Nidhi Gera, Ehrin J. Armstrong, and David E. Golan
Physiologic and Pharmacologic Effects of Endogenous Catecholamines |

Adrenergic pharmacology involves the study of agents that act on pathways mediated by the endogenous catecholamines norepinephrine, epinephrine, and dopamine. The sympathetic nervous system is the major source of endogenous catecholamine production and release. Signaling through catecholamine receptors mediates diverse physiologic effects, including increasing the rate and force of cardiac contraction, modifying the peripheral resistance of the arterial system, inhibiting the release of insulin, stimulating hepatic release of glucose, and increasing adipocyte release of free fatty acids. Drugs that target the synthesis, storage, reuptake, and metabolism of norepinephrine and epinephrine or that directly target the postsynaptic receptors for these transmitters are frequent therapies for many major diseases, including hypertension, shock, asthma, and angina. This chapter examines the biochemical and physiologic basis for adrenergic action and then discusses the action of the different classes of adrenergic drugs.
 The year is 1960. Ms. S has felt depressed for a number of years. She has tried several different medications to alleviate her feelings of hopelessness and lack of motivation, but nothing seems to help. Recently, however, her doctor has prescribed iproniazid, a new medication reported to be of benefit in many cases of depression. He tells her that scientists believe that the drug has beneficial effects in depression by inhibiting an enzyme in the brain called monoamine oxidase (MAO). MAO is one of the enzymes responsible for catecholamine degradation, and its inhibition significantly increases the available concentrations of catecholamines. Because iproniazid is a new drug, its potential adverse effects are not well defined, so her doctor advises Ms. S to report any unusual effects of the medication.
The year is 1960. Ms. S has felt depressed for a number of years. She has tried several different medications to alleviate her feelings of hopelessness and lack of motivation, but nothing seems to help. Recently, however, her doctor has prescribed iproniazid, a new medication reported to be of benefit in many cases of depression. He tells her that scientists believe that the drug has beneficial effects in depression by inhibiting an enzyme in the brain called monoamine oxidase (MAO). MAO is one of the enzymes responsible for catecholamine degradation, and its inhibition significantly increases the available concentrations of catecholamines. Because iproniazid is a new drug, its potential adverse effects are not well defined, so her doctor advises Ms. S to report any unusual effects of the medication.
Hopeful, but not expecting significant changes, Ms. S takes the medication. Within a few weeks, she begins to feel motivated and energetic for the first time in 20 years. Exuberant at her new sense of energy, Ms. S reclaims her past life as a socialite by hosting a gala wine and cheese reception. The best and brightest of the city turn up, expecting a fine evening. As she stands up to give thanks to her attendees, Ms. S celebrates with a large swig of her favorite 1954 Chianti. By the end of the party, Ms. S has a severe headache and nausea. Recalling her doctor’s warning, Ms. S has a friend rush her to the nearest hospital. In the emergency department, the attending physician records a blood pressure of 230/160 mm Hg. Recognizing that Ms. S is experiencing a hypertensive emergency, the doctor administers phentolamine (an α-adrenoceptor antagonist). Ms. S’s blood pressure quickly normalizes, and the doctor’s subsequent clinical investigation identifies a new, and now famous, drug–food interaction involving MAO inhibitors. This potential adverse interaction is shared by some other MAO inhibitors; more recent work on subtype-selective and reversible MAO inhibitors has minimized the incidence of this interaction.
Questions
1. Which enzymes metabolize catecholamines? What are the specificities of isoforms of these enzymes for the various catecholamines?
2. What is the mechanistic explanation for the interaction of MAO inhibitors with red wine and aged cheese?
3. How did phentolamine lower Ms. S’s blood pressure?
 BIOCHEMISTRY AND PHYSIOLOGY OF ADRENERGIC FUNCTION
BIOCHEMISTRY AND PHYSIOLOGY OF ADRENERGIC FUNCTION
The autonomic nervous system contributes to homeostasis through the concerted action of its sympathetic and parasympathetic branches. Catecholamines are the major effectors of sympathetic signaling. The following discussion presents the biochemistry of catecholamine action, from synthesis to metabolism to receptor activation. The physiologic roles of the endogenous catecholamines epinephrine (adrenaline), norepinephrine (noradrenaline), and dopamine are then discussed, with emphasis on the specificity of receptor expression in different organ systems.
Catecholamine Synthesis, Storage, and Release
Catecholamines are synthesized by sequential chemical modifications of the amino acid tyrosine. This synthesis occurs primarily at sympathetic nerve endings and in chromaffin cells. Epinephrine is predominantly synthesized in chromaffin cells of the adrenal medulla; sympathetic neurons produce norepinephrine as their primary neurotransmitter (Fig. 11-1). Tyrosine, the precursor for catecholamine synthesis, is transported into neurons via an aromatic amino acid transporter that uses the Na+ gradient across the neuronal membrane to concentrate tyrosine (as well as phenylalanine, tryptophan, and histidine). The first step in catecholamine synthesis, the oxidation of tyrosine to dihydroxyphenylalanine (DOPA), is mediated by the enzyme tyrosine hydroxylase (TH). TH is the rate-limiting enzyme in catecholamine synthesis, and it is regulated by feedback (end-product) inhibition and by kinase-mediated phosphorylation of the enzyme. DOPA is converted to dopamine by a relatively nonspecific aromatic amino acid decarboxylase. Dopamine is then hydroxylated by dopamine-β-hydroxylase to yield norepinephrine. In tissues that produce epinephrine, norepinephrine is then methylated on its amino group by phenylethanolamine N-methyltransferase (PNMT). Expression of PNMT in the adrenal medulla is largely dependent on the high concentrations of cortisol that flow into the medulla via veins draining the adrenal cortex.
