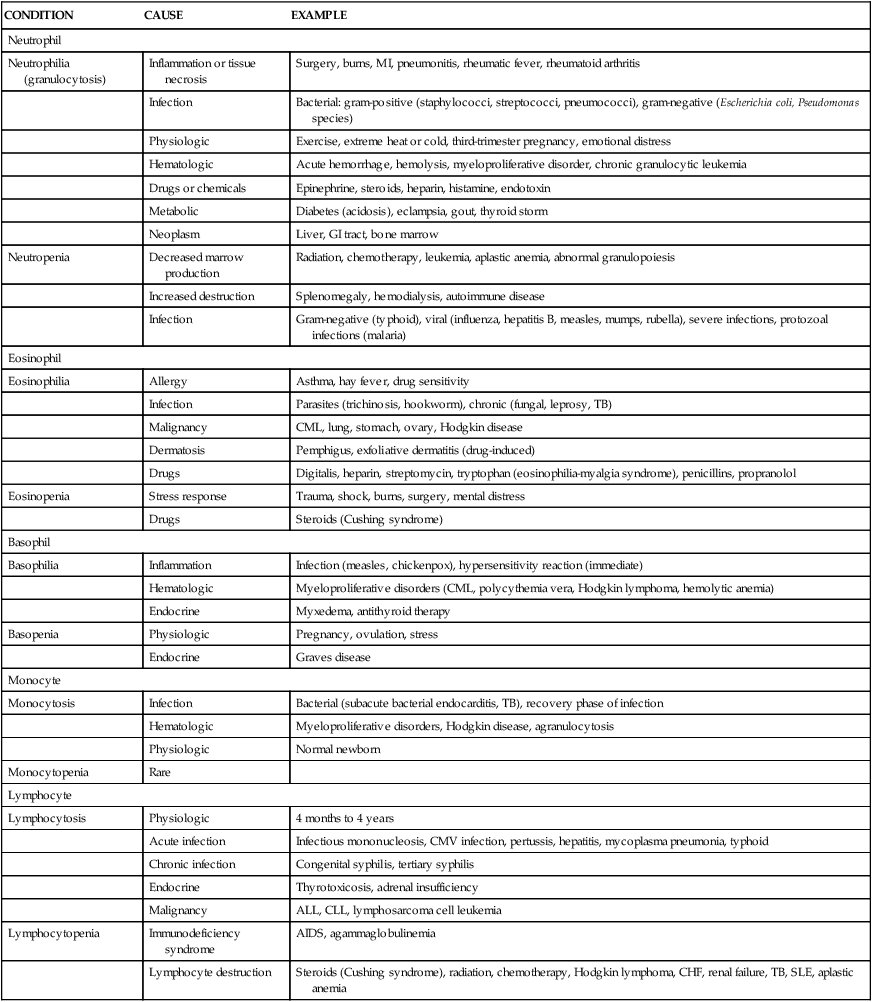
The blood of affected individuals contains an increased number of atypical lymphocytes (Figure 29-1). Diagnosis of IM is commonly based on Hoagland’s criteria of at least 50% lymphocytes and at least 10% atypical lymphocytes in the blood in the presence of fever, pharyngitis, and adenopathy confirmed by a positive serologic test. Serologic tests are used to determine a heterophile antibody response.3 Heterophile antibodies are a heterogeneous group of immunoglobulin M (IgM) antibodies that are agglutinins against nonhuman red blood cells (e.g., sheep, horse) and are detected by qualitative (Monospot) or quantitative methods (heterophile antibody test).
Low power (A) shows moderately high white blood cell count and high number of reactive, or “atypical,” lymphocytes. Higher power (B-G) illustrates the spectrum of lymphoid morphology, including small resting lymphocyte (B) for comparison, large granular lymphocyte (C), atypical forms (D-F), also referred to as “reactive” lymphs, and circulating plasma cell (G). (From Hoffman R et al: Hematology: basic principles and practice, ed 6, Philadelphia, 2013, Churchill Livingstone.)
Leukemias
Leukemia is a clonal malignant disorder of leukocytes in the blood and blood-forming organs. The common feature of all forms of leukemia is an uncontrolled proliferation of malignant leukocytes, causing an overcrowding of bone marrow and decreased production and function of normal hematopoietic cells. The first description of a “leukemic” individual was written by Velpeau in 1827.4 Virchow, a pathologist, coined the term white blood (Weissus blut) and later originated the term leukemia. Since Virchow’s initial discovery, the overall classification of leukemia has become increasingly complex and undergone several permutations.
