Chapter 9
Alterations in Immunity and Inflammation
Neal S. Rote and Kathryn L. McCance

Hypersensitivity: Allergy, Autoimmunity, and Alloimmunity
Hypersensitivity is an altered immunologic response to an antigen that results in disease or damage to the host. Hypersensitivity reactions can be classified in two ways: by the source of the antigen that the immune system is attacking (allergy, autoimmunity, alloimmunity; Table 9-1) and by the mechanism that causes disease (types I, II, III, and IV; see Table 9-3). The term allergy originally denoted both facets of the immune response: immunity, which is beneficial, and hypersensitivity, which is harmful. Allergy has now come to mean the deleterious effects of hypersensitivity to environmental antigens, and immunity means the protective responses to antigens expressed by disease-causing agents.
TABLE 9-1
RELATIVE INCIDENCES AND EXAMPLES OF HYPERSENSITIVITY REACTIONS∗
| TARGET ANTIGEN | MECHANISM | |||
| TYPE I (IMMUNOGLOBULIN E–[IgE] MEDIATED) | TYPE II (TISSUE SPECIFIC) | TYPE III (IMMUNE COMPLEX) | TYPE IV (CELL MEDIATED) | |
| Allergy | ++++ | + | + | ++ |
| Environmental antigens | Hay fever | Hemolysis in drug allergies | Gluten (wheat) allergy | Poison ivy allergy |
| Autoimmunity | ± | ++ | +++ | + |
| Self-antigens | May contribute to some type III reactions | Autoimmune thrombocytopenia | Systemic lupus erythematosus | Hashimoto thyroiditis |
| Alloimmunity | ± | ++ | + | ++ |
| Another person’s antigens | May contribute to some type III reactions | Hemolytic disease of the newborn | Anaphylaxis to IgA in IV gamma globulin | Graft rejection |
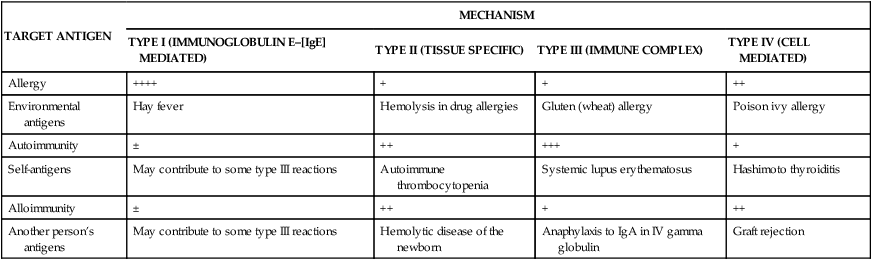
∗The frequency of each reaction is indicated in a range from rare (±) to very common (++++). An example of each reaction is given.
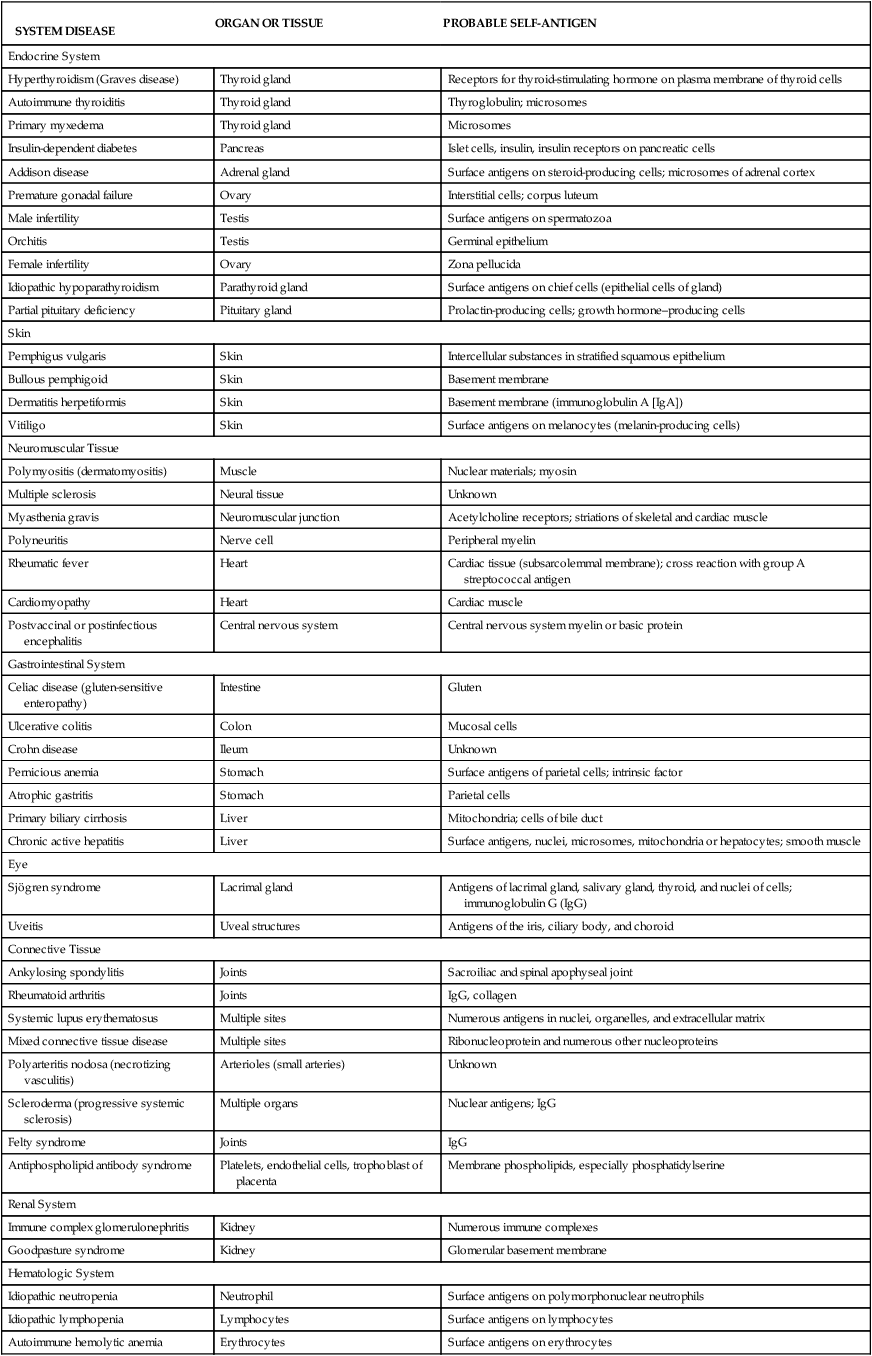
Autoimmunity is a disturbance in the immunologic tolerance of self-antigens. The immune system normally does not strongly recognize the individual’s own antigens. Healthy individuals of all ages, but particularly older adults, may produce low quantities of antibodies against their own antigens (autoantibodies), without development of overt autoimmune disease. Therefore, the presence of low quantities of autoantibodies does not necessarily indicate a disease state. Autoimmune diseases occur when the immune system reacts against self-antigens to such a degree that the person’s own tissues are damaged by autoantibodies or autoreactive T cells. Many clinical disorders are associated with autoimmunity and are collectively referred to as autoimmune diseases (Table 9-2).
