14
Agents Used in Cardiac Arrhythmias
CASE STUDY
A 69-year-old retired teacher presents with a 1-month history of palpitations, intermittent shortness of breath, and fatigue. She has a history of hypertension. An ECG shows atrial fibrillation with a ventricular response of 122 beats/min (bpm) and signs of left ventricular hypertrophy. She is anticoagulated with warfarin and started on sustained-release metoprolol, 50 mg/d. After 7 days, her rhythm reverts to normal sinus rhythm spontaneously. However, over the ensuing month, she continues to have intermittent palpitations and fatigue. Continuous ECG recording over a 48-hour period documents paroxysms of atrial fibrillation with heart rates of 88–114 bpm. An echocardiogram shows a left ventricular ejection fraction of 38% with no localized wall motion abnormality. At this stage, would you initiate treatment with an antiarrhythmic drug to maintain normal sinus rhythm, and if so, what drug would you choose?
Cardiac arrhythmias are a common problem in clinical practice, occurring in up to 25% of patients treated with digitalis, 50% of anesthetized patients, and over 80% of patients with acute myocardial infarction. Arrhythmias may require treatment because rhythms that are too rapid, too slow, or asynchronous can reduce cardiac output. Some arrhythmias can precipitate more serious or even lethal rhythm disturbances; for example, early premature ventricular depolarizations can precipitate ventricular fibrillation. In such patients, antiarrhythmic drugs may be lifesaving. On the other hand, the hazards of antiarrhythmic drugs—and in particular the fact that they can precipitate lethal arrhythmias in some patients—has led to a reevaluation of their relative risks and benefits. In general, treatment of asymptomatic or minimally symptomatic arrhythmias should be avoided for this reason.
Arrhythmias can be treated with the drugs discussed in this chapter and with nonpharmacologic therapies such as pacemakers, cardioversion, catheter ablation, and surgery. This chapter describes the pharmacology of drugs that suppress arrhythmias by a direct action on the cardiac cell membrane. Other modes of therapy are discussed briefly (see Box: The Nonpharmacologic Therapy of Cardiac Arrhythmias, later in the chapter).
ELECTROPHYSIOLOGY OF NORMAL CARDIAC RHYTHM
The electrical impulse that triggers a normal cardiac contraction originates at regular intervals in the sinoatrial (SA) node (Figure 14–1), usually at a frequency of 60–100 bpm. This impulse spreads rapidly through the atria and enters the atrioventricular (AV) node, which is normally the only conduction pathway between the atria and ventricles. Conduction through the AV node is slow, requiring about 0.15 seconds. (This delay provides time for atrial contraction to propel blood into the ventricles.) The impulse then propagates over the His-Purkinje system and invades all parts of the ventricles, beginning with the endocardial surface near the apex and ending with the epicardial surface at the base of the heart. Ventricular activation is complete in less than 0.1 seconds; therefore, contraction of all of the ventricular muscle is normally synchronous and hemodynamically effective.
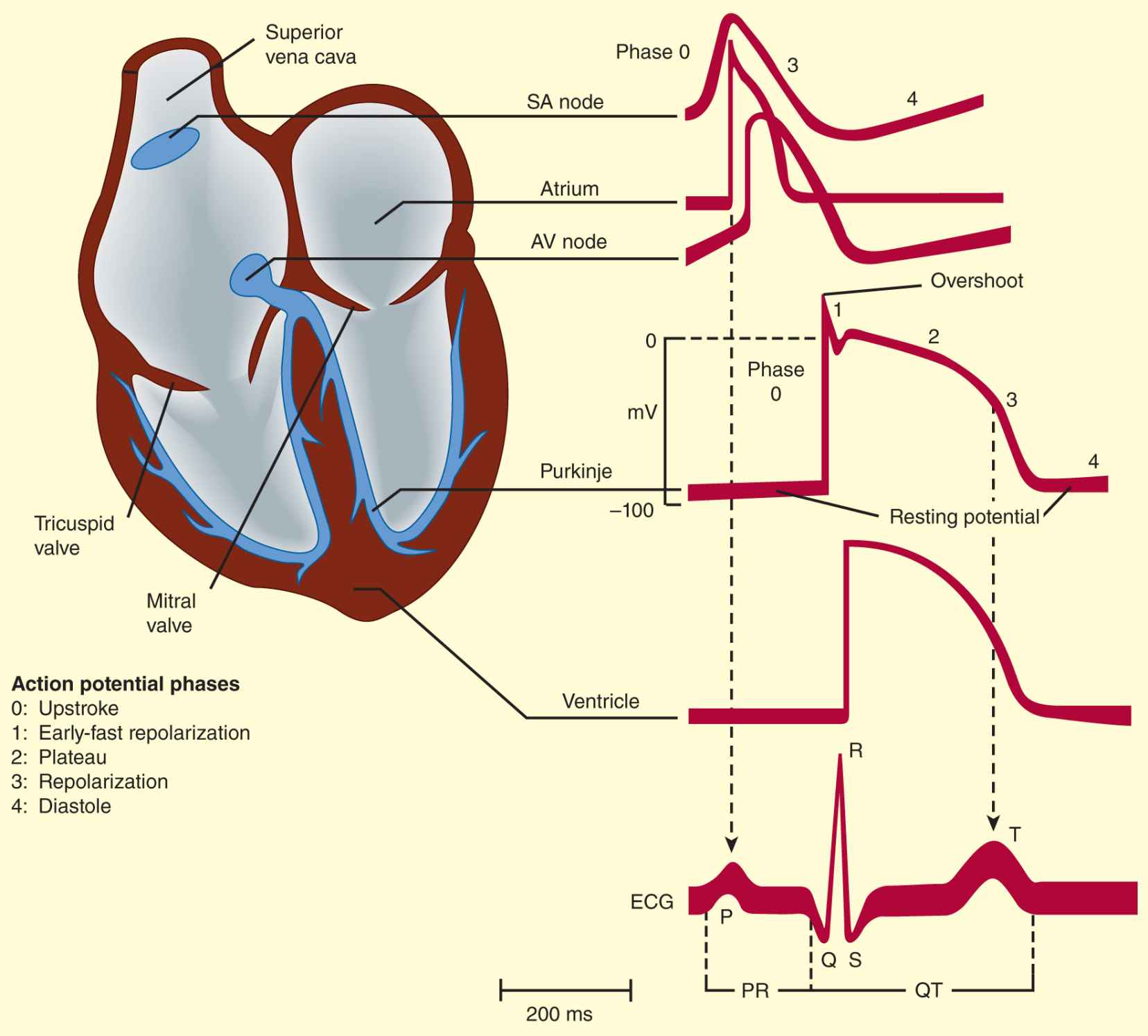
FIGURE 14–1 Schematic representation of the heart and normal cardiac electrical activity (intracellular recordings from areas indicated and ECG). Sinoatrial (SA) node, atrioventricular (AV) node, and Purkinje cells display pacemaker activity (phase 4 depolarization). The ECG is the body surface manifestation of the depolarization and repolarization waves of the heart. The P wave is generated by atrial depolarization, the QRS by ventricular muscle depolarization, and the T wave by ventricular repolarization. Thus, the PR interval is a measure of conduction time from atrium to ventricle, and the QRS duration indicates the time required for all of the ventricular cells to be activated (ie, the intraventricular conduction time). The QT interval reflects the duration of the ventricular action potential.
