Chapter 17
Alterations in Cognitive Systems, Cerebral Hemodynamics, and Motor Function
Barbara J. Boss and Sue E. Huether

A person achieves functional adequacy (competence) through complex integrated processes. Three major neural systems account for this functional adequacy: cognitive systems, sensory systems, and motor systems. Alterations in any or all of these affect functional adequacy. Alterations in cognitive and sensory systems and motor function are associated with many central and peripheral nervous system injuries and pathologies. The purpose of this chapter is to present the concepts and processes of these alterations as an approach to understanding the manifestation of neurologic dysfunction. Some specific diseases are also presented (i.e., Parkinson disease, Huntington disease, and amyotrophic lateral sclerosis) because they fit best here. The manifestations of these concepts and processes are integrated with specific central and peripheral nervous system disorders and are presented in Chapter 18. Alterations in sensory function are presented in Chapter 16.
Alterations in Cognitive Systems
Consciousness has two distinct components: arousal (state of awakeness) and awareness (content of thought). Arousal is mediated by the reticular activating system, which regulates aspects of attention and information processing and maintains consciousness (see Figure 15-6). Cognitive cerebral functions require a functioning reticular activating system. Awareness encompasses all cognitive functions and is mediated by attentional systems, memory systems, language systems, and executive systems.
Alterations in Arousal
Pathophysiology
Structural alterations in arousal are divided according to original location of the pathologic condition or lesion: supratentorial (above the tentorium cerebelli); infratentorial (subtentorial, below the tentorium cerebelli); subdural (below the dura mater [see Figure 17-17]); extracerebral (outside the brain tissue); and intracerebral (within the brain tissue). Structural alterations can be caused by infectious, vascular, neoplastic, traumatic, congenital (developmental), degenerative, genetic, and metabolic disorders.
Psychogenic alterations in arousal (unresponsiveness), although uncommon, may signal general psychiatric disorders (see Chapter 19). Despite apparent unconsciousness, the person actually is physiologically awake and the neurologic examination reflects a normal response.
Clinical Manifestations and Evaluation
Patterns of clinical manifestations assist in determining the extent of brain dysfunction and serve as indexes for identifying increasing or decreasing central nervous system (CNS) function. Distinctions are made between metabolic and structurally induced manifestations (Table 17-1). The types of manifestations suggest the cause of the altered arousal state (Table 17-2). Five categories of neurologic function are critical to the evaluation process: (1) level of consciousness, (2) pattern of breathing, (3) pupillary changes, (4) oculomotor responses, and (5) motor responses.
TABLE 17-1
CLINICAL MANIFESTATIONS OF METABOLIC AND STRUCTURAL CAUSES OF ALTERED AROUSAL
| MANIFESTATION | METABOLICALLY INDUCED | STRUCTURALLY INDUCED |
| Blink to threat (cranial nerves II, VII) | Equal | Asymmetric |
| Discs (cranial nerve II) | Flat, good pulsation | Papilledema |
| Extraocular movement (cranial nerves III, IV, VI) | Roving eye movements; normal doll’s eyes and calorics | Gaze paresis, nerve III palsy, medial longitudinal fasciculus (MLF) syndrome (internuclear ophthalmoplegia) |
| Pupils (cranial nerves II, III) | Equal and reactive; may be large (e.g., atropine), pinpoint (e.g., opiates), or midposition and fixed (e.g., glutethimide [Doriden]) | Asymmetric and/or nonreactive; may be midposition (midbrain injury), pinpoint (pons injury), large (tectal injury) |