TABLE 9-2
DISORDERS ASSOCIATED WITH AUTOIMMUNITY
SYSTEM DISEASE | ORGAN OR TISSUE | PROBABLE SELF-ANTIGEN |
| Endocrine System | ||
| Hyperthyroidism (Graves disease) | Thyroid gland | Receptors for thyroid-stimulating hormone on plasma membrane of thyroid cells |
| Autoimmune thyroiditis | Thyroid gland | Thyroglobulin; microsomes |
| Primary myxedema | Thyroid gland | Microsomes |
| Insulin-dependent diabetes | Pancreas | Islet cells, insulin, insulin receptors on pancreatic cells |
| Addison disease | Adrenal gland | Surface antigens on steroid-producing cells; microsomes of adrenal cortex |
| Premature gonadal failure | Ovary | Interstitial cells; corpus luteum |
| Male infertility | Testis | Surface antigens on spermatozoa |
| Orchitis | Testis | Germinal epithelium |
| Female infertility | Ovary | Zona pellucida |
| Idiopathic hypoparathyroidism | Parathyroid gland | Surface antigens on chief cells (epithelial cells of gland) |
| Partial pituitary deficiency | Pituitary gland | Prolactin-producing cells; growth hormone–producing cells |
| Skin | ||
| Pemphigus vulgaris | Skin | Intercellular substances in stratified squamous epithelium |
| Bullous pemphigoid | Skin | Basement membrane |
| Dermatitis herpetiformis | Skin | Basement membrane (immunoglobulin A [IgA]) |
| Vitiligo | Skin | Surface antigens on melanocytes (melanin-producing cells) |
| Neuromuscular Tissue | ||
| Polymyositis (dermatomyositis) | Muscle | Nuclear materials; myosin |
| Multiple sclerosis | Neural tissue | Unknown |
| Myasthenia gravis | Neuromuscular junction | Acetylcholine receptors; striations of skeletal and cardiac muscle |
| Polyneuritis | Nerve cell | Peripheral myelin |
| Rheumatic fever | Heart | Cardiac tissue (subsarcolemmal membrane); cross reaction with group A streptococcal antigen |
| Cardiomyopathy | Heart | Cardiac muscle |
| Postvaccinal or postinfectious encephalitis | Central nervous system | Central nervous system myelin or basic protein |
| Gastrointestinal System | ||
| Celiac disease (gluten-sensitive enteropathy) | Intestine | Gluten |
| Ulcerative colitis | Colon | Mucosal cells |
| Crohn disease | Ileum | Unknown |
| Pernicious anemia | Stomach | Surface antigens of parietal cells; intrinsic factor |
| Atrophic gastritis | Stomach | Parietal cells |
| Primary biliary cirrhosis | Liver | Mitochondria; cells of bile duct |
| Chronic active hepatitis | Liver | Surface antigens, nuclei, microsomes, mitochondria or hepatocytes; smooth muscle |
| Eye | ||
| Sjögren syndrome | Lacrimal gland | Antigens of lacrimal gland, salivary gland, thyroid, and nuclei of cells; immunoglobulin G (IgG) |
| Uveitis | Uveal structures | Antigens of the iris, ciliary body, and choroid |
| Connective Tissue | ||
| Ankylosing spondylitis | Joints | Sacroiliac and spinal apophyseal joint |
| Rheumatoid arthritis | Joints | IgG, collagen |
| Systemic lupus erythematosus | Multiple sites | Numerous antigens in nuclei, organelles, and extracellular matrix |
| Mixed connective tissue disease | Multiple sites | Ribonucleoprotein and numerous other nucleoproteins |
| Polyarteritis nodosa (necrotizing vasculitis) | Arterioles (small arteries) | Unknown |
| Scleroderma (progressive systemic sclerosis) | Multiple organs | Nuclear antigens; IgG |
| Felty syndrome | Joints | IgG |
| Antiphospholipid antibody syndrome | Platelets, endothelial cells, trophoblast of placenta | Membrane phospholipids, especially phosphatidylserine |
| Renal System | ||
| Immune complex glomerulonephritis | Kidney | Numerous immune complexes |
| Goodpasture syndrome | Kidney | Glomerular basement membrane |
| Hematologic System | ||
| Idiopathic neutropenia | Neutrophil | Surface antigens on polymorphonuclear neutrophils |
| Idiopathic lymphopenia | Lymphocytes | Surface antigens on lymphocytes |
| Autoimmune hemolytic anemia | Erythrocytes | Surface antigens on erythrocytes |
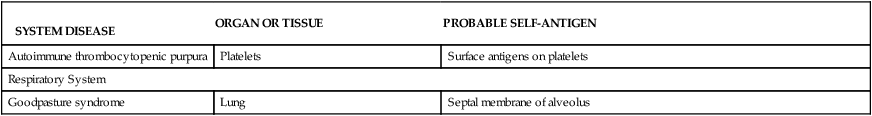
| Autoimmune thrombocytopenic purpura | Platelets | Surface antigens on platelets |
| Respiratory System | ||
| Goodpasture syndrome | Lung | Septal membrane of alveolus |
Mechanisms of Hypersensitivity
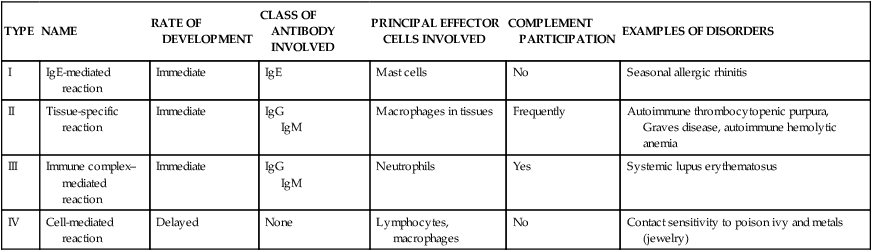
Diseases caused by hypersensitivity reactions can be characterized also by the particular immune mechanism that results in the disease (see Table 9-1). These mechanisms are apparent in most hypersensitivity reactions and have been divided into four distinct types: type I (immunoglobulin E [IgE]–mediated) hypersensitivity reactions, type II (tissue-specific) hypersensitivity reactions, type III (immune complex–mediated) hypersensitivity reactions, and type IV (cell-mediated) hypersensitivity reactions (Table 9-3).1 This classification is artificial and seldom is a particular disease associated with only a single mechanism. The four mechanisms are interrelated, and in most hypersensitivity reactions, several mechanisms can be at work simultaneously or sequentially. Some of the mechanisms are secondary to the disease and not directly involved in the pathologic process, whereas others are the primary cause of tissue destruction.
