35
Agents Used in Dyslipidemia
CASE STUDY
A 42-year-old man with moderately severe coronary artery disease has a body mass index (BMI) of 29, increased abdominal girth, and hypertension that is well controlled. In addition to medicine for hypertension, he is taking 40 mg atorvastatin. Current lipid panel (mg/dL): cholesterol 184, triglycerides 200, LDL-C 110, HDL-C 34, non–HDL-C 150. Lipoprotein(a) (Lp[a]) is twice normal. Fasting glucose is 102 mg/dL, HbA1C is 6%, and fasting insulin is 38 μU/mL. Liver enzymes are normal. Creatine kinase level is mildly elevated. The patient is referred for help with management of his dyslipidemia. You advise dietary measures, exercise, and weight loss. Which additional drugs would help him achieve his lipoprotein treatment goals: LDL-C 50–60 mg/dL; triglycerides < 120 mg/dL; HDL-C > 45 mg/dL; and reduced level of Lp(a)? Would this patient also benefit from a drug to manage insulin resistance? If so, which drug?
Plasma lipids are transported in complexes called lipoproteins. Metabolic disorders that involve elevations in any lipoprotein species are termed hyperlipoproteinemias or hyperlipidemias. Hyperlipemia denotes increased levels of triglycerides.
The two major clinical sequelae of hyperlipidemias are acute pancreatitis and atherosclerosis. The former occurs in patients with marked hyperlipemia. Control of triglycerides can prevent recurrent attacks of this life-threatening disease.
Atherosclerosis is the leading cause of death for both genders in the USA and other Western countries. Lipoproteins that contain apolipoprotein (apo) B-100 convey lipids into the artery wall. These are low-density (LDL), intermediate-density (IDL), very-low-density (VLDL), and lipoprotein(a) (Lp[a]). Remnant lipoproteins formed during the catabolism of chylomicrons that contain the B-48 protein (apo B-48) can also enter the artery wall, contributing to atherosclerosis.
Cellular components in atherosclerotic plaques include foam cells, which are transformed macrophages, and smooth muscle cells filled with cholesteryl esters. These cellular alterations result from endocytosis of modified lipoproteins via at least four species of scavenger receptors. Chemical modification of lipoproteins by free radicals creates ligands for these receptors. The atheroma grows with the accumulation of foam cells, collagen, fibrin, and frequently calcium. Whereas such lesions can slowly occlude coronary vessels, clinical symptoms are more frequently precipitated by rupture of unstable atheromatous plaques, leading to activation of platelets and formation of occlusive thrombi.
Although treatment of hyperlipidemia can cause slow physical regression of plaques, the well-documented reduction in acute coronary events that follows vigorous lipid-lowering treatment is attributable chiefly to mitigation of the inflammatory activity of macrophages and is evident within 2–3 months after starting therapy.
High-density lipoproteins (HDL) exert several antiatherogenic effects. They participate in retrieval of cholesterol from the artery wall and inhibit the oxidation of atherogenic lipoproteins. Low levels of HDL (hypoalphalipoproteinemia) are an independent risk factor for atherosclerotic disease and thus are a potential target for intervention.
Cigarette smoking is a major risk factor for coronary disease. It is associated with reduced levels of HDL, impairment of cholesterol retrieval, cytotoxic effects on the endothelium, increased oxidation of lipoproteins, and stimulation of thrombogenesis. Diabetes, also a major risk factor, is another source of oxidative stress.
Normal coronary arteries can dilate in response to ischemia, increasing delivery of oxygen to the myocardium. This process is mediated by nitric oxide, acting on smooth muscle cells of the arterial media. This function is impaired by atherogenic lipoproteins, thus aggravating ischemia. Reducing levels of atherogenic lipoproteins and inhibiting their oxidation restores endothelial function.
Because atherogenesis is multifactorial, therapy should be directed toward all modifiable risk factors. Atherogenesis is a dynamic process. Quantitative angiographic trials have demonstrated net regression of plaques during aggressive lipid-lowering therapy. Primary and secondary prevention trials have shown significant reduction in mortality from new coronary events and in all-cause mortality.