| Corneal reflex (cranial nerves V, VII) | Symmetric response | Asymmetric response |
| Grimace to pain (cranial nerve VII) | Symmetric response | Asymmetric response |
| Motor function movement | Symmetric | Asymmetric |
| Tone | Symmetric | Paratonic, spastic, flaccid, especially if asymmetric |
| Posture | Symmetric | Decorticate, especially if symmetric; decerebrate, especially if asymmetric |
| Deep tendon reflexes | Symmetric | Asymmetric |
| Babinski sign | Absent or symmetric response | Present |
| Sensation | Symmetric | Asymmetric |
TABLE 17-2
DIFFERENTIAL CHARACTERISTICS OF DISORDERS CAUSING ALTERED AROUSAL
| MECHANISM | MANIFESTATIONS |
| Supratentorial mass lesions compressing or displacing diencephalons or brainstem | Initiating signs usually of focal cerebral dysfunction Signs of dysfunction progress rostral to caudal Neurologic signs at any given time point to one anatomic area (e.g., diencephalon, mesencephalon, medulla) Motor signs often asymmetric |
| Infratentorial mass of destruction, causing coma | History of preceding brainstem dysfunction or sudden onset of coma Localizing brainstem signs precede or accompany onset of coma and always include oculovestibular abnormality Cranial nerve palsies; usually manifest “bizarre” respiratory patterns that appear at onset |
| Metabolic coma Exogenous toxins (drugs) Endogenous toxins (organ system failure) | Confusion and stupor commonly precede motor signs Motor signs usually are symmetric Pupillary reactions usually are preserved Asterixis, myoclonus, tremor, and seizures are common Acid-base imbalance with hyperventilation or hypoventilation is common |
| Psychiatric unresponsiveness | Lids close actively Pupils reactive or dilated (cycloplegics) Oculocephalic reflexes are unpredictable; oculovestibular reflexes are physiologic (nystagmus is present) Motor tone is inconsistent or normal Eupnea or hyperventilation is usual No pathologic reflexes are present Electroencephalogram (EEG) is normal |
Level of Consciousness
Level of consciousness is the most critical clinical index of nervous system function or dysfunction. Changes can indicate either improvement or deterioration of the individual’s condition and state of awakeness. A person who is alert and oriented to self, others, place, and time is considered to be functioning at the highest level of consciousness, which implies full use of all the person’s cognitive capacities. From this normal alert state, levels of consciousness diminish in stages from confusion to coma, each of which is clinically defined (Table 17-3).
TABLE 17-3
LEVELS OF ALTERED CONSCIOUSNESS
| STATE | DEFINITION |
| Confusion | Loss of ability to think rapidly and clearly; impaired judgment and decision making |
| Disorientation | Beginning loss of consciousness; disorientation to time followed by disorientation to place and impaired memory; lost last is recognition of self |
| Lethargy | Limited spontaneous movement or speech; easy arousal with normal speech or touch; may not be oriented to time, place, or person |
| Obtundation | Mild to moderate reduction in arousal (awakeness) with limited response to the environment; falls asleep unless stimulated verbally or tactilely; answers questions with minimum response |
| Stupor | A condition of deep sleep or unresponsiveness from which the person may be aroused or caused to open eyes only by vigorous and repeated stimulation; response is often withdrawal or grabbing at stimulus |
| Coma | No verbal response to the external environment or to any stimuli; noxious stimuli such as deep pain or suctioning yields motor movement |
| Light coma | Associated with purposeful movement on stimulation |
| Coma | Associated with nonpurposeful movement only on stimulation |
| Deep coma | Associated with unresponsiveness or no response to any stimulus |
Pattern of Breathing