TABLE 9-3
IMMUNOLOGIC MECHANISMS OF TISSUE DESTRUCTION
| TYPE | NAME | RATE OF DEVELOPMENT | CLASS OF ANTIBODY INVOLVED | PRINCIPAL EFFECTOR CELLS INVOLVED | COMPLEMENT PARTICIPATION | EXAMPLES OF DISORDERS |
| I | IgE-mediated reaction | Immediate | IgE | Mast cells | No | Seasonal allergic rhinitis |
| II | Tissue-specific reaction | Immediate | IgG IgM | Macrophages in tissues | Frequently | Autoimmune thrombocytopenic purpura, Graves disease, autoimmune hemolytic anemia |
| III | Immune complex–mediated reaction | Immediate | IgG IgM | Neutrophils | Yes | Systemic lupus erythematosus |
| IV | Cell-mediated reaction | Delayed | None | Lymphocytes, macrophages | No | Contact sensitivity to poison ivy and metals (jewelry) |
The most rapid and severe immediate hypersensitivity reaction is anaphylaxis.2 Anaphylaxis occurs within minutes of reexposure to the antigen and can be either systemic (generalized) or cutaneous (localized).3 Symptoms of systemic anaphylaxis include itching, erythema, headaches, vomiting, abdominal cramps, diarrhea, and breathing difficulties. In severe cases, contraction of bronchial smooth muscle, laryngeal edema, and vascular collapse may result in respiratory distress, decreased blood pressure, shock, and death. Examples of systemic anaphylaxis are allergic reactions to bee stings, peanuts, and fish. Cutaneous anaphylaxis causes the less severe symptom of local inflammation.
Type I: IgE-Mediated Hypersensitivity Reactions
Type I reactions are mediated by antigen-specific IgE and the products of tissue mast cells4 (Figure 9-1). Most common allergies (e.g., pollen allergies) are type I reactions. In addition, most type I reactions occur against environmental antigens and are therefore allergic. Because of this strong association, many healthcare professionals use the term allergy to indicate only IgE-mediated reactions. However, IgE can contribute to a few autoimmune and alloimmune diseases, and many common allergies (e.g., poison ivy) are not mediated by IgE.
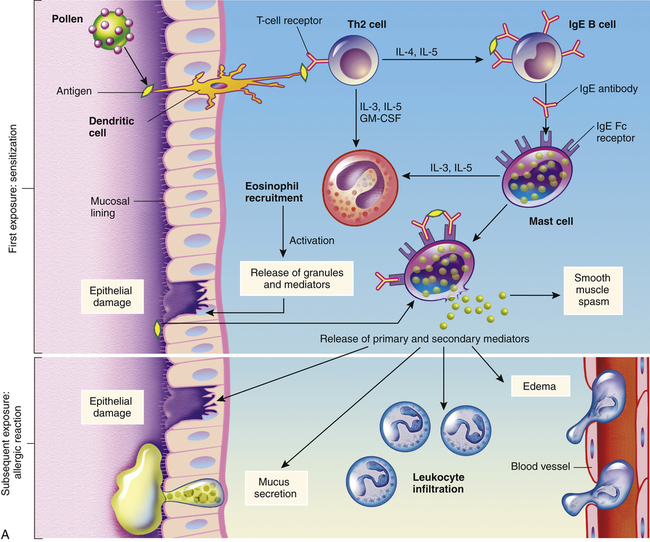
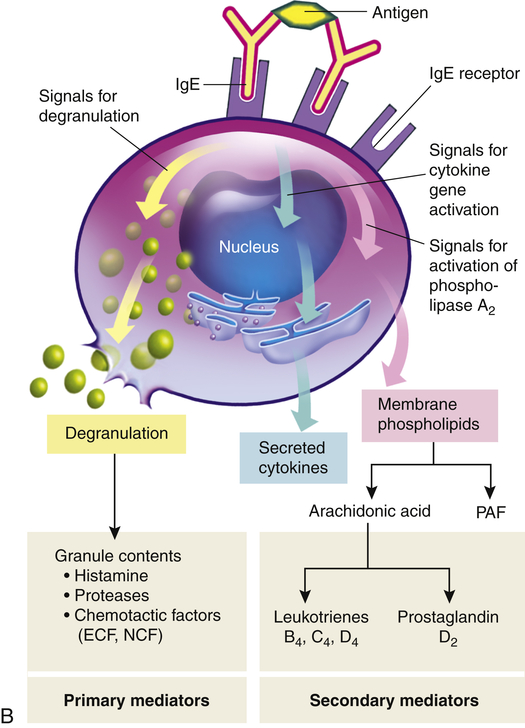
A, Th2 cells are activated by antigen-presenting dendritic cells to produce cytokines, including IL-3, IL-4, IL-5, and granulocyte-macrophage colony-stimulating factor (GM-CSF). IL-3, IL-5, and GM-CSF attract and promote the survival of eosinophils. Other cytokines (e.g., IL-4) induce B cells to class-switch to IgE-producing plasma cells. The IgE coats the surface of the mast cell by binding with IgE-specific Fc receptors on the mast cell’s plasma membrane (sensitization). Further exposure to the same allergen cross-links the surface-bound IgE and activates signals from the cytoplasmic portion of the IgE Fc receptors. These signals initiate two parallel and interdependent processes: mast cell degranulation and discharge of preformed mediators (e.g., histamine, eosinophil-chemotactic factor of anaphylaxis) and production of newly formed mediators such as arachidonic metabolites (leukotrienes, prostaglandins). Many local type I hypersensitivity reactions have two well-defined phases. The initial phase is characterized by vasodilation, vascular leakage, and, depending on the location, smooth muscle spasm or glandular secretions. These changes usually become evident within 5 to 30 minutes after exposure to the antigen. The late phase occurs 2 to 8 hours later without additional exposure to the antigen. The late phase has more intense infiltration of tissues with eosinophils, neutrophils, basophils, monocytes, and Th cells and tissue destruction in the form of mucosal epithelial cell damage. B, Activation of mast cells leading to degranulation of preformed mediators (primary mediators) and synthesis of newly formed (de novo) mediators (secondary mediators). ECF, Eosinophilic chemotactic factor; NCF, neutrophil chemotactic factor; PAF, platelet-activating factor.