 PATHOPHYSIOLOGY OF HYPERLIPOPROTEINEMIA
PATHOPHYSIOLOGY OF HYPERLIPOPROTEINEMIA
NORMAL LIPOPROTEIN METABOLISM
Structure
Lipoproteins have hydrophobic core regions containing cholesteryl esters and triglycerides surrounded by unesterified cholesterol, phospholipids, and apoproteins. Certain lipoproteins contain very high-molecular-weight B proteins that exist in two forms: B-48, formed in the intestine and found in chylomicrons and their remnants; and B-100, synthesized in liver and found in VLDL, VLDL remnants (IDL), LDL (formed from VLDL), and Lp(a) lipoproteins. HDL consist of at least 20 discrete molecular species containing apolipoprotein A-I (apo A-I). About 100 other proteins are known to be distributed variously among the HDL species.
Synthesis & Catabolism
A. Chylomicrons
Chylomicrons are formed in the intestine and carry triglycerides of dietary origin, unesterified cholesterol, and cholesteryl esters. They transit the thoracic duct to the bloodstream.
ACRONYMS
Triglycerides are removed in extrahepatic tissues through a pathway shared with VLDL that involves hydrolysis by the lipoprotein lipase (LPL) system. Decrease in particle diameter occurs as triglycerides are depleted. Surface lipids and small apoproteins are transferred to HDL. The resultant chylomicron remnants are taken up by receptor-mediated endocytosis into hepatocytes.
B. Very-Low-Density Lipoproteins
VLDL are secreted by liver and export triglycerides to peripheral tissues (Figure 35–1). VLDL triglycerides are hydrolyzed by LPL, yielding free fatty acids for storage in adipose tissue and for oxidation in tissues such as cardiac and skeletal muscle. Depletion of triglycerides produces remnants (IDL), some of which undergo endocytosis directly into hepatocytes. The remainder is converted to LDL by further removal of triglycerides mediated by hepatic lipase. This process explains the “beta shift” phenomenon, the increase of LDL (beta-lipoprotein) in serum as hypertriglyceridemia subsides. Increased levels of LDL can also result from increased secretion of VLDL and from decreased LDL catabolism.
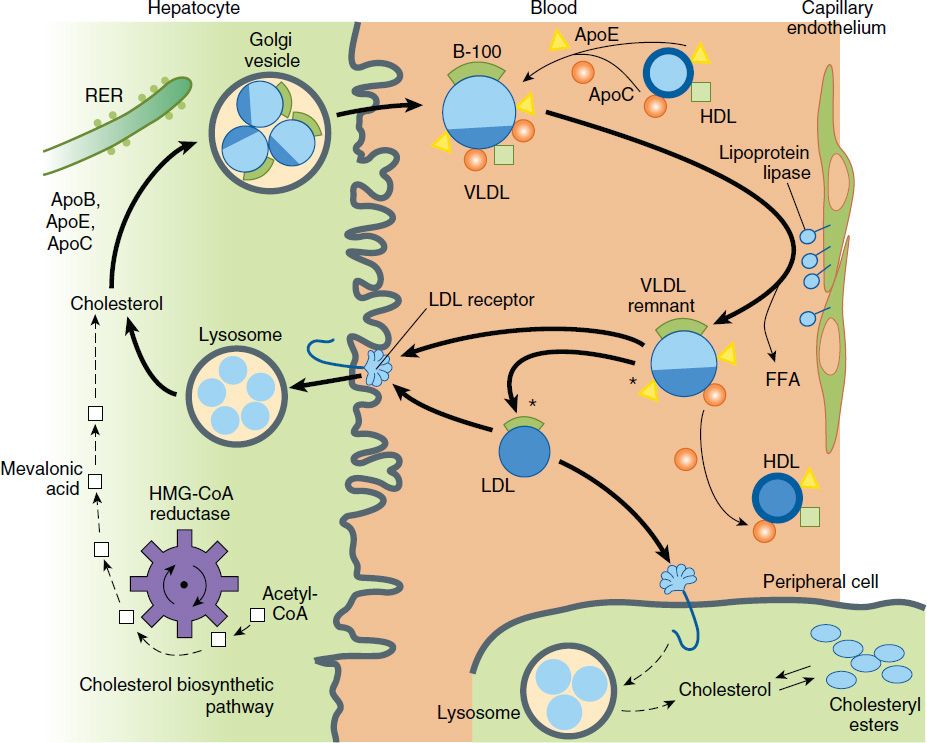
FIGURE 35–1 Metabolism of lipoproteins of hepatic origin. The heavy arrows show the primary pathways. Nascent VLDL are secreted via the Golgi apparatus. They acquire additional apo C lipoproteins and apo E from HDL. Very-low-density lipoproteins (VLDL) are converted to VLDL remnants (IDL) by lipolysis via lipoprotein lipase in the vessels of peripheral tissues. In the process, C apolipoproteins and a portion of the apo E are given back to high-density lipoproteins (HDL). Some of the VLDL remnants are converted to LDL by further loss of triglycerides and loss of apo E. A major pathway for LDL degradation involves the endocytosis of LDL by LDL receptors in the liver and the peripheral tissues, for which apo B-100 is the ligand. Dark color denotes cholesteryl esters; light color denotes triglycerides; the asterisk denotes a functional ligand for LDL receptors; triangles indicate apo E; circles and squares represent C apolipoproteins. FFA, free fatty acid; RER, rough endoplasmic reticulum. (Adapted, with permission, from Kane J, Malloy M: Disorders of lipoproteins. In: Rosenberg RN et al [editors]: The Molecular and Genetic Basis of Neurological Disease. 2nd ed. Butterworth-Heinemann, 1997.)