Respiratory patterns help to evaluate level of brain dysfunction and level of coma. Rate, rhythm, and pattern of breathing should be assessed. The breathing patterns can be categorized as hemispheric or brainstem breathing patterns (Table 17-4 and Figure 17-1).
TABLE 17-4
| BREATHING PATTERN | DESCRIPTION | LOCATION OF INJURY |
| Hemispheric Breathing Patterns | ||
| Normal | After a period of hyperventilation that lowers the arterial carbon dioxide pressure (Paco2), the individual continues to breathe regularly but with a reduced depth. | Response of the nervous system to an external stressor—not associated with injury to the CNS |
| Posthyperventilation apnea | Respirations stop after hyperventilation has lowered the Pco2 level below normal. Rhythmic breathing returns when the Pco2 level returns to normal. (Usually an intact cerebral cortex will trigger breathing within 10 seconds regardless of Pco2.) | Associated with diffuse bilateral metabolic or structural disease of the cerebrum |
| Cheyne-Stokes respirations | Breathing pattern has a smooth increase (crescendo) in the rate and depth of breathing (hyperpnea), which peaks and is followed by a gradual smooth decrease (decrescendo) in the rate and depth of breathing to the point of apnea when the cycle repeats itself. The hyperpneic phase lasts longer than the apneic phase (represents an amplitude change). | Bilateral dysfunction of the deep cerebral or diencephalic structures; seen with supratentorial injury and metabolically induced coma states unrelated to neurologic dysfunction; may see also in CHF |
| Brainstem Breathing Patterns | ||
| Central reflex hyperpnea (central neurogenic hyperventilation) | A sustained deep rapid but regular pattern (hyperpnea) occurs, with a decreased Paco2 and a corresponding increase in pH and increased Po2. | May result from CNS damage or disease that involves the lower midbrain and upper pons; seen after increased intracranial pressure and blunt head trauma |
| Apneusis | A prolonged inspiratory cramp (a pause at full inspiration) occurs. A common variant of this is a brief end-inspiratory pause of 2 or 3 seconds, often alternating with an end-expiratory pause. | Indicates damage to the respiratory control mechanism located at the pontine level; most commonly associated with pontine infarction but documented with hypoglycemia, anoxia, and meningitis |
| Cluster breathing | A cluster of breaths has a disordered sequence with irregular pauses between breaths. | Dysfunction in the lower pontine and high medullary areas |
| Ataxic breathing | Completely irregular breathing occurs, with random shallow and deep breaths and irregular pauses. Often the rate is slow. | Originates from a primary dysfunction of the lower pons or upper medulla |
| Gasping breathing pattern (agonal gasps) | A pattern of deep “all-or-none” breaths is accompanied by a slow respiratory rate. | Indicative of a failing medullary respiratory center |
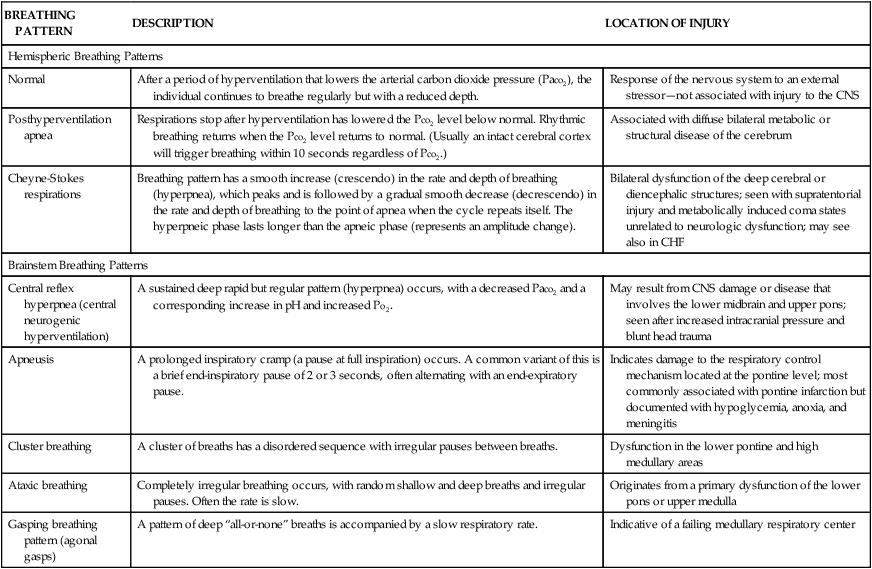
Central neurogenic hyperventilation is a respiratory pattern of sustained hyperventilation caused by a lesion in the central pons. Apneustic respirations have prolonged inspiratory and expiratory phases caused by injury to the pons or upper medulla. Cluster respirations are characterized by periods or clusters of rapid respirations of near equal depth resulting from trauma or compression to the medulla or from chronic opioid abuse. Ataxic respirations are irregular respirations with prolonged periods of apnea associated with damage to the medulla (see Figure 17-1).
Oculomotor Responses
Resting, spontaneous, and reflexive eye movements and oculovestibular (caloric) reflexes change at various levels of brain dysfunction (Table 17-5). Persons with metabolically induced coma, except in cases of barbiturate-hypnotic and phenytoin (Dilantin) poisoning, generally retain ocular reflexes, however, even when other signs of brainstem damage, such as central neurogenic hyperventilation, are present.