Arrhythmias consist of cardiac depolarizations that deviate from the above description in one or more aspects: there is an abnormality in the site of origin of the impulse, its rate or regularity, or its conduction.
Ionic Basis of Membrane Electrical Activity
The transmembrane potential of cardiac cells is determined by the concentrations of several ions—chiefly sodium (Na+), potassium (K+), calcium (Ca2+), and chloride (Cl–)—on either side of the membrane and the permeability of the membrane to each ion. These water-soluble ions are unable to freely diffuse across the lipid cell membrane in response to their electrical and concentration gradients; they require aqueous channels (specific pore-forming proteins) for such diffusion. Thus, ions move across cell membranes in response to their gradients only at specific times during the cardiac cycle when these ion channels are open. The movements of the ions produce currents that form the basis of the cardiac action potential. Individual channels are relatively ion-specific, and the flux of ions through them is controlled by “gates” (flexible portions of the peptide chains that make up the channel proteins). Each type of channel has its own type of gate (sodium, calcium, and some potassium channels are each thought to have two types of gates). The channels primarily responsible for the cardiac action potential (sodium, calcium, and several potassium) are opened and closed (“gated”) by voltage changes across the cell membrane; that is, they are voltage-sensitive. Most are also modulated by ion concentrations and metabolic conditions, and some potassium channels are primarily ligand- rather than voltage-gated.
The ionic currents that are thought to contribute to the cardiac action potential are illustrated in Figure 14–2. At rest, most cells are not significantly permeable to sodium, but at the start of each action potential, they become quite permeable (see below). In electrophysiologic terms, the conductance of the fast sodium channel suddenly increases in response to a depolarizing stimulus. Similarly, calcium enters and potassium leaves the cell with each action potential. Therefore, in addition to ion channels, the cell must have mechanisms to maintain stable transmembrane ionic conditions by establishing and maintaining ion gradients. The most important of these active mechanisms is the sodium pump, Na+/K+-ATPase, described in Chapter 13. This pump and other active ion carriers contribute indirectly to the transmembrane potential by maintaining the gradients necessary for diffusion through channels. In addition, some pumps and exchangers produce net current flow (eg, by exchanging three Na+ for two K+ ions) and hence are termed “electrogenic.”
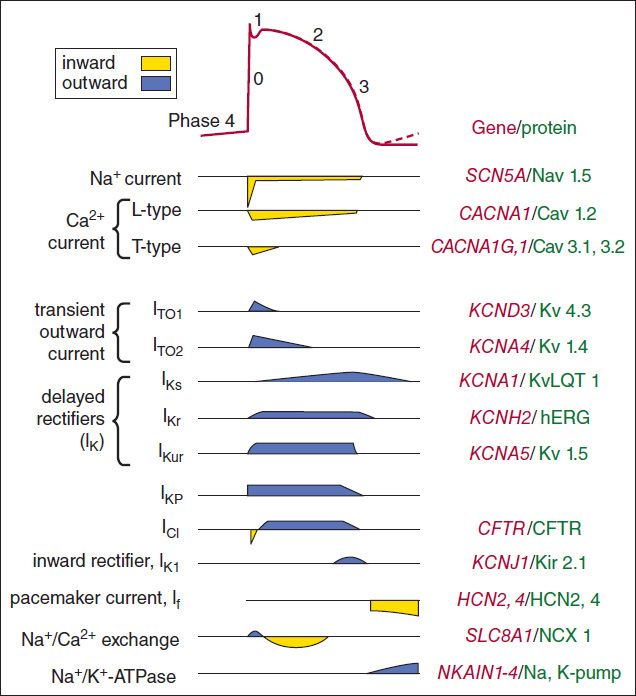
FIGURE 14–2 Schematic diagram of the ion permeability changes and transport processes that occur during an action potential and the diastolic period following it. Yellow indicates inward (depolarizing) membrane currents; blue indicates outward (repolarizing) membrane currents. Multiple subtypes of potassium and calcium currents, with different sensitivities to blocking drugs, have been identified. The right side of the figure lists the genes and proteins responsible for each type of channel or transporter.