C. Low-Density Lipoproteins
LDL are catabolized chiefly in hepatocytes and other cells by receptor-mediated endocytosis. Cholesteryl esters from LDL are hydrolyzed, yielding free cholesterol for the synthesis of cell membranes. Cells also obtain cholesterol by synthesis via a pathway involving the formation of mevalonic acid by HMG-CoA reductase. Production of this enzyme and of LDL receptors is transcriptionally regulated by the content of cholesterol in the cell. Normally, about 70% of LDL is removed from plasma by hepatocytes. Even more cholesterol is delivered to the liver via IDL and chylomicrons. Unlike other cells, hepatocytes can eliminate cholesterol by secretion in bile and by conversion to bile acids.
D. Lp(a) Lipoprotein
Lp(a) lipoprotein is formed from LDL and the (a) protein, linked by a disulfide bridge. The (a) protein is highly homologous with plasminogen but is not activated by tissue plasminogen activator. It occurs in a number of isoforms of different molecular weights. Levels of Lp(a) vary from nil to over 2000 nM/L and are determined chiefly by genetic factors. Lp(a) can be found in atherosclerotic plaques and may also contribute to coronary disease by inhibiting thrombolysis. Levels are elevated in certain inflammatory states. The risk of coronary disease is strongly related to the level of Lp(a). A common variant (I4399M) in the coding region is associated with elevated levels.
E. High-Density Lipoproteins
The apoproteins of HDL are secreted by the liver and intestine. Much of the lipid comes from the surface monolayers of chylomicrons and VLDL during lipolysis. HDL also acquires cholesterol from peripheral tissues, protecting the cholesterol homeostasis of cells. Free cholesterol is transported from the cell membrane chiefly by a transporter, ABCA1, acquired by a small particle termed prebeta-1 HDL, and then esterified by lecithin:cholesterol acyltransferase (LCAT), leading to the formation of larger HDL species. Cholesterol is also exported from macrophages by the ABCG1 transporter and the docking scavenger receptor, SR-BI, to large HDL particles. The cholesteryl esters are transferred to VLDL, IDL, LDL, and chylomicron remnants with the aid of cholesteryl ester transfer protein (CETP). Much of the cholesteryl ester thus transferred is ultimately delivered to the liver by endocytosis of the acceptor lipoproteins. HDL can also deliver cholesteryl esters directly to the liver via SR-BI that does not cause endocytosis of the lipoproteins. At the population level, HDL-C levels relate inversely to atherosclerosis risk. Among individuals, the capacity to accept exported cholesterol can vary widely at identical levels of HDL-C. The ability of peripheral tissues to export cholesterol via the transporter mechanism and the acceptor capacity of HDL are emerging as major determinants of coronary atherosclerosis.