TABLE 17-5
CHANGES IN OCULOMOTOR RESPONSES
| STATE | RESTING AND SPONTANEOUS EYE MOVEMENTS | REFLEXIVE EYE MOVEMENTS |
| Full consciousness | Eyes at rest, still (cortical gaze centers inhibit spontaneous roving eye movements) | Eyes move as the head turns Oculocephalic responses not elicited or inconsistently elicited (frontal gaze centers inhibit brainstem reflexes that fix gaze straight ahead) Oculovestibular (caloric) stimulation produces nystagmus |
| Cortical dysfunction or disruption of efferent pathways | Conjugate, horizontal, roving eye movements may well be present (cortical gaze centers no longer inhibit these brainstem-generated roving eye movements) | Gaze fixed straight ahead regardless of head position—positive doll’s eyes reaction (normal oculocephalic reflexes are no longer inhibited by frontal gaze centers) |
| Diffuse anoxic damage to cortex | “Ocular dipping”—slow, dysrhythmic downward movement followed by faster, upward movement | Nystagmus is no longer induced by caloric stimulation (normally a cold-water stimulus produces deviation of the eyes opposite the irrigated ear; a warm-water stimulus deviates the eyes to the same [ipsilateral] side) |
| With an injury that depresses cortical gaze center function, the eyes (and often the entire head) deviate or appear to look toward the side of the injured hemisphere | ||
| With an injury that irritates (stimulates) the neurons of the cortical gaze center, the eyes (and often the entire head) deviate away from the injured hemisphere (all fibers from the frontal gaze centers decussate and therefore control the function of the contralateral pontine gaze center, which moves the eyes in the ipsilateral direction) | ||
| Mesencephalon dysfunction | Roving eye movements cease and the eyes become immobile and directed ahead (roving eye movements require an intact brainstem) Eyes may turn down and inward | Oculovestibular reflexes become inconsistent and abnormal Loss of Bell phenomenon (upward deviation of eyes on stimulation) (requires intact eye movement pathways from the mesencephalon to pons) |
| Pontine dysfunction | Loss of spontaneous blinking (requires an intact pons) “Ocular bobbing”—brisk, conjugate, downward movement of eyes with loss of horizontal eye movements |
The presence of brisk oculocephalic reflexes and roving eye movements, as well as the failure to elicit nystagmus with instillation of cold or warm water into the external ear canal, indicates a decrease in consciousness (loss of cortical influence) but an intact brainstem (Figures 17-3 and 17-4).
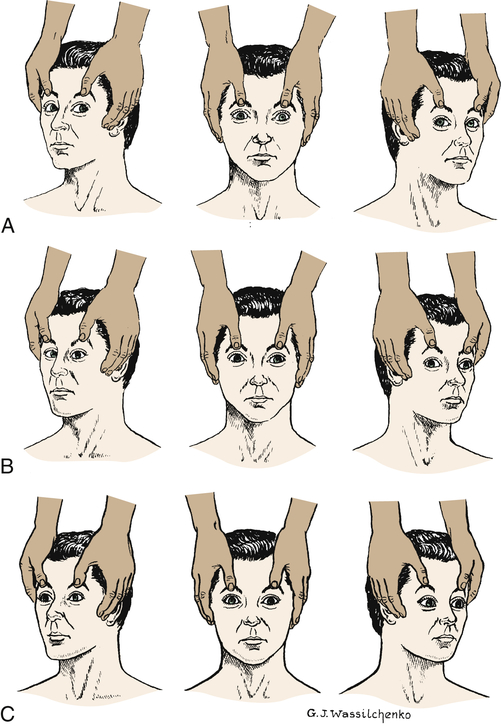
A, Normal response—eyes turn together to side opposite from turn of head. B, Abnormal response—eyes do not turn in conjugate manner. C, Absent response—eyes do not turn as head position changes. (A and C from Rudy EB: Advanced neurological and neurosurgical nursing, St Louis, 1984, Mosby.)
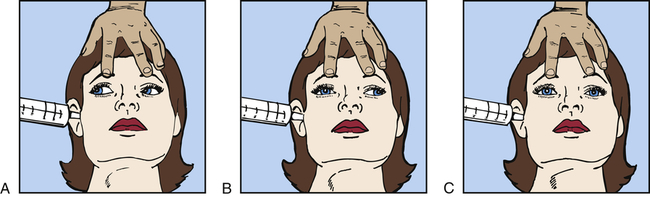
A, Normal response—conjugate eye movements. B, Abnormal response—dysconjugate or asymmetric eye movements. C, Absent response—no eye movements.
Motor Responses
Motor signs indicating loss of cortical inhibition are commonly associated with decreased consciousness. These signs include contralateral or bilateral (depending on whether the process is localized or diffuse) reflex grasping, reflex sucking, snout reflex, palmomental reflex, and rigidity (paratonia) (Figure 17-5). Abnormal flexor and extensor responses, particularly in the upper extremities, are defined in Table 17-6 and illustrated in Figure 17-6.