When the cardiac cell membrane becomes permeable to a specific ion (ie, when the channels selective for that ion are open), movement of that ion across the cell membrane is determined by Ohm’s law: current = voltage ÷ resistance, or current = voltage × conductance. Conductance is determined by the properties of the relevant ion channel protein. The voltage term is the difference between the actual membrane potential and the reversal potential for that ion (the membrane potential at which no current would flow even if channels were open). For example, in the case of sodium in a cardiac cell at rest, there is a substantial concentration gradient (140 mmol/L Na+ outside; 10–15 mmol/L Na+ inside) and an electrical gradient (0 mV outside; −90 mV inside) that would drive Na+ into cells. Sodium does not enter the cell at rest because sodium channels are closed; when sodium channels open, the very large influx of Na+ accounts for phase 0 depolarization of the action potential. The situation for K+ in the resting cardiac cell is quite different. Here, the concentration gradient (140 mmol/L inside; 4 mmol/L outside) would drive the ion out of the cell, but the electrical gradient would drive it in; that is, the inward gradient is in equilibrium with the outward gradient. In fact, certain potassium channels (“inward rectifier” channels) are open in the resting cell, but little current flows through them because of this balance. The equilibrium, or reversal potential, for ions is determined by the Nernst equation:
![]()
where Ce and Ci are the extracellular and intracellular concentrations, respectively, multiplied by their activity coefficients. Note that raising extracellular potassium makes EK less negative. When this occurs, the membrane depolarizes until the new EK is reached. Thus, extracellular potassium concentration and inward rectifier K+ channel function are the major factors determining the membrane potential of the resting cardiac cell. The conditions required for application of the Nernst equation are approximated at the peak of the overshoot (using sodium concentrations) and during rest (using potassium concentrations) in most nonpacemaker cardiac cells. If the permeability (P) is significant for both potassium and sodium, the Nernst equation is not a good predictor of membrane potential, but the Goldman-Hodgkin-Katz equation may be used:
![]()
In pacemaker cells (whether normal or ectopic), spontaneous depolarization (the pacemaker potential) occurs during diastole (phase 4, Figure 14–1). This depolarization results from a gradual increase of depolarizing current through special hyperpolarization-activated ion channels (If, also called Ih) in SA node cells. If was initially referred to as the “funny” current since it has the unusual property of being an inward current activated by hyperpolarization. The hyperpolarization-activated channel in the sinus node belongs to a superfamily of voltage-gated channels (HCN1–HCN4). They have a cyclic nucleotide-binding domain and their activity is regulated by cAMP. HCN4 is the principal isoform expressed in the sinus node and co-localizes with the β2-adrenergic receptor. The close association with the β2 receptor may play a role in the autonomic regulation of heart rate. The effect of changing extracellular potassium is more complex in a pacemaker cell than it is in a nonpacemaker cell because the effect on permeability to potassium is much more important in a pacemaker (see Box: Effects of Potassium). In a pacemaker—especially an ectopic one—the end result of an increase in extracellular potassium is usually to slow or stop the pacemaker. Conversely, hypokalemia often facilitates ectopic pacemakers.
The Active Cell Membrane
In normal atrial, Purkinje, and ventricular cells, the action potential upstroke (phase 0) is dependent on sodium current. From a functional point of view, it is convenient to describe the behavior of the sodium current in terms of three channel states (Figure 14–3). The cardiac sodium channel protein has been cloned, and it is now recognized that these channel states actually represent different protein conformations. In addition, regions of the protein that confer specific behaviors, such as voltage sensing, pore formation, and inactivation, are now being identified. The gates described below and in Figure 14–3 represent such regions.
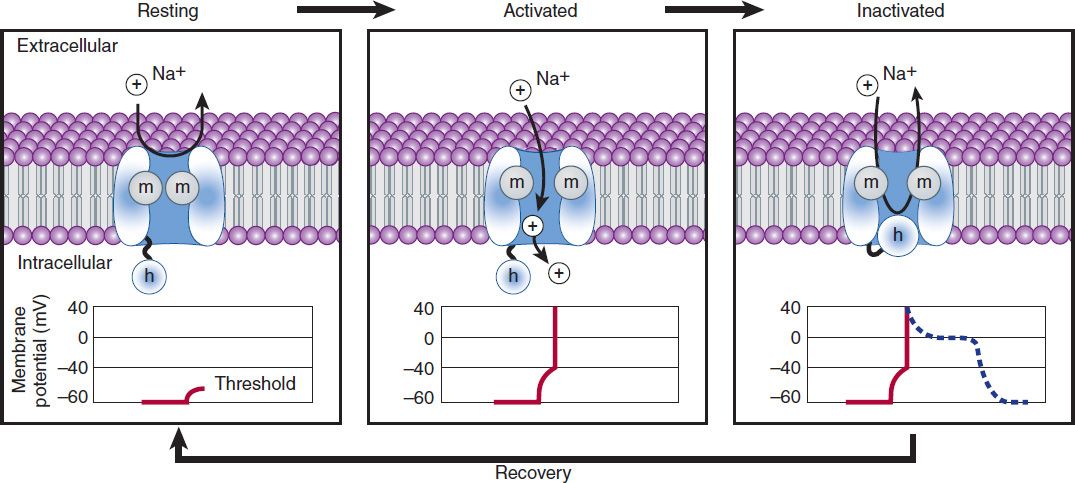
FIGURE 14–3 A schematic representation of Na+ channels cycling through different conformational states during the cardiac action potential. Transitions between resting, activated, and inactivated states are dependent on membrane potential and time. The activation gate is shown as m and the inactivation gate as h. Potentials typical for each state are shown under each channel schematic as a function of time. The dashed line indicates that part of the action potential during which most Na+ channels are completely or partially inactivated and unavailable for reactivation.