In some individuals, exposure to an environmental antigen causes primarily IgE production. Repeated exposure to the antigen usually is required to elicit enough IgE so that the person becomes “sensitized.” IgE has a relatively short life span in the blood because it rapidly binds to very-high-affinity Fc receptors on the plasma membranes of mast cells (see Figure 9-1). The subclass IgG4 also has specific receptors on the mast cell and may contribute to the type I mechanism. Antibody that binds to mast cells is termed cytotropic antibody (able to bind to cell surfaces) or reagin (skin-sensitizing antibody). Unlike Fc receptors on phagocytes, which bind IgG that has reacted with antigen, the Fc receptors on mast cells bind with IgE that has not previously interacted with antigen.
If further exposure of a sensitized individual to the antigen occurs, one molecule of antigen may bind simultaneously to two molecules of IgE-Fc receptor complexes on the mast cell’s surface (cross-link) resulting in activation of intracellular signaling pathways and mast cell degranulation (see Figure 9-1, B, and Chapter 7). The antigen that triggers cross-linking must have at least two antigenic determinants on the same molecule. Sometimes an IgE-mediated response is beneficial to the host, as is the case of some immune reactions against parasites. (This mechanism is described in Chapter 8 and illustrated in Figure 8-26.)
The products of mast cell degranulation can modulate almost all aspects of an acute inflammatory response. (The effects of biochemical mediators released by mast cells are illustrated in Figure 7-11). The most potent mediator is histamine, which affects several key target cells. Acting through the H1 receptors, histamine contracts bronchial smooth muscles, causing bronchial constriction; increases vascular permeability, causing edema; and causes vasodilation, increasing blood flow into the affected area (see Figures 7-3 and 7-12). The interaction of histamine with H2 receptors results in increased gastric acid secretion and a decrease of histamine released from mast cells and basophils. The action of histamine through H2 receptors suggests an important negative-feedback mechanism that stops degranulation. That is, the released histamine inhibits release of additional histamine by interacting with H2 receptors on the mast cells. Histamine also may affect control of the immune response through H2 receptors on most cells of the immune system. Another important activity of histamine is enhancement of the chemotactic activity of other factors, such as eosinophil chemotactic factor of anaphylaxis (ECF-A), which attracts eosinophils into sites of allergic inflammatory reactions and prevents them from migrating out of the inflammatory site. (The role of the eosinophil in inflammation is discussed in Chapter 7.)
Type II: Tissue-Specific Hypersensitivity Reactions
Type II hypersensitivity reactions are generally characterized by a specific cell or tissue being the target of an immune response. In addition to major histocompatibility locus antigens (HLAs; discussed in Chapter 8), most cells have other antigens on their surfaces. Some of these other antigens are called tissue-specific antigens because they are expressed on the plasma membranes of only certain cells in specific tissues. Platelets, for example, have groups of antigens that are found on no other cells of the body. The symptoms of many type II diseases are determined by which tissue or organ expresses the particular antigen. Environmental antigens (e.g., drugs or their metabolites) may bind to the plasma membranes of specific cells (especially erythrocytes and platelets) and function as targets of type II reactions.
The five general mechanisms by which type II hypersensitivity reactions can affect cells are shown in Figure 9-2. All of these mechanisms begin with antibody binding to tissue-specific antigens or antigens that have attached to particular tissues. First, the cell can be destroyed by antibody (IgG or IgM) and activation of the complement cascade through the classical pathway. Formation of the membrane attack complex (C5-9) damages the membrane and may result in lysis of the cell (see Figure 9-2, A). For example, erythrocytes are destroyed by complement-mediated lysis in individuals with autoimmune hemolytic anemia (see Chapter 28) or as a result of an alloimmune reaction to ABO-mismatched transfused blood cells.
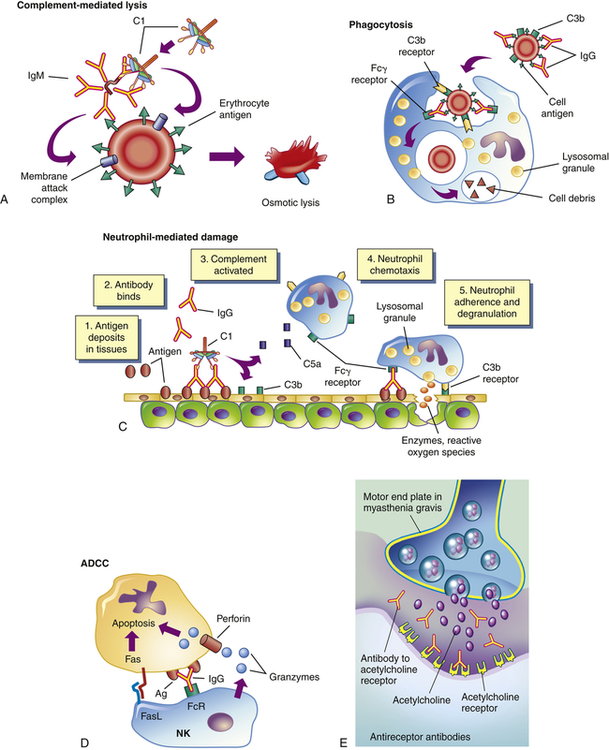
Antigens on the target cell bind with antibody and are destroyed or prevented from functioning by A, complement-mediated lysis (an erythrocyte target is illustrated here); B, clearance (phagocytosis) by macrophages in the tissue; C, neutrophil-mediated immune destruction; D, antibody-dependent cell-mediated cytotoxicity (ADCC) (apoptosis of target cells is induced by granzymes and perforin produced by natural killer [NK] cells and interactions of Fas ligand [FasL] on the surface of NK cells with Fas on the surface of target cells); or E, modulation or blocking the normal function of receptors by antireceptor antibody. This example of mechanism E depicts myasthenia gravis in which acetylcholine receptor antibodies block acetylcholine from attaching to its receptors on the motor end plates of skeletal muscle, thereby impairing neuromuscular transmission and causing muscle weakness. C1, Complement component C1; C3b, complement fragment produced from C3, which acts as an opsonin; C5a, complement fragment produced from C5, which acts as a chemotactic factor for neutrophils; Fcγ receptor, cellular receptor for the Fc portion of IgG; FcR, Fc receptor.
Second, antibody may cause cell destruction through phagocytosis by macrophages. IgG and also C3b of the complement system are opsonins that bind to receptors on the macrophage (see Figure 9-2, B). Phagocytosis of the target cell follows. (Phagocytosis is illustrated in Figures 7-15 and 7-16.) For example, antibodies against platelet-specific antigens or against red blood cell antigens of the Rh system coat those cells at low density, resulting in their preferential removal by phagocytosis in the spleen, rather than by complement-mediated lysis.
Third, antibody and complement may attract neutrophils. Either antigen expressed normally on the vessel walls or soluble antigen in the circulation (e.g., released from cells within the body or from infectious agents or by way of drugs or medications) that has been deposited on the surface of endothelial cells may bind antibody (see Figure 9-2, C). The antibody initiates the complement cascade, resulting in the release of C3a and C5a, which are chemotactic for neutrophils, and deposition of complement component C3b. Neutrophils bind to the tissues through receptors for the Fc portion of antibody (Fc receptor) or for C3b and attempt to phagocytose the tissue. Because the tissue is large, phagocytosis cannot be completed; even so, neutrophils release their granules onto the healthy tissue. The components of neutrophil granules, as well as the several toxic oxygen products produced by these cells, will damage the tissue.