TABLE 17-6
ABNORMAL MOTOR RESPONSES WITH DECREASED RESPONSIVENESS
MOTOR RESPONSE | DESCRIPTION OF MOTOR RESPONSES | LOCATION OF INJURY |
| Decorticate posturing/rigidity: upper extremity flexion, lower extremity extension (Figure 17-6) | Slowly developing flexion of the arm, wrist, and fingers with abduction in the upper extremity and extension, internal rotation, and plantar flexion of the lower extremity | Hemispheric damage above midbrain releasing medullary and pontine reticulospinal systems |
| Decerebrate posturing/rigidity: upper and lower extremity extensor responses (Figure 17-6) | Opisthotonos (hyperextension of the vertebral column) with clenching of the teeth; extension, abduction, and hyperpronation of the arms; and extension of the lower extremities | Associated with severe damage involving midbrain or upper pons |
| In acute brain injury, shivering and hyperpnea may accompany unelicited recurrent decerebrate spasms | Acute brain injury may cause limb extension regardless of location | |
| Extensor responses in the upper extremities accompanied by flexion in the lower extremities | Pons | |
| Flaccid state with little or no motor response to stimuli | Lower pons and upper medulla |
Data from Goetz CG: Textbook of clinical neurology, ed 3, Philadelphia, 2007, Saunders; Nadeau SE et al: Medical neuroscience, Philadelphia, 2004, Saunders.
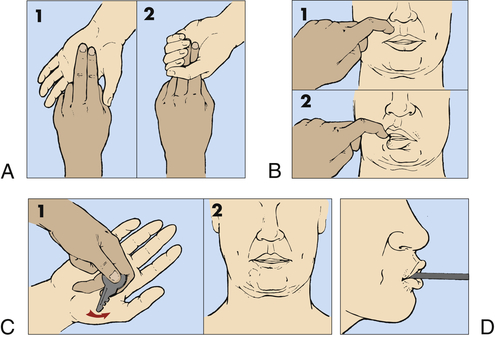
A, Grasp reflex. B, Snout reflex. C, Palmomental reflex. D, Suck reflex.
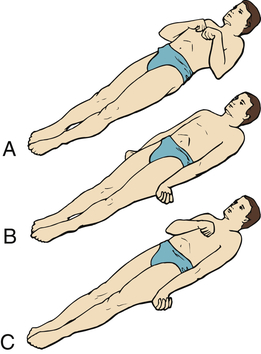
A, Decorticate response (flexor posturing): bilateral flexion of elbows and wrists with shoulder adduction in upper extremities. Extension, internal rotation of lower extremities. B, Decerebrate response (extensor posturing): all four extremities in rigid extension, internal rotation of shoulders with hyperpronation of forearms. C, Decorticate response on right side of body and decerebrate response on left side of body: the most pronounced response differences are in the upper body. (From Rudy EB: Advanced neurological and neurosurgical nursing, St Louis, 1984, Mosby.)
Outcomes of Alterations in Arousal
Clinical criteria for brain death include the absence of discernible evidence of cerebral hemisphere function or function of the brainstem’s vital centers for an extended period. There is no detectable function above the level of the foramen magnum so there is whole brain death. In addition, the abnormality of brain function must result from structural or known metabolic disease and not be caused by a depressant drug, alcohol poisoning, neuromuscular blockage, or hypothermia. An isoelectric, or flat, electroencephalogram (EEG) (electrocerebral silence) for a period of 6 to 12 hours in a person who is not hypothermic and has not ingested depressant drugs indicates that no mental recovery is possible. This usually means that the brain is already dead. A task force to determine brain death in children recommended the same criteria as those used for adults1 but with a longer observation period.
The following summary of medical criteria determines brain death2–5:
1. Completion of all appropriate and therapeutic procedures with no possibility of brain function recovery
2. Unresponsive coma (absence of motor and reflex movements)
3. No spontaneous respiration (apnea)—a Paco2 that rises above 60 mmHg without breathing efforts, providing evidence of a nonfunctioning respiratory center (apnea challenge)
4. No brainstem function (ocular responses to head turning or caloric stimulation; dilated, fixed pupils; no gag or corneal reflex)
5. Isoelectric (flat) EEG (electrocerebral silence)
6. Persistence of these signs for an appropriate observation period
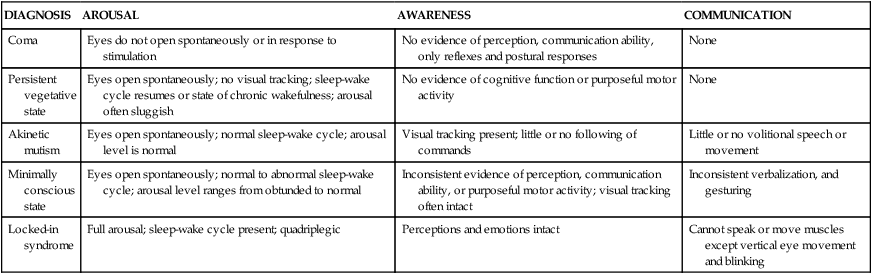
The recovery spectrum after severe brain injury includes: (1) coma, (2) vegetative state, (3) akinetic mutism, (4) minimally conscious state, and (5) locked-in syndrome (Table 17-7). The survivor of cerebral death may remain in a coma or emerge into a vegetative state. In coma (a state of unarousable neurobehavioral unresponsiveness) the eyes are usually closed with no evidence of eye opening either spontaneously or in response to external stimuli. The person does not follow commands, does not verbalize or mouth words, and has no goal-directed or volitional behavior. There are no sustained visual pursuit movements beyond a 45-degree arc.