Effects of Potassium
The effects of changes in serum potassium on cardiac action potential duration, pacemaker rate, and arrhythmias can appear somewhat paradoxical if changes are predicted based solely on a consideration of changes in the potassium electrochemical gradient. In the heart, however, changes in serum potassium concentration have the additional effect of altering potassium conductance (increased extracellular potassium increases potassium conductance) independent of simple changes in electrochemical driving force, and this effect often predominates. As a result, the actual observed effects of hyperkalemia include reduced action potential duration, slowed conduction, decreased pacemaker rate, and decreased pacemaker arrhythmogenesis. Conversely, the actual observed effects of hypokalemia include prolonged action potential duration, increased pacemaker rate, and increased pacemaker arrhythmogenesis. Furthermore, pacemaker rate and arrhythmias involving ectopic pacemaker cells appear to be more sensitive to changes in serum potassium concentration, compared with cells of the sinoatrial node. These effects of serum potassium on the heart probably contribute to the observed increased sensitivity to potassium channel-blocking antiarrhythmic agents (quinidine or sotalol) during hypokalemia, eg, accentuated action potential prolongation and a tendency to cause torsades de pointes.
Depolarization to the threshold voltage results in opening of the activation (m) gates of sodium channels (Figure 14–3, middle). If the inactivation (h) gates of these channels have not already closed, the channels are now open or activated, and sodium permeability is markedly increased, greatly exceeding the permeability for any other ion. Extracellular sodium therefore diffuses down its electrochemical gradient into the cell, and the membrane potential very rapidly approaches the sodium equilibrium potential, ENa (about +70 mV when Nae = 140 mmol/L and Nai = 10 mmol/L). This intense sodium current is very brief because opening of the m gates upon depolarization is promptly followed by closure of the h gates and inactivation of the sodium channels (Figure 14–3, right).
Most calcium channels become activated and inactivated in what appears to be the same way as sodium channels, but in the case of the most common type of cardiac calcium channel (the “L” type), the transitions occur more slowly and at more positive potentials. The action potential plateau (phases 1 and 2) reflects the turning off of most of the sodium current, the waxing and waning of calcium current, and the slow development of a repolarizing potassium current.
Final repolarization (phase 3) of the action potential results from completion of sodium and calcium channel inactivation and the growth of potassium permeability, so that the membrane potential once again approaches the potassium equilibrium potential. The major potassium currents involved in phase 3 repolarization include a rapidly activating potassium current (IKr) and a slowly activating potassium current (IKs). These two potassium currents are sometimes discussed together as “IK.” It is noteworthy that a different potassium current, distinct from IKr and IKs, may control repolarization in SA nodal cells. This explains why some drugs that block either IKr or IKs may prolong repolarization in Purkinje and ventricular cells, but have little effect on SA nodal repolarization (see Box: Molecular & Genetic Basis of Cardiac Arrhythmias).
The Effect of Resting Potential on Action Potentials
A key factor in the pathophysiology of arrhythmias and the actions of antiarrhythmic drugs is the relation between the resting potential of a cell and the action potentials that can be evoked in it (Figure 14–4, left panel). Because the inactivation gates of sodium channels in the resting membrane close over the potential range from −75 mV to −55 mV, fewer sodium channels are “available” for diffusion of sodium ions when an action potential is evoked from a resting potential of −60 mV than when it is evoked from a resting potential of −80 mV. Important consequences of the reduction in peak sodium permeability include reduced maximum upstroke velocity (called ![]() max, for maximum rate of change of membrane voltage), reduced action potential amplitude, reduced excitability, and reduced conduction velocity.
max, for maximum rate of change of membrane voltage), reduced action potential amplitude, reduced excitability, and reduced conduction velocity.