LIPOPROTEIN DISORDERS
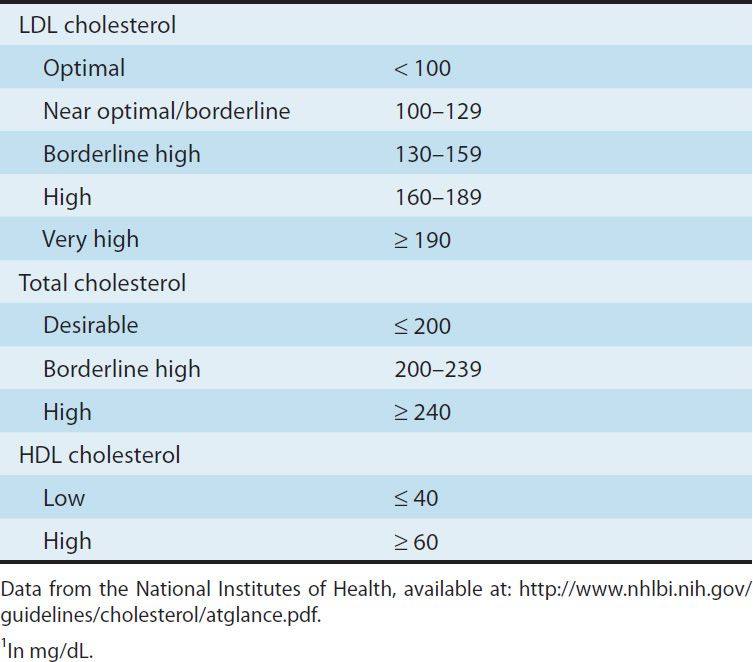
Lipoprotein disorders are detected by measuring lipids in serum after a 10-hour fast. Risk of heart disease increases with concentrations of the atherogenic lipoproteins, is inversely related to levels of HDL, and is modified by other risk factors (Table 35–1). Evidence from clinical trials suggests that LDL cholesterol levels of 60 mg/dL may be optimal for patients with coronary disease. Ideally, triglycerides should be below 120 mg/dL. Although LDL-C is still the primary target of treatment, reducing the levels of VLDL and IDL is also important. Calculation of non-HDL cholesterol provides a means of assessing levels of all the lipoproteins in the VLDL to LDL cascade. Differentiation of the disorders requires identification of the lipoproteins involved (Table 35–2). Diagnosis of a primary disorder usually requires further clinical and genetic data as well as ruling out secondary hyperlipidemias (Table 35–3).
TABLE 35–1 Current blood lipid guidelines.1
Phenotypes of abnormal lipoprotein distribution are described in this section. Drugs mentioned for use in these conditions are described in the following section on basic and clinical pharmacology.
THE PRIMARY HYPERTRIGLYCERIDEMIAS
Hypertriglyceridemia is associated with increased risk of coronary disease. VLDL and IDL have been found in atherosclerotic plaques. These patients tend to have cholesterol-rich VLDL of small-particle diameter and small, dense LDL. Hypertriglyceridemic patients with coronary disease or risk equivalents should be treated aggressively. Patients with triglycerides above 700 mg/dL should be treated to prevent acute pancreatitis because the LPL clearance mechanism is saturated at about this level.
Hypertriglyceridemia is an important component of the metabolic syndrome, which also includes low levels of HDL-C, insulin resistance, hypertension, and abdominal obesity. Hyperuricemia is frequently present. Insulin resistance appears to be central to this disorder. Management of these patients frequently requires, in addition to a fibrate, the use of metformin, another agent, or both (see Chapter 41). The severity of hypertriglyceridemia of any cause is increased in the presence of the metabolic syndrome or type 2 diabetes.