The current classification of leukemia is based on (1) the predominant cell of origin (either myeloid or lymphoid) and (2) the rate of progression, which usually reflects the degree at which cell differentiation was arrested when the cell became malignant (acute or chronic) (Figure 29-2). Acute leukemia is characterized by undifferentiated or immature cells, usually a blast cell, and the onset of disease is abrupt and rapid with a short survival time. In chronic leukemia the predominant cell is more differentiated but does not function normally, with a relatively slow progression. There are four types of leukemia: acute lymphocytic (ALL), acute myelogenous (AML), chronic lymphocytic (CLL), and chronic myelogenous (CML). In 1976 the French-American-British Cooperative Group developed more extensive criteria for the classification of acute leukemias. This system is based on characteristics that may provide significant therapeutic prognostic information, such as structure, number of cells, genetics, identification of surface markers, and histochemical staining. Since this time, the World Health Organization has developed a classification that incorporates more recent research on the genetics and clinical features of AML that have prognostic and therapeutic relevance.5
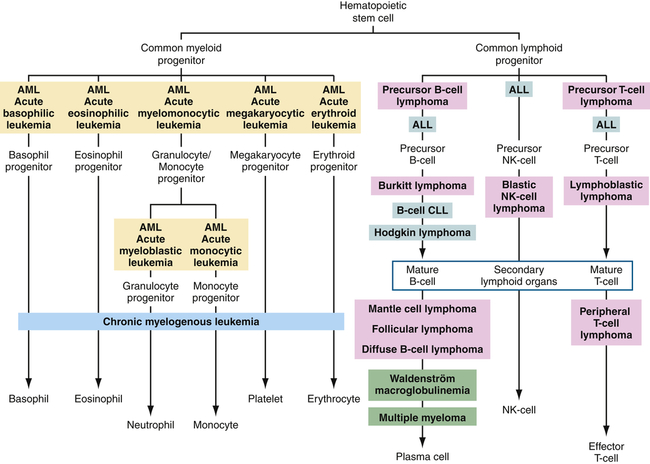
Differentiation pathways of blood-forming cells and reported sites from which specific leukemias and lymphomas originate. Tumors of similar types are given the same background coloring. ALL, Acute lymphocytic leukemia; AML, acute myelogenous leukemia; CLL, chronic lymphocytic leukemia; NK, natural killer.
Leukemia occurs with varying frequencies at different ages and is more common in adults than children (Figure 29-3). It is estimated that more than 44,600 cases of leukemia were newly diagnosed in 2011, with males having a slightly higher incidence than females (Table 29-2).6 In all types of leukemia males have a higher incidence rate (56%) as do Americans of European descent. White children have higher rates of leukemia than children of other racial groups. ALL is the least common type overall, but is the most common in children (approximately 66% of ALL cases are diagnosed before the age of 20). Leukemia accounts for about 34% of all childhood cancers, and ALL accounts for almost 78% of all new cases of leukemia in children. CLL and AML are the most common types in adults. CML is found mostly in adults.