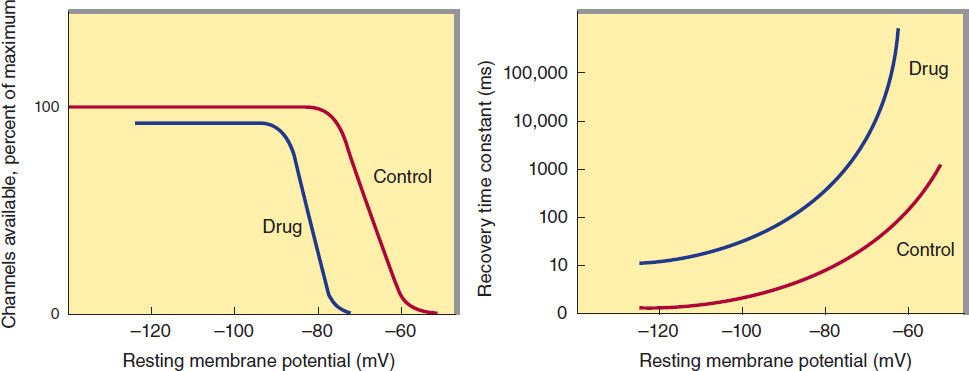
FIGURE 14–4 Dependence of sodium channel function on the membrane potential preceding the stimulus. Left: The fraction of sodium channels available for opening in response to a stimulus is determined by the membrane potential immediately preceding the stimulus. The decrease in the fraction available when the resting potential is depolarized in the absence of a drug (control curve) results from the voltage-dependent closure of h gates in the channels. The curve labeled Drug illustrates the effect of a typical local anesthetic antiarrhythmic drug. Most sodium channels are inactivated during the plateau of the action potential. Right: The time constant for recovery from inactivation after repolarization also depends on the resting potential. In the absence of drug, recovery occurs in less than 10 ms at normal resting potentials (−85 to −95 mV). Depolarized cells recover more slowly (note logarithmic scale). In the presence of a sodium channel-blocking drug, the time constant of recovery is increased, but the increase is far greater at depolarized potentials than at more negative ones.
During the plateau of the action potential, most sodium channels are inactivated. Upon repolarization, recovery from inactivation takes place (in the terminology of Figure 14–3, the h gates reopen), making the channels again available for excitation. The time between phase 0 and sufficient recovery of sodium channels in phase 3 to permit a new propagated response to an external stimulus is the refractory period. Changes in refractoriness (determined by either altered recovery from inactivation or altered action potential duration) can be important in the genesis or suppression of certain arrhythmias. Another important effect of less negative resting potential is prolongation of this recovery time, as shown in Figure 14–4 (right panel). The prolongation of recovery time is reflected in an increase in the effective refractory period.
A brief, sudden, depolarizing stimulus, whether caused by a propagating action potential or by an external electrode arrangement, causes the opening of large numbers of activation gates before a significant number of inactivation gates can close. In contrast, slow reduction (depolarization) of the resting potential, whether brought about by hyperkalemia, sodium pump blockade, or ischemic cell damage, results in depressed sodium currents during the upstrokes of action potentials. Depolarization of the resting potential to levels positive to −55 mV abolishes sodium currents, since all sodium channels are inactivated. However, such severely depolarized cells have been found to support special action potentials under circumstances that increase calcium permeability or decrease potassium permeability. These “slow responses”—slow upstroke velocity and slow conduction—depend on a calcium inward current and constitute the normal electrical activity in the SA and AV nodes, because these tissues have a normal resting potential in the range of −50 to −70 mV. Slow responses may also be important for certain arrhythmias.
Modern techniques of molecular biology and electrophysiology can identify multiple subtypes of calcium and potassium channels. One way in which such subtypes may differ is in sensitivity to drug effects, so drugs targeting specific channel subtypes may be developed in the future.
MECHANISMS OF ARRHYTHMIAS
Many factors can precipitate or exacerbate arrhythmias: ischemia, hypoxia, acidosis or alkalosis, electrolyte abnormalities, excessive catecholamine exposure, autonomic influences, drug toxicity (eg, digitalis or antiarrhythmic drugs), overstretching of cardiac fibers, and the presence of scarred or otherwise diseased tissue. However, all arrhythmias result from (1) disturbances in impulse formation, (2) disturbances in impulse conduction, or (3) both.
Disturbances of Impulse Formation
The interval between depolarizations of a pacemaker cell is the sum of the duration of the action potential and the duration of the diastolic interval. Shortening of either duration results in an increase in pacemaker rate. The more important of the two, diastolic interval, is determined primarily by the slope of phase 4 depolarization (pacemaker potential). Vagal discharge and β-receptor-blocking drugs slow normal pacemaker rate by reducing the phase 4 slope (acetylcholine also makes the maximum diastolic potential more negative). Acceleration of pacemaker discharge is often brought about by increased phase 4 depolarization slope, which can be caused by hypokalemia, β-adrenoceptor stimulation, positive chronotropic drugs, fiber stretch, acidosis, and partial depolarization by currents of injury.
Molecular & Genetic Basis of Cardiac Arrhythmias
It is now possible to define the molecular basis of several congenital and acquired cardiac arrhythmias. The best example is the polymorphic ventricular tachycardia known as torsades de pointes (Figure 14–8), which is associated with prolongation of the QT interval (especially at the onset of the tachycardia), syncope, and sudden death. This must represent prolongation of the action potential of at least some ventricular cells (Figure 14–1). The effect can, in theory, be attributed to either increased inward current (gain of function) or decreased outward current (loss of function) during the plateau of the action potential. In fact, recent molecular genetic studies have identified up to 300 different mutations in at least eight ion channel genes that produce the congenital long QT (LQT) syndrome (Table 14–1), and different mutations may have different clinical implications. Loss-of-function mutations in potassium channel genes produce decreases in outward repolarizing current and are responsible for LQT subtypes 1, 2, 5, 6, and 7. HERG and KCNE2 (MiRP1) genes encode subunits of the rapid delayed rectifier potassium current (IKr), whereas KCNQ1 and KCNE1 (minK) encode subunits of the slow delayed rectifier potassium current (IKs). KCNJ2 encodes an inwardly rectifying potassium current (IKir). In contrast, gain-of-function mutations in the sodium channel gene (SCN5A) or calcium channel gene (CACNA1c) cause increases in inward plateau current and are responsible for LQT subtypes 3 and 8, respectively.