TABLE 17-7
COMPARATIVE CLINICAL FEATURES OF ALTERATIONS IN LEVELS OF AROUSAL
| DIAGNOSIS | AROUSAL | AWARENESS | COMMUNICATION |
| Coma | Eyes do not open spontaneously or in response to stimulation | No evidence of perception, communication ability, only reflexes and postural responses | None |
| Persistent vegetative state | Eyes open spontaneously; no visual tracking; sleep-wake cycle resumes or state of chronic wakefulness; arousal often sluggish | No evidence of cognitive function or purposeful motor activity | None |
| Akinetic mutism | Eyes open spontaneously; normal sleep-wake cycle; arousal level is normal | Visual tracking present; little or no following of commands | Little or no volitional speech or movement |
| Minimally conscious state | Eyes open spontaneously; normal to abnormal sleep-wake cycle; arousal level ranges from obtunded to normal | Inconsistent evidence of perception, communication ability, or purposeful motor activity; visual tracking often intact | Inconsistent verbalization, and gesturing |
| Locked-in syndrome | Full arousal; sleep-wake cycle present; quadriplegic | Perceptions and emotions intact | Cannot speak or move muscles except vertical eye movement and blinking |
Data from Giacino JT et al: Neurology 58(3):349-353, 2002; Owen AM: Ann N Y Acad Sci 1125:225–238, 2008.
A persistent vegetative state (VS) is complete unawareness of the self or surrounding environment and complete loss of cognitive function. The Multi-Society Task Force on Persistent Vegetative States (MSTF) identified the diagnostic criteria for VS as: (1) periods of eye opening (spontaneous or following stimulation); (2) the potential for subcortical responses to external stimuli, including generalized physiologic responses to pain, such as posturing, tachycardia, and diaphoresis, and subcortical motor responses, such as grasp reflex; (3) return of so-called vegetative (autonomic) functions, including sleep-wake cycles and normalization of respiratory and digestive system functions; and (4) occasional roving eye movements without concomitant visual tracking ability.6 The person’s eyes open spontaneously or following stimulation, or both. There may be random hand, extremity, or head movements. The individual maintains blood pressure and breathing without support. Brainstem reflexes (pupillary, oculocephalic, chewing, swallowing) are intact. No discrete localizing motor responses are present, and the individual does not speak any comprehensible words or follow commands.7
In a minimally conscious state (MCS), an individual demonstrates minimal but defined behavioral evidence of self or environmental awareness.6,8 The clinical features include: (1) following simple commands, (2) manipulating objects, (3) responding with gestural or verbal “yes/no” responses, (4) demonstrating intelligible verbalization, and (5) blinking and smiling that occur in a meaningful relationship to the eliciting stimulus and are not attributable to reflexive activity.
Akinetic mutism (AM) is a neurobehavioral state characterized by a severe disturbance in behavioral drive (motivation). Generally, these individuals evidence eye opening with visual tracking and have little or no spontaneous speech or following of commands. Little movement is present. This is not attributable to decreased wakefulness or motor weakness or impairment. The pathology involves damage to the frontal lobe or cingulate gyrus (a component of the limbic system) (also see p. 538).9
With locked-in syndrome there is complete paralysis of voluntary muscles with the exception of eye movement. Content of thought and level of arousal are intact but the efferent pathways are disrupted (injury at the base of the pons with the reticular formation intact, often caused by basilar artery occlusion).10 Thus the individual cannot communicate either through speech or through body movement but is fully conscious, with intact cognitive function. The upper cranial nerves (I through IV) often are preserved, so that the person possesses vertical eye movement and blinking as a means of communication.