TABLE 29-2
ESTIMATED NEW CASES AND DEATHS: LEUKEMIA AND LYMPHOMA IN THE UNITED STATES IN 2013∗
| TYPE | ESTIMATED NUMBER AND PROPORTION (%) OF NEW CASES | ESTIMATED NUMBER OF DEATHS | 5-YEAR SURVIVAL RATE (2005-2009) | |||||
| TOTAL | MALE | FEMALE | TOTAL | MALE | FEMALE | OVERALL | <5 YEARS OF AGE | |
| Leukemias | 48,610 | 27,880 | 20,730 | 23,720 | 13,660 | 10,060 | 58% | |
| Acute lymphocytic leukemia | 6050 | 3450 | 2600 | 1440 | 820 | 620 | 65.2% | 90%∗ |
| Chronic lymphocytic leukemia | 15,680 | 9720 | 5960 | 4580 | 2750 | 1830 | 78.8% | |
| Acute myelogenous leukemia | 14,590 | 7820 | 6770 | 10,370 | 5930 | 4440 | 23.5% | 55.2% |
| Chronic myelogenous leukemia | 5920 | 3420 | 2500 | 610 | 340 | 270 | 59.1% | |
| Other leukemia | 6350 | 3570 | 2780 | 6730 | 3820 | 2910 | ||
| Lymphomas | 79,030 | 42,670 | 36,360 | 20,200 | 11,250 | 8950 | 70.6% | |
| Hodgkin lymphoma | 69,740 | 5070 | 4220 | 1180 | 660 | 520 | 87% | |
| Non-Hodgkin lymphoma | 19,020 | 37,600 | 32,140 | 19,020 | 10,590 | 8430 | 71% | |
| Multiple myeloma | 22,350 | 12,440 | 9910 | 10,710 | 6070 | 4640 | 41.1% | |
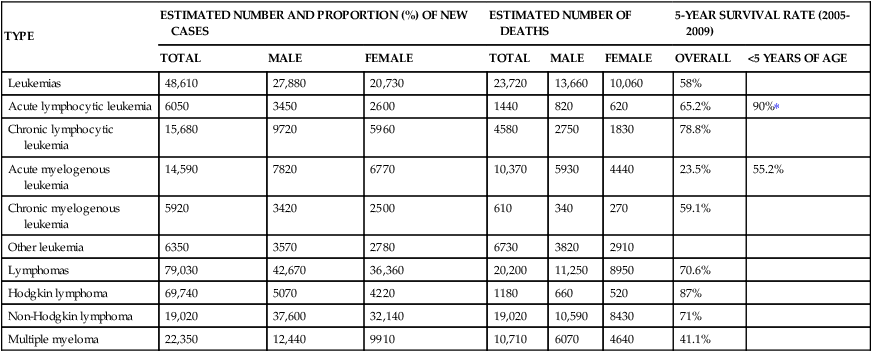
∗Based on SEER Stat Fact Sheet, posted to the SEER website, NCI, 2013.
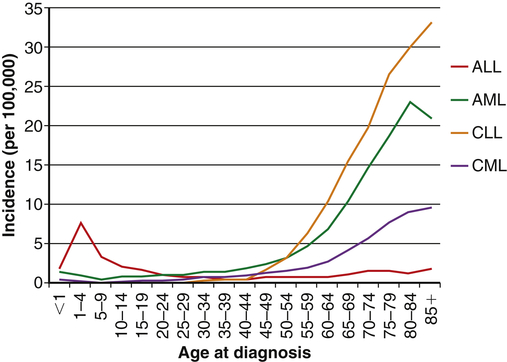
The incidences of acute myelogenous leukemia (AML), chronic lymphocytic leukemia (CLL), and chronic myelogenous leukemia (CML) are relatively stable until middle age and then increase dramatically. The incidence of acute lymphocytic leukemia (ALL) peaks in childhood, and then diminishes until middle age when the incidence begins rising slowly with age. (Data obtained from Howlader N et al, editors: SEER cancer statistics review, 1975-2009 [vintage 2009 populations], Bethesda, MD, 2012, National Cancer Institute. Available at http://seer.cancer.gov/csr/1975_2009_pops09/.)
Over the past two decades the rates of induced remission and survival in most forms of leukemia have increased. Current survival rates range from 24% for AML to 81% for CLL, and as high as 91% for children and adolescents younger than 15 years of age with ALL.7
Pathophysiology
All leukemias have certain pathophysiologic features in common. Although the exact cause of leukemia is unknown, several risk factors and related genetic aberrations are associated with the onset of malignancy. There is a statistically significant tendency for leukemia to reappear in families. There is also an increased incidence of leukemia in association with other hereditary abnormalities such as Down syndrome, Fanconi aplastic anemia, Bloom syndrome, trisomy 13, Patau syndrome, and some immune deficiencies (i.e., ataxia-telangiectasia, Wiskott-Aldrich syndrome, and congenital X-linked agammaglobulinemia; see Chapter 9).