Molecular genetic studies have identified the reason why congenital and acquired cases of torsades de pointes can be so strikingly similar. The potassium channel IKr (encoded by HERG) is blocked or modified by many drugs (eg, quinidine, sotalol) or electrolyte abnormalities (hypokalemia, hypomagnesemia, hypocalcemia) that also produce torsades de pointes. Thus, the identification of the precise molecular mechanisms underlying various forms of the LQT syndromes now raises the possibility that specific therapies may be developed for individuals with defined molecular abnormalities. Indeed, preliminary reports suggest that the sodium channel blocker mexiletine can correct the clinical manifestations of congenital LQT subtype 3 syndrome. It is likely that torsades de pointes originates from triggered upstrokes arising from early afterdepolarizations (Figure 14–5). Thus, therapy is directed at correcting hypokalemia, eliminating triggered upstrokes (eg, by using β blockers or magnesium), or shortening the action potential (eg, by increasing heart rate with isoproterenol or pacing)—or all of these.
The molecular basis of several other congenital cardiac arrhythmias associated with sudden death has also recently been identified. Three forms of short QT syndrome have been identified that are linked to gain-of-function mutations in three different potassium channel genes (KCNH2, KCNQ1, and KCNJ2). Catecholaminergic polymorphic ventricular tachycardia, a disease that is characterized by stress- or emotion-induced syncope, can be caused by genetic mutations in two different proteins in the sarcoplasmic reticulum that control intracellular calcium homeostasis. Mutations in two different ion channel genes (HCN4 and SCN5A) have been linked to congenital forms of sick sinus syndrome. The Brugada syndrome, which is characterized by ventricular fibrillation associated with persistent ST-segment elevation, and progressive cardiac conduction disorder (PCCD), characterized by impaired conduction in the His-Purkinje system and right or left bundle block leading to complete AV block, have both been linked to several loss-of-function mutations in the sodium channel gene, SCN5A. At least one form of familial atrial fibrillation is caused by a gain-of-function mutation in the potassium channel gene, KCNQ1.
TABLE 14–1 Molecular and genetic basis of some cardiac arrhythmias.
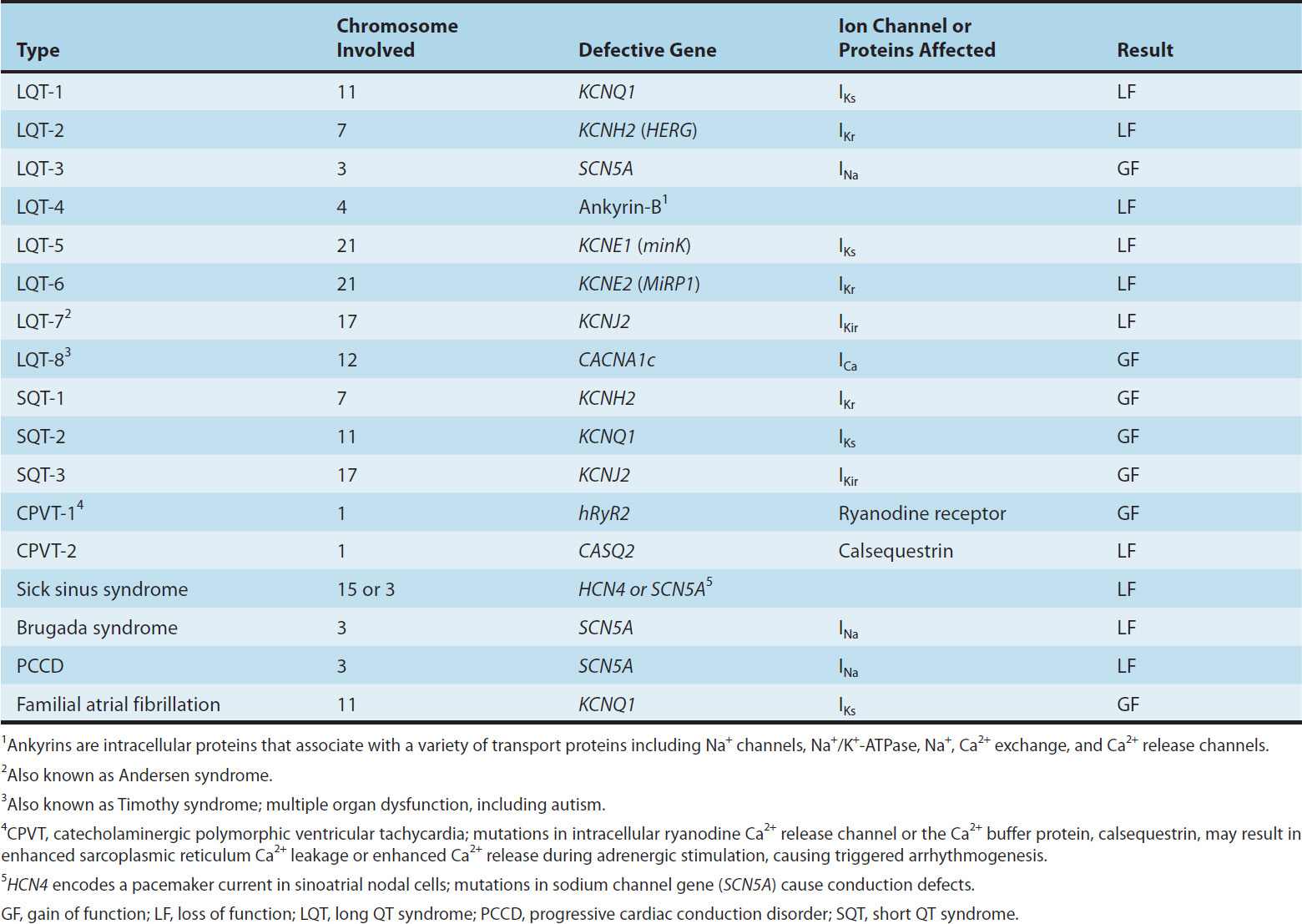
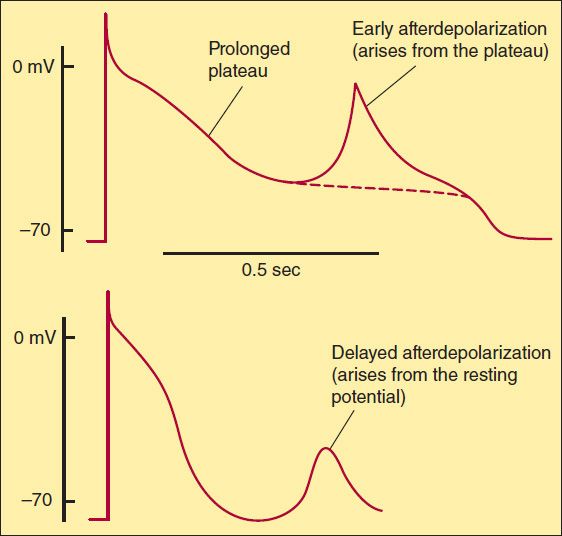