Primary Chylomicronemia
Chylomicrons are not present in the serum of normal individuals who have fasted 10 hours. The recessive traits of deficiency of LPL or its cofactor, apo C-II, are usually associated with severe lipemia (2000–3000 mg/dL of triglycerides when the patient is consuming a typical American diet). These disorders might not be diagnosed until an attack of acute pancreatitis occurs. Patients may have eruptive xanthomas, hepatosplenomegaly, hypersplenism, and lipid-laden foam cells in bone marrow, liver, and spleen. The lipemia is aggravated by estrogens because they stimulate VLDL production, and pregnancy may cause marked increases in triglycerides despite strict dietary control. Although these patients have a predominant chylomicronemia, they may also have moderately elevated VLDL, presenting with a pattern called mixed lipemia (fasting chylomicronemia and elevated VLDL). LPL deficiency is diagnosed by assay of lipolytic activity after intravenous injection of heparin. A presumptive diagnosis is made by demonstrating a pronounced decrease in triglycerides a few days after reduction of daily fat intake below 15 g. Marked restriction of total dietary fat is the basis of effective long-term treatment. Niacin, a fibrate, or marine omega-3 fatty acids may be of some benefit if VLDL levels are increased. Genetic variants at other loci that participate in intravascular lipolysis, including LMF1, apo A-V, GPI-HDL BP1, and apo C-III, can have profound effects on triglyceride levels.
Familial Hypertriglyceridemia
A. Severe (Usually Mixed Lipemia)
Mixed lipemia usually results from impaired removal of triglyceride-rich lipoproteins. Factors that increase VLDL production aggravate the lipemia because VLDL and chylomicrons are competing substrates for LPL. The primary mixed lipemias probably reflect a variety of genetic determinants. Most patients have centripetal obesity with insulin resistance. Other factors that increase secretion of VLDL also worsen the lipemia. Eruptive xanthomas, lipemia retinalis, epigastric pain, and pancreatitis are variably present depending on the severity of the lipemia. Treatment is primarily dietary, with restriction of total fat, avoidance of alcohol and exogenous estrogens, weight reduction, exercise, and supplementation with marine omega-3 fatty acids. Most patients also require treatment with a fibrate. If insulin resistance is not present, niacin may be useful.
B. Moderate
Increased levels of VLDL can also reflect a genetic predisposition and are worsened by factors that increase the rate of VLDL secretion from liver, ie, obesity, alcohol, diabetes, and estrogens. Treatment includes addressing these issues and the use of fibrates or niacin as needed. Marine omega-3 fatty acids are a valuable adjuvant.
Familial Combined Hyperlipoproteinemia (FCH)
In this common disorder associated with an increased incidence of coronary disease, individuals may have elevated levels of VLDL, LDL, or both, and the pattern may change with time. Familial combined hyperlipoproteinemia involves an approximate doubling in VLDL secretion and appears to be transmitted as a dominant trait. Triglycerides can be increased by the factors noted above. Elevations of cholesterol and triglycerides are generally moderate, and xanthomas are absent. Diet alone does not normalize lipid levels. A reductase inhibitor alone, or in combination with niacin or fenofibrate, is usually required to treat these patients. When fenofibrate is combined with a reductase inhibitor, either pravastatin or rosuvastatin is recommended because neither is metabolized via CYP3A4. Marine omega-3 fatty acids may be useful.
Familial Dysbetalipoproteinemia
In this disorder, remnants of chylomicrons and VLDL accumulate and levels of LDL are decreased. Because remnants are rich in cholesteryl esters, the level of total cholesterol may be as high as that of triglycerides. Diagnosis is confirmed by the absence of the ε3 and ε4 alleles of apo E, the ε2/ε2 genotype. Other apo E isoforms that lack receptor ligand properties can also be associated with this disorder. Patients often develop tuberous or tuberoeruptive xanthomas, or characteristic planar xanthomas of the palmar creases. They tend to be obese, and some have impaired glucose tolerance. These factors, as well as hypothyroidism, can aggravate the lipemia. Coronary and peripheral atherosclerosis occurs with increased frequency. Weight loss, together with decreased fat, cholesterol, and alcohol consumption, may be sufficient, but a fibrate or niacin is usually needed to control the condition. These agents can be given together in more resistant cases. Reductase inhibitors are also effective because they increase hepatic LDL receptors that participate in remnant removal.