Genetic translocations (mitotic errors) are observed in leukemic cells. The most common genetic abnormality is the reciprocal translocation between chromosomes 9 and 22 t(9;22)(q34;q11), the Philadelphia chromosome.8
The Philadelphia chromosome was first observed in persons with CML, and is present in 95% of those with CML, 3% of individuals with AML, and 25% to 30% of adults with ALL and 2% to 10% of children with ALL.9 This translocation results in the novel fusion of the BCR1 gene region from chromosome 22 and the proto-oncogene ABL1 from chromosome 9 (Figure 29-4). The BCR-ABL1 joining results in the expression of a unique fused oncoprotein, BCR-ABL1.8 The ABL1 protein is a tyrosine kinase in the signaling pathway that promotes cell proliferation. The BCR-ABL1 variant possesses greater tyrosine kinase activity and has proven to be essential for transformation into leukemic cells. BCR-ABL1 appears to excessively activate intracellular pathways, leading to increased proliferation, decreased sensitivity to apoptosis, and premature release of immature cells into the circulation. In most leukemias and lymphomas a single major genetic abnormality, such as the t(9;22) translocation, does not lead to an aggressive malignancy. The initial event is usually followed by a series of secondary genetic changes.10 Thus the original tumor becomes genetically unstable and diverse.
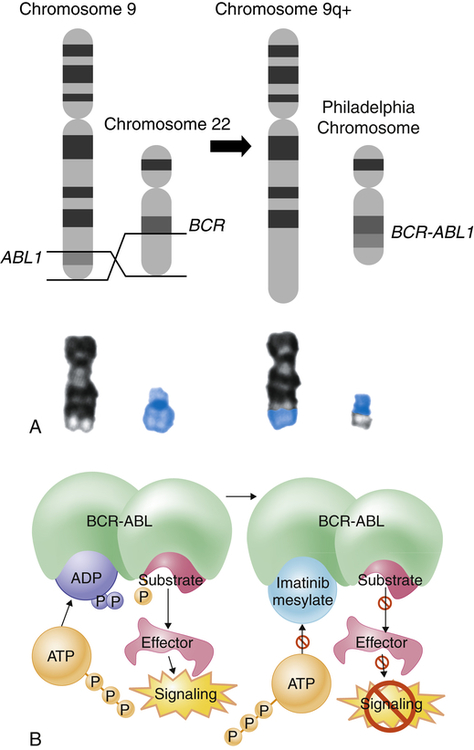
The Philadelphia chromosome is an example of a reciprocal chromosomal translocation that results in an abnormal gene product responsible for a clinical disorder. A, An exchange occurs between the long arm of chromosome 9 (black chromosome) and the long arm of chromosome 22 (blue chromosome); t(9;22)(q34;q11). B, Mechanism of action of imatinib. By occupying the ATP-binding pocket of the ABL kinase domain, imatinib prevents substrate phosphorylation and downstream activation of signals, thus inhibiting the leukemogenic effects of BCR-ABL on cells in chronic myelogenous leukemia. ADP, Adenosine diphosphate; ATP, adenosine triphosphate; P, phosphate group. (A, Top portion from Rakel R, Bope E: Conn’s current therapy 2008, Philadelphia, 2008, Saunders. Lower portion from Yanoff M, Duker J: Ophthalmology, ed 3, Edinburgh, 2009, Mosby. B from Goldman L, Schafer AI: Goldman’s Cecil medicine, ed 24, Philadelphia, 2012, Saunders.)
Risk factors for the onset of leukemia include environmental factors as well as other diseases. Increased risk in adults has been linked to exposure to cigarette smoke, benzene, and ionizing radiation. Large doses of ionizing radiation particularly result in an increased incidence of myelogenous leukemia. There is growing concern about the effect of low-dose radiation on subsequent risk of leukemia.11 Infections with HIV or hepatitis C virus increase the risk for leukemia, and it is now widely accepted that some types of leukemia are caused by infection with the human T-cell leukemia/lymphoma virus type 1 (HTLV-1). Drugs that cause bone marrow depression (e.g., chloramphenicol, phenylbutazone, and certain alkylating agents, such as cytoxan) also can predispose an individual to leukemia. AML is the most frequently reported secondary cancer after high doses of chemotherapy for Hodgkin lymphoma, non-Hodgkin lymphoma, multiple myeloma, ovarian cancer, and breast cancer. Acute leukemia also may develop secondary to certain acquired disorders, including CML, CLL, polycythemia vera, myelofibrosis, Hodgkin lymphoma, multiple myeloma, ovarian cancer, and sideroblastic anemia.