FIGURE 14–5 Two forms of abnormal activity, early (top) and delayed afterdepolarizations (bottom). In both cases, abnormal depolarizations arise during or after a normally evoked action potential. They are therefore often referred to as “triggered” automaticity; that is, they require a normal action potential for their initiation.
Latent pacemakers (cells that show slow phase 4 depolarization even under normal conditions, eg, some Purkinje fibers) are particularly prone to acceleration by the above mechanisms. However, all cardiac cells, including normally quiescent atrial and ventricular cells, may show repetitive pacemaker activity when depolarized under appropriate conditions, especially if hypokalemia is also present.
Afterdepolarizations (Figure 14–5) are transient depolarizations that interrupt phase 3 (early afterdepolarizations, EADs) or phase 4 (delayed afterdepolarizations, DADs). EADs are usually exacerbated at slow heart rates and are thought to contribute to the development of long QT-related arrhythmias (see Box: Molecular & Genetic Basis of Cardiac Arrhythmias). DADs, on the other hand, often occur when intracellular calcium is increased (see Chapter 13). They are exacerbated by fast heart rates and are thought to be responsible for some arrhythmias related to digitalis excess, to catecholamines, and to myocardial ischemia.
Disturbances of Impulse Conduction
Severely depressed conduction may result in simple block, eg, AV nodal block or bundle branch block. Because parasympathetic control of AV conduction is significant, partial AV block is sometimes relieved by atropine. Another common abnormality of conduction is reentry (also known as “circus movement”), in which one impulse reenters and excites areas of the heart more than once (Figure 14–6).
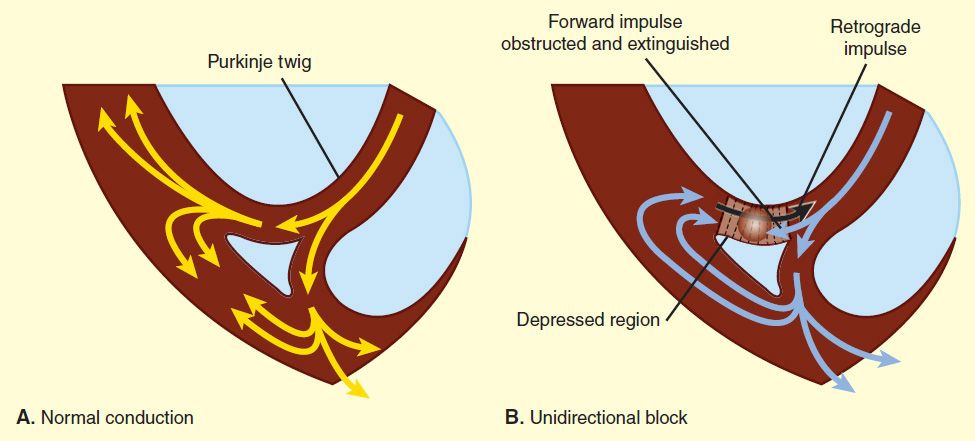
FIGURE 14–6 Schematic diagram of a reentry circuit that might occur in small bifurcating branches of the Purkinje system where they enter the ventricular wall. A: Normally, electrical excitation branches around the circuit, is transmitted to the ventricular branches, and becomes extinguished at the other end of the circuit due to collision of impulses. B: An area of unidirectional block develops in one of the branches, preventing anterograde impulse transmission at the site of block, but the retrograde impulse may be propagated through the site of block if the impulse finds excitable tissue; that is, the refractory period is shorter than the conduction time. This impulse then reexcites tissue it had previously passed through, and a reentry arrhythmia is established.