Alterations in Awareness
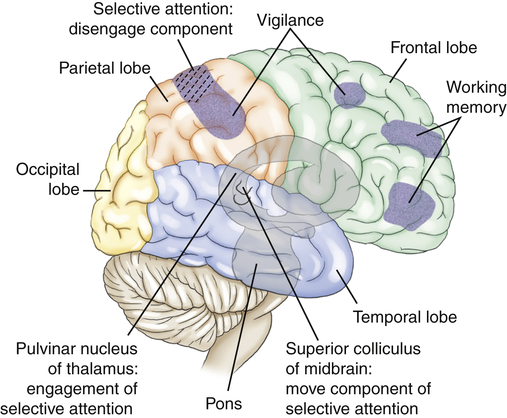
Selective attention (orienting) refers to the ability to select specific information to be processed from available, competing environmental and internal stimuli and to focus on those stimuli.11 Frontal and parietal regions of the right hemisphere contribute to selective attention. The engage component is mediated by the pulvinar nucleus of the thalamus (Figure 17-7). The disengagement mechanism is mediated by the right parietal lobe. The move component is mediated by the superior colliculi for visual orienting. A weak orienting network results in a neglect syndrome.
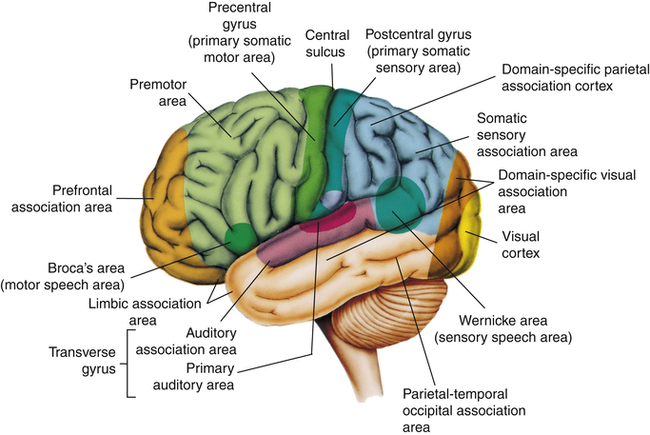
Memory is the recording, retention, and retrieval of knowledge. Two types of memory exist: declarative and nondeclarative (Figure 17-10). Declarative memory involves the learning and remembrance of episodic memories (personal history, events, and experiences) and semantic memories (facts and information). Declarative memory is mediated by domain-specific cortical areas of the association areas. This includes: (1) areas of the temporal, parietal, and occipital lobes (Figure 17-8), where long-term memories are thought to be stored; and (2) domain-independent areas of the medial temporal lobe (i.e., hippocampus), the diencephalon (thalamic structures and hypothalamus), and the basal forebrain (located ventral to the striatum and produces acetylcholine) (Figure 17-9), where it is thought distinct domain-specific features of an experience are related or bound.12
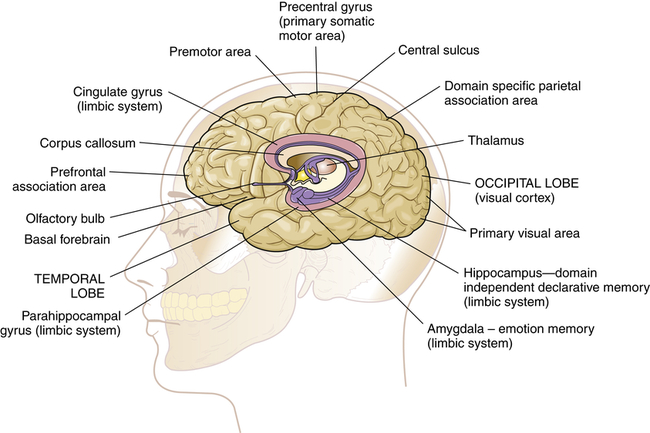
Nondeclarative memory (nonconscious), also called reflexive, procedural, or implicit memory, is the memory for actions, behaviors (habits), skills, and outcomes.13 It is not a language memory but a motor memory. Nondeclarative memory involves the construction of the motor pattern for the motor performance so that the action, behavior, or skill becomes increasingly more automatic. The striatum of the basal ganglia supports this learning across trials (stimulus-response learning), as well as probabilistic classification learning, which supports outcome prediction.14 All skills and habits are stored in this memory network. Cerebellar memory was originally thought to be related to only motor learning but it is now believed to involve nonmotor functions of cognition, emotion, and memory.15 Emotional memory is mediated by the amygdala (located on the inner surface of the temporal lobe) (see Figure 17-9). The amygdala attaches positive (e.g., pleasure) or negative (e.g., fear) dispositions to stimuli in the absence of conscious recollection of the circumstances of the emotional experience. Additionally, the amygdala modulates the event memory during and after the event (memory-enhancing effect).16 The reflex pathways mediate nonassociative learning (e.g., habituation [decreased response to a stimulus] and sensitization [increased response to a stimulus]) (see Figure 17-10).