The fourth mechanism is antibody-dependent cell-mediated cytotoxicity (ADCC) (see Figure 9-2, D). This mechanism involves a subpopulation of cytotoxic cells that are not antigen specific (natural killer [NK] cells). Antibody on the target cell is recognized by Fc receptors on the NK cells, which release toxic substances that destroy the target cell.
The fifth mechanism does not destroy the target cell, but rather causes it to malfunction. In this mechanism of type II injury, the antibody is usually directed against antigenic determinants associated with specific cell surface receptors, and the symptoms of the disease are a result of a direct effect of antibody binding alone (see Figure 9-2, E). The antibody reacts with the receptors on the target cell surface and modulates the function of the receptor by preventing interactions with their normal ligands, replacing the ligand and inappropriately stimulating the receptor, or destroying the receptor. For example, in the hyperthyroidism (excessive thyroid activity) of Graves disease, autoantibody binds to and activates receptors for thyroid-stimulating hormone (TSH) (a pituitary hormone that controls the production of the hormone thyroxine by the thyroid). In this way the antibody stimulates the thyroid cells to produce thyroxine.5 Under normal conditions, the increasing levels of thyroxine in the blood would signal the pituitary to decrease TSH production, which would result in less stimulation of the TSH receptor in the thyroid and a concomitant decrease in thyroxine production. Because the level of anti-TSH receptor antibody is not controlled by the pituitary, increasing amounts of thyroxine in the blood have no effect on antibody levels, and thyroxine production continues to increase despite decreasing amounts of TSH (see Chapter 22).
Type III: Immune Complex–Mediated Hypersensitivity Reactions
Mechanisms of Type III Hypersensitivity
Most type III hypersensitivity diseases are caused by antigen-antibody (immune) complexes that are formed in the circulation and deposited later in vessel walls or extravascular tissues (Figure 9-3). The primary difference between type II and type III mechanisms is that in type II hypersensitivity antibody binds to the antigen on the cell surface, whereas in type III the antibody binds to soluble antigen that was released into the blood or body fluids, and the complex is then deposited in the tissues. Type III reactions are not organ specific, and symptoms have little to do with the particular antigenic target of the antibody. The harmful effects of immune complex deposition are caused by complement activation, particularly through the generation of chemotactic factors for neutrophils. The neutrophils bind to antibody and C3b contained in the complexes and attempt to ingest the immune complexes.
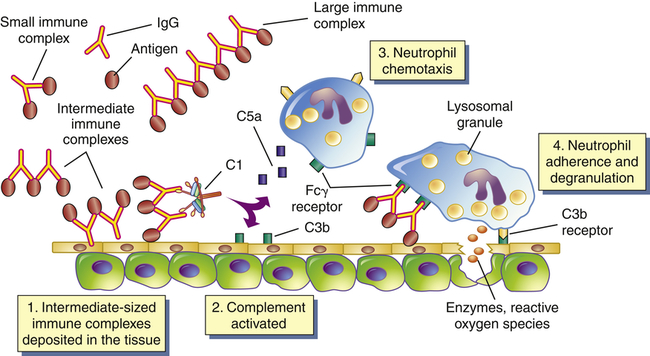
Immune complexes form in the blood from circulating antigen and antibody. Both small and large immune complexes are removed successfully from the circulation and do not cause tissue damage. Intermediate-sized complexes are deposited in certain target tissues in which the circulation is slow or filtration of the blood occurs. The complexes activate the complement cascade through C1 and generate fragments including C5a and C3b. C5a is chemotactic for neutrophils, which migrate into the inflamed area and attach to the IgG and C3b in the immune complexes. The neutrophils attempt unsuccessfully to phagocytose the tissue and in the process release a variety of degradative enzymes that destroy the healthy tissues. Fcγ receptor is the cellular receptor for the Fc portion of IgG.
Immune Complex Disease
Serum Sickness
The systemic prototype of immune complex–mediated disease is called serum sickness because it was initially described as being caused by the therapeutic administration of foreign serum, such as horse serum that contained antibody against tetanus toxin.6 Foreign serum generally is not administered to individuals today, although serum sickness reactions can be caused by the repeated intravenous administration of other antigens, such as drugs, and the characteristics of serum sickness are observed in systemic type III autoimmune diseases. Serum sickness–type reactions are caused by the formation of immune complexes in the blood and their subsequent generalized deposition in target tissues. Typically affected tissues are the blood vessels, joints, and kidneys. Other symptoms include fever, enlarged lymph nodes, rash, and pain at sites of inflammation.
Arthus Reaction
An Arthus reaction is the prototypic example of a localized immune complex–mediated inflammatory response.7 It is caused by repeated local exposure to an antigen that reacts with preformed antibody and forms immune complexes in the walls of the local blood vessels. Symptoms of an Arthus reaction begin within 1 hour of exposure and peak 6 to 12 hours later. The lesions are characterized by a typical inflammatory reaction, with increased vascular permeability, an accumulation of neutrophils, edema, hemorrhage, clotting, and tissue damage.
Type IV: Cell-Mediated Hypersensitivity Reactions
Whereas types I, II, and III hypersensitivity reactions are mediated by antibody, type IV reactions are mediated by T lymphocytes and do not involve antibody (Figure 9-4). Type IV mechanisms occur through either cytotoxic T lymphocytes (Tc cells) or lymphokine-producing Th1 and Th17 cells. Tc cells attack and destroy cellular targets directly. Th1 and Th17 cells produce cytokines that recruit and activate phagocytic cells, especially macrophages. Destruction of the tissue is usually caused by direct killing by toxins from Tc cells or by the release of soluble factors, such as lysosomal enzymes and toxic reactive oxygen species (ROS), from activated macrophages.
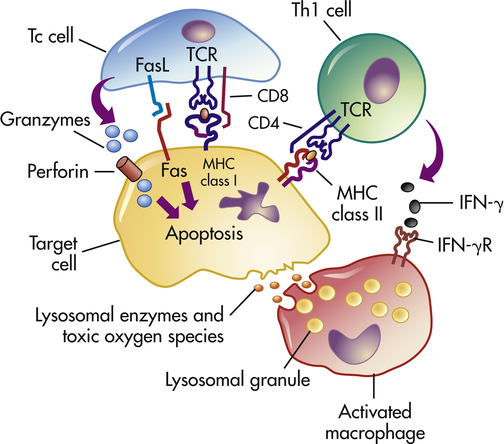
Antigens from target cells stimulate T cells to differentiate into cytotoxic T cells (Tc cells), which have direct cytotoxic activity, and helper T cells (Th1 cells) involved in delayed hypersensitivity. The Th1 cells produce lymphokines (especially interferon-gamma [IFN-γ]) that activate the macrophage through specific receptors (e.g., IFN-γ receptor [IFN-γR]). The macrophages can attach to targets and release enzymes and reactive oxygen species that are responsible for most of the tissue destruction.