The path of the reentering impulse may be confined to very small areas, eg, within or near the AV node, or it may involve large portions of the atrial or ventricular walls. Some forms of reentry are strictly anatomically determined; for example, in Wolff-Parkinson-White syndrome, the reentry circuit consists of atrial tissue, the AV node, ventricular tissue, and an accessory AV connection (bundle of Kent, a bypass tract). In other cases (eg, atrial or ventricular fibrillation), multiple reentry circuits, determined by the varying properties of the cardiac tissue, may meander through the heart in apparently random paths. The circulating impulse often gives off “daughter impulses” that can spread to the rest of the heart. Depending on how many round trips through the pathway the reentrant impulse makes before dying out, the arrhythmia may be manifest as one or a few extra beats or as a sustained tachycardia.
For reentry to occur, three conditions must coexist, as indicated in Figure 14–6. (1) There must be an obstacle (anatomic or physiologic) to homogeneous conduction, thus establishing a circuit around which the reentrant wavefront can propagate. (2) There must be unidirectional block at some point in the circuit; that is, conduction must die out in one direction but continue in the opposite direction (as shown in Figure 14–6, the impulse can gradually decrease as it invades progressively more depolarized tissue until it finally blocks—a process known as decremental conduction). (3) Conduction time around the circuit must be long enough that the retrograde impulse does not enter refractory tissue as it travels around the obstacle; that is, the conduction time must exceed the effective refractory period. It is important to note that reentry depends on conduction that has been depressed by some critical amount, usually as a result of injury or ischemia. If conduction velocity is too slow, bidirectional block rather than unidirectional block occurs; if the reentering impulse is too weak, conduction may fail, or the impulse may arrive so late that it collides with the next regular impulse. On the other hand, if conduction is too rapid—ie, almost normal—bidirectional conduction rather than unidirectional block will occur. Even in the presence of unidirectional block, if the impulse travels around the obstacle too rapidly, it will reach tissue that is still refractory. Representative electrocardiograms of important arrhythmias are shown in Figures 14–7 and 14–8.
FIGURE 14–7 Electrocardiograms of normal sinus rhythm and some common arrhythmias. Major deflections (P, Q, R, S, and T) are labeled in each electrocardiographic record except in panel 5, in which electrical activity is completely disorganized and none of these deflections is recognizable. (Adapted, with permission, from Goldman MJ: Principles of Clinical Electrocardiography, 11th ed. McGraw-Hill, 1982. Copyright © The McGraw-Hill Companies, Inc.)
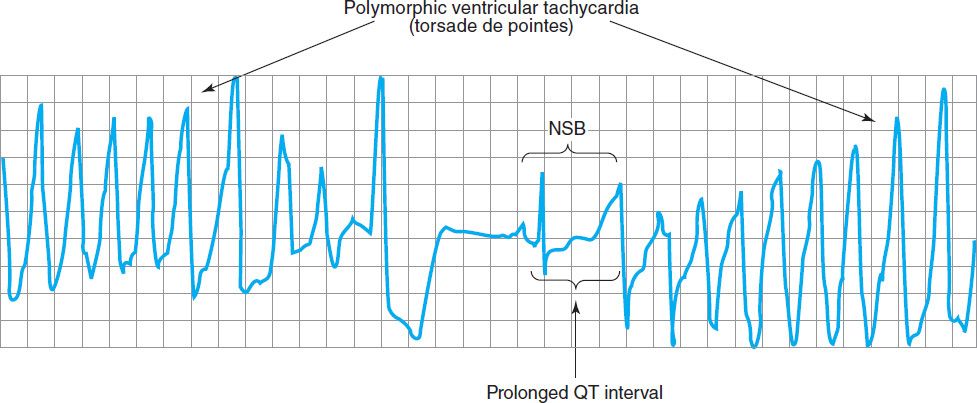
FIGURE 14–8 Electrocardiogram from a patient with the long QT syndrome during two episodes of torsades de pointes. The polymorphic ventricular tachycardia is seen at the start of this tracing and spontaneously halts at the middle of the panel. A single normal sinus beat (NSB) with an extremely prolonged QT interval follows, succeeded immediately by another episode of ventricular tachycardia of the torsades type. The usual symptoms include dizziness or transient loss of consciousness. (Reproduced, with permission, from Basic and Clinical Pharmacology, 10th edition, McGraw-Hill, 2007. Copyright © The McGraw-Hill Companies, Inc.)
Slowing of conduction may be due to depression of sodium current, depression of calcium current (the latter especially in the AV node), or both. Drugs that abolish reentry usually work by further slowing depressed conduction (by blocking the sodium or calcium current) and causing bidirectional block. In theory, accelerating conduction (by increasing sodium or calcium current) would also be effective, but only under unusual circumstances does this mechanism explain the action of any available drug.
Lengthening (or shortening) of the refractory period may also make reentry less likely. The longer the refractory period in tissue near the site of block, the greater the chance that the tissue will still be refractory when reentry is attempted. (Alternatively, the shorter the refractory period in the depressed region, the less likely it is that unidirectional block will occur.) Thus, increased dispersion of refractoriness is one contributor to reentry, and drugs may suppress arrhythmias by reducing such dispersion.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