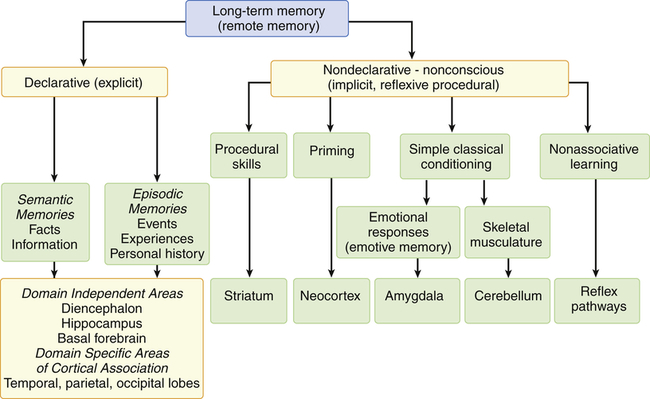
Dysmnesia is a disorder of the domain-independent declarative memory network. Dysmnesia is defined as the loss of past memories (retrograde amnesia) coupled with an inability to form new memories (anterograde amnesia) despite intact attentional networks. The hippocampus and other temporal lobe structures are often involved. Isolated (pure) domain-independent dysmnesia is caused by only a limited number of conditions, such as transient global dysmnesia (episodic global dysmnesia), amnestic stroke, and Korsakoff psychosis (amnestic or dysmnesic syndrome), as well as after temporal lobectomy. Many disorders may temporarily or permanently produce domain-independent dysmnesia that accompanies other deficits of the cognitive systems.16a A temporary domain-independent dysmnesia is found during complex partial seizures that persist for a time in the postictal state, in postconcussive states, and in mild posttraumatic brain injury states. A permanent domain-independent dysmnesia may be seen in several disorders including subarachnoid hemorrhage or moderate or severe posttraumatic brain injury states; carbon dioxide poisoning and other hypoxic or anoxic states; Wernicke encephalopathy, viral encephalitis, and granulomatous meningitides; tumors; and Alzheimer and Pick diseases.
A pure auditory or visual domain–specific declarative memory deficit manifests as an isolated agnosia (see Table 17-9). An isolated (pure) domain-specific declarative memory deficit of tactile sensations rarely occurs clinically because selective attention would likely be affected as well. A temporary auditory, visual, or tactile pattern recognition (remote memory) deficit may appear as a result of seizure activity or a postictal state. A temporary or permanent deficit can occur with temporal, occipital, or parietal lobe contusion; with subdural hematoma or ischemic stroke; and in encephalitis. A progressive domain-specific declarative memory deficit may occur in temporal, occipital, or parietal gliomas; in metastatic tumors; and in Alzheimer and Pick diseases.
TABLE 17-9
TYPES OF AGNOSIA (CONCEPT DISORDERS)
| TYPE OF AGNOSIA | DEFINITION | LOCATION OF INJURY |
| Tactile agnosia (astereognosis) | Inability to recognize objects by touch | Parietal lobe |
| Spatial agnosia | Incapacity to find one’s way around familiar places; disturbance of perception of space (disorders of [1] topographic [extrapersonal] orientation or [2] topographic and geographic memory [construction]) | Parietal lobe |
| Gerstmann syndrome | Loss of spatial orientation of fingers, body, sides, and numbers | Left angular gyrus (parietal lobe) |
| Finger agnosia (digital agnosia) | Inability to identify the names of one’s fingers | |
| Right-left confusion | Inability to distinguish right from left | |
| Agraphia | Inability to write | |
| Acalculia | Inability to perform mathematic calculations | |
| Visual agnosia | ||
| Object agnosia | Inability to recognize objects and pictures | Temporo-occipital area |
| Prosopagnosia | Inability to recognize faces | Temporo-occipital ventromesial region |
| Color agnosia | Inability to understand colors as qualities of objects; faulty color concepts and inability to evoke color images in the absence of color blindness; specific types: (1) “hue” problem, (2) color anomia (cannot name color) | Inferior occipital cortex in left hemisphere |
| Body image agnosias (may be spatial) | ||
| Anosognosia | Ignorance or denial of existence of the disease | Right parietal lobe |
| Autotopagnosia | Loss of ability to identify the body, in whole or in part, or to recognize relationships among various parts | Right parietal lobe |
| Word blindness (alexia/dyslexia) | Inability to recognize written symbols | Left parietotemporal region |
| Auditory agnosia (pure word deafness) | Inability to recognize speech sounds | Superior temporal area |
| Amusia (music deafness) | Loss of capacity to recognize tones and melodies | Right superior temporal area |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