Clinical examples of type IV hypersensitivity reactions include graft rejection and allergic reactions resulting from contact with such substances as poison ivy and metals. A type IV component also may be present in many autoimmune diseases. For example, T cells against type II collagen (a protein present in joint tissues) contribute to the destruction of joints in rheumatoid arthritis; T cells against a thyroid cell surface antigen contribute to the destruction of the thyroid in autoimmune thyroiditis (Hashimoto disease); and T cells against an antigen on the surface of pancreatic beta cells (the cell that normally produces insulin) are responsible for beta-cell destruction in insulin-dependent (type 1) diabetes mellitus.8
A type IV hypersensitivity reaction in the skin was thoroughly described first by Ehrlich in 1891 and led to the development of a diagnostic skin test for tuberculosis.9 The reaction follows an intradermal injection of tuberculin antigen into a suitably sensitized individual and is called a delayed hypersensitivity skin test because of its slow onset—24 to 72 hours to reach maximum intensity. The reaction site is infiltrated with T lymphocytes and macrophages, resulting in a clear hard center (induration) and a reddish surrounding area (erythema).
Antigenic Targets of Hypersensitivity Reactions
Allergy
Allergy is a hypersensitivity response against an environmental antigen (allergen). Although the most common allergies are type I hypersensitivities, any of the other three mechanisms may cause allergic responses.10
Genetic Predisposition
Certain individuals are genetically predisposed to develop allergies, particularly type I allergies, and are called atopic.11 In families in which one parent has an allergy, allergies develop in about 40% of the offspring. If both parents have allergies, the incidence in the offspring may be as high as 80%. (Principles of genetic inheritance are discussed in Chapter 4.)
Clinical Symptoms of Type I Allergies
The clinical manifestations of type I reactions are attributable mostly to the biologic effects of histamine. Tissues most commonly affected contain large numbers of mast cells and are sensitive to the effects of histamine released from them. These tissues are found in the gastrointestinal tract, the skin, and the respiratory tract (Figure 9-5 and Table 9-4). The particular symptoms frequently reflect the main portal of entry for the allergen. For instance, pollens and other airborne allergens usually cause respiratory symptoms.
TABLE 9-4
CAUSES OF CLINICAL MANIFESTATIONS OF ALLERGY
| TYPICAL ALLERGEN | MECHANISM OF HYPERSENSITIVITY | CLINICAL MANIFESTATION |
| Ingestants | ||
| Foods | Type I | Gastrointestinal allergy |
| Drugs | Types I, II, III | Urticaria, immediate drug reaction, hemolytic anemia, serum sickness |
| Inhalants | ||
| Pollens, dust, molds | Type I | Allergic rhinitis, bronchial asthma |
| Aspergillus fumigatus | Types I, III | Allergic bronchopulmonary aspergillosis |
| Thermophilic actinomycetes∗ | Types III, IV | Extrinsic allergic alveolitis |
| Injectants | ||
| Drugs | Types I, II, III | Immediate drug reaction, hemolytic anemia, serum sickness |
| Bee venom | Type I | Anaphylaxis |
| Vaccines | Type III | Localized Arthus reaction |
| Serum | Types I, III | Anaphylaxis, serum sickness |
| Contactants | ||
| Poison ivy, metals | Type IV | Contact dermatitis |
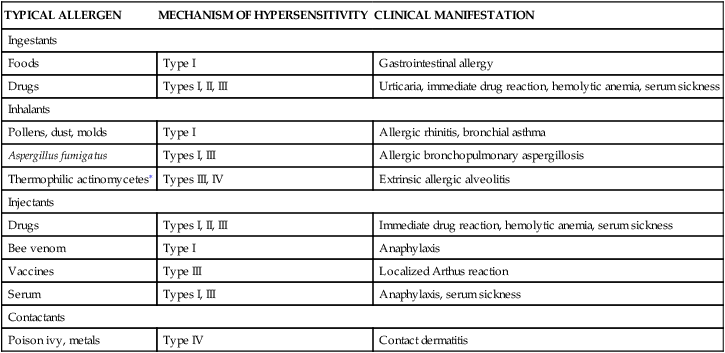
∗An order of fungi that is stimulated by warmth to grow and proliferate.
Modified from Bellanti JA: Immunology III, Philadelphia, 1985, Saunders.
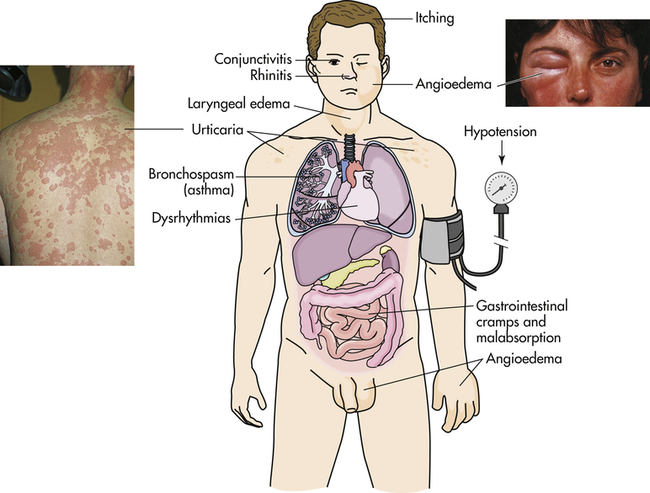
Manifestations of allergic reactions as a result of type I hypersensitivity include itching, angioedema (swelling caused by exudation), edema of the larynx, urticaria (hives), bronchospasm (constriction of airways in the lungs), hypotension (low blood pressure), and dysrhythmias (irregular heartbeat) because of anaphylactic shock, and gastrointestinal cramping caused by inflammation of the gastrointestinal mucosa. Photographic inserts show a diffuse allergic-like eye and skin reaction on an individual. The skin lesions have raised edges and develop within minutes or hours, with resolution occurring after about 12 hours. (Inserts from Male D et al: Immunology, ed 8, St Louis, 2013, Mosby.)
Effects of allergens on the mucosa of the eyes, nose, and respiratory tract include conjunctivitis (inflammation of the membranes lining the eyelids), rhinitis (inflammation of the mucous membranes of the nose), and asthma (constriction of the bronchi).12 Symptoms are caused by vasodilation, hypersecretion of mucus, edema, and swelling of the respiratory mucosa. Because the mucous membranes lining the respiratory tract (accessory sinuses, nasopharynx, and upper and lower respiratory tracts) are continuous, they are all adversely affected. The degree to which each is affected determines the symptoms of the disease.