Acute Leukemias
Acute leukemias consist of two types: acute lymphocytic leukemia (ALL) and acute myelogenous leukemia (AML). Acute leukemias are seen in both genders and in all ages, with the incidence increasing dramatically in individuals older than 50 years. Mortality for all acute leukemias in the United States is about 7 per 100,000. In children younger than 15 years, leukemia accounts for one third of all deaths from cancer. North American and Scandinavian countries have the highest mortality; Eastern European countries, Asia (except Japan), and Central America have the lowest mortality. Japan’s higher mortality is the result of the atomic bombs dropped in World War II. Blacks have consistently shown a lower mortality than whites. More than 6070 new cases of ALL and 14,590 cases of AML are estimated in 2013, with more than 1430 deaths attributed to ALL and 10,370 to AML.6,12,13
Pathophysiology
ALL is a progressive neoplasm defined by the presence of greater than 30% lymphoblasts in the bone marrow or blood. Most cases of ALL occur in children (80% of ALL), and it is the most common leukemia in children, most often occurring in the first decade. The median age of diagnosis of ALL is age 13. Although adults with ALL account for only 20% of all cases, their mortality is significantly higher (see Table 29-2). The 5-year survival rate for individuals 20 to 59 years old is about 30% to 40%, about 15% to 16% for persons older than 60 years, and 5% for persons older than 70 years. The survival rate in children is about 78%. The significant difference between the incidence of ALL in adults and children is thought to be determined by differences in the biology of the disease. Children with the highest survival rate (82% to 83%) have no radiographic manifestations, whereas children with five or more skeletal lesions have a survival rate of about 72% to 73%.14
Immunotyping of leukemic blast cells allows for the identification of subtypes of ALL. Approximately 75% of ALL cases in children originate from transformed precursor B cells, whereas adult ALL is a mixture of cancers of precursor B-cell or precursor T-cell origin (Table 29-3). A small percentage of ALL cases have neither B- nor T-cell origination and are called null cell. Precursor B-cell ALL can be subdivided into different phenotypes, depending on their progression through the B-cell maturation process before becoming malignant.15,16 The general phenotype of precursor B-cell ALL expresses CD19, human leukocyte antigen DR (HLA-DR), and other B-cell–associated antigens in the cytoplasm. The most immature form (pro–B-cell ALL) occurs in about 5% of precursor B-cell ALL and is characterized by lack of expression of CD10. CD10 (common acute lymphocytic leukemia antigen [CALLA]) is a cell surface metalloprotease. Lack of CD10 is frequently associated with translocation of the myeloid/lymphoid leukemia (MLL) gene and a poor prognosis. The common precursor B-cell ALL comprises approximately 80% of precursor B-cell ALL cases; these express surface CD10, but have not yet undergone rearrangement of the immunoglobulin genes. The remaining individuals have a more mature form of precursor B-cell ALL (pre–B-cell ALL) in which the cells express immunoglobulin molecules in the cytoplasm. Less common variations include cells that are intermediate between the common precursor and pre–B-cell phenotypes and express immunoglobulin heavy chain, but no light chain, and cells that are more mature than the pre–B-cell ALL and express surface immunoglobulin and do not stain for the enzyme terminal deoxynucleotidyl transferase (TdT).
TABLE 29-3
IMMUNOPHENOTYPE OF ADULT ACUTE LYMPHOCYTIC LEUKEMIA
| Lineage | TdT | HLA-DR | CD34 | CD19 | CD22 | CD79a | CD10 | Cyµ | cgκ/λ | slgH/L | cyCD3 | CD7 | CD1a | CD2 | CD5 | sCD3 | Frequency (%) |
| Precursor B-cell ALL | 5-10 | ||||||||||||||||
| Pro-B ALL | + | + | + | + | + | + | − | − | − | − | 40-50 | ||||||
| CALL | + | + | − | + | + | − | + | − | − | − | 10 | ||||||
| Pre-B ALL | + | + | − | + | + | − | ± | + | − | − | 1 | ||||||
| Transitional precursor B-cell ALL | ± | + | − | + | + | − | − | − | − | +∗ | 1 | ||||||
| Mature B-cell ALL | − | + | − | + | ± | − | − | − | + | + | 5 | ||||||
| T-lineage ALL | |||||||||||||||||
| Pro-T ALL | + | ± | ± | + | + | − | − | − | − | 5 | |||||||
| Pre-T ALL | + | ± | ± | + | + | − | + | + | − | ||||||||
| Cortical-T ALL | + | − | − | + | + | + | + | + | − | 10-15 | |||||||
| Mature-T ALL | + | − | − | + | + | − | + | + | + | 5-10 |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