THE PRIMARY HYPERCHOLESTEROLEMIAS
Familial Hypercholesterolemia (FH)
Familial hypercholesterolemia is an autosomal dominant trait. Although levels of LDL tend to increase throughout childhood, the diagnosis can often be made on the basis of elevated umbilical cord blood cholesterol. In most heterozygotes, cholesterol levels range from 260 to 500 mg/dL. Triglycerides are usually normal. Tendon xanthomas are often present. Arcus corneae and xanthelasma may appear in the third decade. Coronary disease tends to occur prematurely. In homozygous familial hypercholesterolemia, which can lead to coronary disease in childhood, levels of cholesterol often exceed 1000 mg/dL and early tuberous and tendinous xanthomas occur. These patients may also develop elevated plaque-like xanthomas of the aortic valve, digital webs, buttocks, and extremities.
Defects of LDL receptors underlie familial hypercholesterolemia. Some individuals have combined heterozygosity for alleles producing nonfunctional and kinetically impaired receptors. In heterozygous patients, LDL can be normalized with reductase inhibitors or combined drug regimens (Figure 35–2). Homozygotes and those with combined heterozygosity whose receptors retain even minimal function may partially respond to niacin, ezetimibe, and reductase inhibitors. Emerging therapies for these patients include mipomersen, employing an antisense strategy targeted at apo B-100, and lomitapide, a small molecule inhibitor of microsomal triglyceride transfer protein (MTP). LDL apheresis is effective in medication-refractory patients.
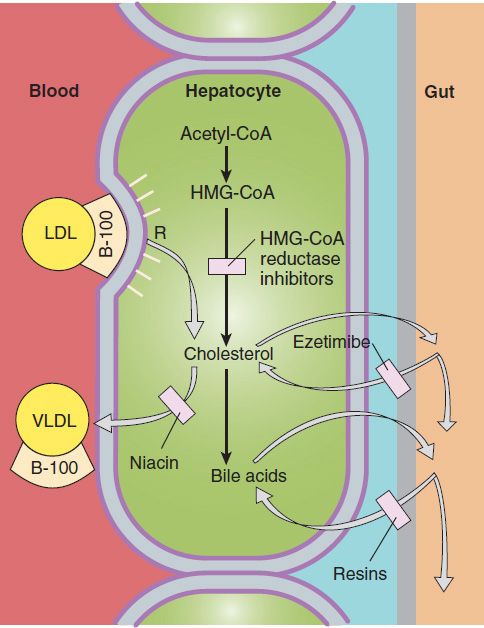
FIGURE 35–2 Sites of action of HMG-CoA reductase inhibitors, niacin, ezetimibe, and resins used in treating hyperlipidemias. Low-density lipoprotein (LDL) receptors are increased by treatment with resins and HMG-CoA reductase inhibitors. VLDL, very-low-density lipoproteins; R, LDL receptor.
Familial Ligand-Defective Apolipoprotein B-100
Defects in the domain of apo B-100 that binds to the LDL receptor impair the endocytosis of LDL, leading to hypercholesterolemia of moderate severity. Tendon xanthomas may occur. Response to reductase inhibitors is variable. Up-regulation of LDL receptors in liver increases endocytosis of LDL precursors but does not increase uptake of ligand-defective LDL particles. Niacin often has beneficial effects by reducing VLDL production.
Familial Combined Hyperlipoproteinemia (FCH)
As described, some persons with familial combined hyperlipoproteinemia have only an elevation in LDL. Serum cholesterol is often less than 350 mg/dL. Dietary and drug treatment, usually with a reductase inhibitor, is indicated. It may be necessary to add niacin or ezetimibe to normalize LDL.
Lp(a) Hyperlipoproteinemia
This familial disorder, which is associated with increased atherogenesis and arterial thrombus formation, is determined chiefly by alleles that dictate increased production of the (a) protein moiety. Lp(a) can be secondarily elevated in patients with severe nephrosis and certain other inflammatory states. Niacin reduces levels of Lp(a) in many patients. Reduction of levels of LDL-C below 100 mg/dL decreases the risk attributable to Lp(a), as does the administration of low dose aspirin.
Cholesteryl Ester Storage Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