Gastrointestinal allergies are caused primarily by allergens that enter through the mouth—usually foods or medicines. Symptoms include vomiting, diarrhea, or abdominal pain and may be severe enough to result in malabsorption or protein-losing enteropathy, if the reactions are prolonged or recurrent. Foods most often implicated in gastrointestinal allergies are milk, chocolate, citrus fruits, eggs, wheat, nuts, peanut butter, and fish.13 When food is the allergen, the active immunogen may be a product of food breakdown by digestive enzymes.
Urticaria, or hives, is a dermal (skin) manifestation of type I allergic reactions (see Figure 9-5). The underlying mechanism is the localized release of histamine and increased vascular permeability, resulting in limited areas of edema. Urticaria is characterized by white fluid-filled blisters (wheals) surrounded by areas of redness (flares). The wheal and flare reaction is usually accompanied by itching. Not all urticarial symptoms are caused by allergic (immunologic) reactions. Some, termed nonimmunologic urticaria, result from exposure to cold temperatures, emotional stress, medications, systemic diseases, hyperthyroidism, or malignancies (e.g., lymphomas).
Although some type I allergic responses can be controlled by blocking histamine receptors with antihistamines, the primary mechanism of control is the autonomic nervous system. The autonomic nervous system includes biochemical mediators (e.g., epinephrine, acetylcholine) that, like the mediators of the inflammatory response, have profound effects on cells. These mediators bind to appropriate receptors on mast cells and the target cells of inflammation (e.g., smooth muscle), thereby controlling (1) the release of inflammatory mediators from mast cells and (2) the degree to which target cells respond to inflammatory mediators (see Chapter 7).
Tests of IgE-Mediated Allergy
Allergic reactions can be life threatening; therefore, it is essential that severely allergic individuals be made aware of the specific allergen against which they are sensitized and instructed to avoid contact with that material.14 Several tests are available, including food challenges, skin tests with allergens, and laboratory tests for measurements of total IgE and allergen-specific IgE in the blood.15
Desensitization
Clinical desensitization to allergens can be achieved in some individuals. Minute quantities of the allergen are injected in increasing doses over a prolonged period. The procedure may reduce the severity of the allergic reaction in the treated individual. However, this form of therapy is associated with a risk of systemic anaphylaxis, which can be severe and life threatening. This approach works best for routine respiratory allergens16 and biting insect allergies (80% to 90% rate of desensitization over 5 years of treatment). Food allergies have been very difficult to suppress, but some promising trials are underway to evaluate desensitization by oral or sublingual administration of increasing amounts of allergen.17
Type IV Allergic Hypersensitivities
The allergens that induce a type IV allergic reaction are mostly haptens that react with normal self-proteins in the skin. When presented in this fashion, these antigens are recognized by pattern recognition receptors (PRRs) on antigen-presenting cells in the skin and induce a cell-mediated response.18 (Pattern recognition receptors are discussed in Chapter 7.) The primary result is an allergic contact dermatitis that is confined to the area of contact with the allergen. The best-known example is poison ivy (Figure 9-6). The antigen in that instance is a plant catechol, urushiol, that reacts with normal skin proteins and evokes a cell-mediated immune response.
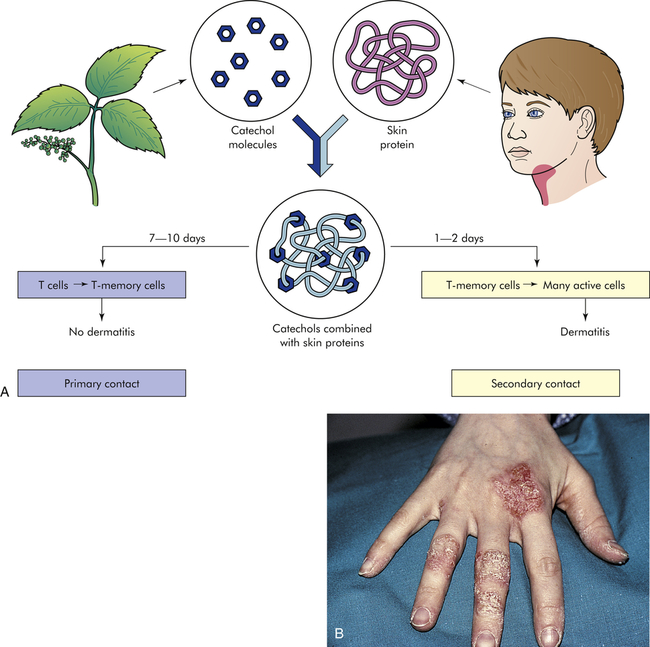
A, Shown here is the development of allergy to catechols from poison ivy. No dermatitis results from the primary contact because the antigens (catechols) are sensitizing the immune response and producing memory T cells. Secondary contact, however, quickly activates a type IV, cell-mediated reaction that causes dermatitis. B, This contact dermatitis was caused by a delayed hypersensitivity reaction that led to vesicles and scaling at the sites of contact. (From Damjanov I, Linder J: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
As noted, type I hypersensitivity reactions may result in a skin reaction (e.g., hives formed during an allergic reaction to a particular food). The distribution of the lesions may suggest whether the reaction is caused by immediate (type I) or delayed (type IV) hypersensitivity mechanisms.19 Immediate hypersensitivity reactions, termed atopic dermatitis, are usually characterized by widely distributed lesions, whereas contact dermatitis (delayed hypersensitivity) consists of lesions only at the site of contact with the allergen, such as a metal allergy to jewelry (see Figure 9-6).
Types II and III Allergic Hypersensitivities
Type II allergic hypersensitivities are usually against allergic haptens that bind to the surface of cells and elicit an IgG or IgM response. For instance, allergic reactions against many drugs (e.g., penicillin, sulfonamides) occur after the drug binds to proteins on the plasma membranes of a person’s cells and becomes immunogenic.20 The immune system attacks the allergen on the cell membrane and destroys the cell as well. In allergic reactions to penicillin, the immunogenic antigen is a metabolite of penicillin catabolism that binds to the plasma membranes of erythrocytes or platelets and induces an antibody response that destroys the cells (type II hypersensitivity), causing anemia or thrombocytopenia. Type II allergic reactions also can occur against antigens of infectious diseases. For instance, encephalitis secondary to a rubella infection may result from damage to cells of the nervous system by an immune response against rubella virus antigen on the cell’s plasma membrane.
Type III allergic reactions occur after the formation of immune complexes containing soluble allergens. For instance, Arthus reactions may be observed after injection, ingestion, or inhalation of allergens. Skin reactions can follow subcutaneous or intradermal inoculation with drugs, fungal extracts, or antigens used in skin tests. Gastrointestinal reactions, such as gluten-sensitive enteropathy (celiac disease), follow ingestion of antigen, usually gluten from wheat products (see Chapter 41). Allergic alveolitis is a type III acute hemorrhagic inflammation of the air sacs (alveoli) of the lungs resulting from inhalation of fungal antigens, usually particles from moldy hay (farmer’s lung) or pigeon feces (pigeon breeder’s disease)21 (see Chapter 35). Circulating drugs (e.g., penicillin) or antigens produced from infectious diseases (e.g., hepatitis B, streptococcal infection) may form circulating immune complexes that are deposited in the circulation (vasculitis) or the kidneys (glomerulonephritis).
Autoimmunity
Breakdown of Tolerance
Self-antigens are usually in a state of tolerance, or immunologic homeostasis, with the host’s own immune system. Central tolerance develops in humans during the embryonic period as autoreactive lymphocytes are either eliminated or suppressed in the primary lymphoid organs during differentiation and proliferation of immature T or B lymphocytes (see Figures 8-10 and 8-12). Clones of cells with antigen receptors for self-antigens are deleted. Peripheral tolerance is maintained in the secondary lymphoid organs through the action of T-regulatory lymphocytes or antigen-presenting dendritic cells. Autoimmunity is a breakdown of tolerance in which the body’s immune system begins to recognize self-antigens as foreign. In most autoimmune conditions the mechanism of tolerance breakdown is unknown, although several potential mechanisms have been suggested.
Infectious Disease
A long-standing hypothesis is that foreign antigens from infectious microorganisms can initiate autoimmune disease through a process of molecular mimicry. Some antigens of infectious agents so closely resemble (mimic) a particular self-antigen that antibodies or T cells produced to protect against the infection also recognize the self-antigen as foreign (cross-reactive antibody or T cell). The relationship between many autoimmune diseases and predisposing infections, such as enterovirus infection and type 1 diabetes, is being investigated.22 However, the only clearly defined example so far is acute rheumatic fever that may occur after a group A streptococcal sore throat (see following).
Neoantigen
In certain situations a neoantigen that induces an allergic reaction may also lead to autoimmunity. Many neoantigens (new antigens) are haptens that become immunogenic after binding to self-proteins. The immune reaction against the neoantigen may lead to an immunologic reaction against normal antigenic determinants on the protein. Other new antigenic determinants may result from posttranslational modifications of a normal protein so that one or more amino acids are chemically altered (e.g., addition or removal of amino acid side groups, phosphorylation) without affecting the protein’s function.23 Many experimental autoimmune diseases (e.g., experimental autoimmune thyroiditis) can be initiated by immunization with molecules containing neoantigenic determinants.
Forbidden Clone
During differentiation and proliferation of lymphoid stem cells into immature T and B lymphocytes (see Figures 8-10 and 8-12), some lymphocytes produce receptors that react with self-antigens. Many autoreactive lymphocytes interact with self-antigens and other co-stimulatory molecules on the surface of thymic epithelial cells and are induced to undergo clonal deletion by a process of apoptosis. Thus lymphocytes reactive against self-antigen are prevented, or “forbidden,” from maturing. Autoimmunity may result from the survival of a forbidden clone and its proliferation later in life.
Original Insult
Although many theories exist, the initial cause of most autoimmune diseases is unknown (see What’s New? Maternal Microchimerism and Autoimmune Disease).
WHAT’S NEW?
Maternal/Fetal Microchimerism and Autoimmune Disease
A rapidly growing body of evidence suggests that we are in a state of microchimerism (Mc); possessing a small number of cells originating from another individual.1 The placenta, which was once considered a cellular “barrier,” is more porous than suspected and routinely allows bidirectional passage of both maternal and fetal cells. Passage results during normal pregnancy, as well as spontaneous miscarriages and elective terminations,2 and includes a broad spectrum of cells, ranging from erythrocytes and mature white blood cells, to stem cells at various stages of differentiation. Most gravid women (having been pregnant) have detectable fetal cells (fetal Mc) in their blood and many organs (e.g., white blood cells, hepatocytes, kidney tubular epithelium, neurons and glia, cardiomyocytes, endothelial cells, thyrocytes, intestinal epithelium, and islet β cells), which apparently originated as fetal stem cells that differentiated in the maternal tissues. As early as 6 weeks gestation, maternal Mc is apparent in fetal blood and tissues (e.g., thymus, lung, heart, pancreas, liver, spleen, kidney, adrenal gland, ovary, testis, and brain).3 Maternal Mc originates predominately from maternal cells, but may also include cells from one’s grandmother, a non-identical twin, or older siblings, all of which were harbored in the mother’s blood and tissues. Both maternal and fetal Mc persists for decades after pregnancy.
The long-term implications of Mc may include both detrimental and beneficial effects. Fetal cells express both maternal and paternal HLA antigens and are recognized as partially foreign by maternal immune cells. Most studies of maternal Mc use assays for the presence of the Y chromosome in maternal blood or organs. Many autoimmune diseases are associated with increased numbers of male cells in the diseased organs (e.g., scleroderma, dermatomyositis, Sjögren syndrome, thyroiditis, primary biliary cirrhosis, hyper- and hypothyroidism, and systemic lupus erythematosus) suggesting that Mc may contribute to the etiology of these diseases.1 Maternal Mc in the offspring also may affect their risk for autoimmune disease. Increased levels of maternal cells in the child’s blood have been reported in cases of juvenile inflammatory myopathy, neonatal lupus syndrome, type 1 diabetes, and biliary atresia. Maternal cells were found as myocytes in the myocardium of infants with fatal neonatal lupus-associated heart block and as insulin-producing cells in the pancreas of diabetics.1 It cannot yet be determined whether the foreign cells are initiators of autoimmune damage or whether injury to the tissue results in increased proliferation. Thus these observations remain intriguing but of unknown significance.
Beneficial effects of Mc are also observed. Treg cells may control immunologic tolerance to maternal cells. A significantly greater success rate of liver transplantation was observed with maternal or sibling donors than paternal donors.4 Thus Mc may modulate the immune response and minimize rejection of organs from selected donors. Fetal Mc occurs in several regions of the brain, but at a significantly lower incidence and concentration in women with Alzheimer’s disease than those without neurologic disease.5 Whether Mc is protective or not remains unknown.
References
1. Nelson, J. L. Trends Immunol. 2012; 33(8):421–427.
2. Peterson, S. E., et al. Human Reprod. 2012; 27(9):2607–2612.
3. Boyon, C., et al. Eur J Obset Gynecol Reprod Biol. 2011; 158(2):148–152.
4. Nijagal, A., Fleck, S., Mackenzie, T. C. Chimerism. 2012; 3(2):37–39.
5. Chan, W. F.N., et al. Plos ONE. 2012:e45592, doi; 7(9): 10.1371/journal.pone.0045592.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