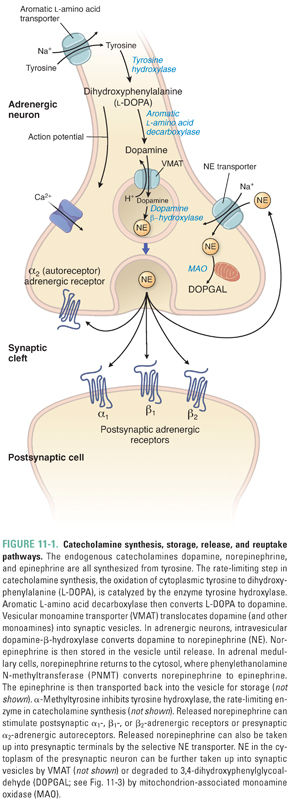
The conversions of tyrosine to DOPA and of DOPA to dopamine occur within the cytoplasm. Dopamine is transported into synaptic vesicles by a 12-helix membrane-spanning proton antiporter called the vesicular monoamine transporter (VMAT). Unlike all the other enzymes in the catecholamine biosynthesis pathways, dopamine-β-hydroxylase is associated with the inner surface of secretory vesicles, and it catalyzes the conversion of dopamine to norepinephrine inside these vesicles.
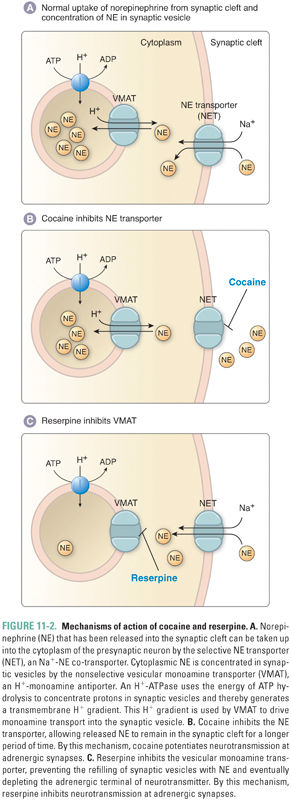
There are three distinct vesicular transporters that differ in substrate specificity and localization. VMAT1 and VMAT2 (also known as Uptake 2 [Fig. 11-2]) both transport serotonin (5-HT), histamine, and all catecholamines. The tissue-specific expression of VMAT1 and VMAT2 is mutually exclusive: VMAT1 expression is restricted mainly to nonneuronal cells (adrenal gland, gastric mucosa, intestine, and sympathetic ganglia) and VMAT2 is expressed primarily in the central nervous system (CNS). In addition to these expression differences, the affinity of VMAT2 for histamine (Km, 3 μM) is significantly higher than that of VMAT1 for histamine (Km, 436 μM). The vesicular acetylcholine transporter (VAChT) is expressed in cholinergic neurons, including motor nerves (see Chapter 10, Cholinergic Pharmacology). These antiporters use the proton gradient generated by a H+-ATPase in the vesicular membrane to concentrate dopamine (or, in the case of VAChT, acetylcholine) inside the vesicle. Norepinephrine concentrations within the vesicle can reach 100 mM. To stabilize the osmotic pressure resulting from the high concentration gradient for norepinephrine across the vesicle membrane, norepinephrine is thought to condense with ATP. Consequently, ATP and norepinephrine are co-released upon vesicle exocytosis.
In adrenal medullary cells, norepinephrine is transported or diffuses from vesicles back into the cytoplasm, where PNMT converts it to epinephrine. Epinephrine is then transported back into vesicles for storage until its eventual release by exocytosis. The nonselective nature of VMAT1 and VMAT2 has important pharmacologic consequences, as discussed below.
Activation of the sympathetic nervous system and subsequent catecholamine release are initiated by signals originating in an array of processing areas in the CNS, especially the limbic system. These CNS neurons project axons that synapse on sympathetic preganglionic neurons in the intermediolateral columns of the spinal cord. The preganglionic axons project to the sympathetic ganglia. The preganglionic neurons use acetylcholine as the neurotransmitter to activate nicotinic acetylcholine (ACh) receptors, which are cation-selective channels that depolarize the neuronal membrane and thereby generate postsynaptic potentials in postganglionic neurons. Ganglionic blockers such as hexamethonium and mecamylamine block the ganglionic nicotinic ACh receptor, without significant effects on skeletal muscle ACh receptors (see Chapter 10). The sympathetic postganglionic axons form varicosities or en passant connections with target organs. The arrival of an action potential at these endings opens voltage-gated neuronal Ca2+ channels, and the ensuing Ca2+ influx triggers exocytosis of the catecholamine-containing synaptic vesicles. Various novel substances—including peptides from sea snails—block these Ca2+ channels; ziconotide is an example of a drug in this class that has efficacy in the treatment of severe pain (see Chapter 18, Pharmacology of Analgesia). Norepinephrine rapidly diffuses away from the sympathetic nerve endings and locally regulates target tissue responses (e.g., smooth muscle tone) by activating adrenergic receptors expressed on target tissues. (An exception is that ACh is the transmitter used at sympathetic nerve endings in sweat glands.) Importantly, adrenergic receptors are also expressed at sympathetic nerve endings; these receptors may serve as an autoregulatory mechanism for modulating the extent of neurotransmitter release.
Reuptake and Metabolism of Catecholamines
The action of a catecholamine molecule at its postsynaptic receptor is terminated by one of three mechanisms: (1) reuptake of catecholamine into the presynaptic neuron, (2) metabolism of catecholamine to an inactive metabolite, and (3) diffusion of the catecholamine away from the synaptic cleft. The first two of these mechanisms require specific transport proteins or enzymes and, therefore, are targets for pharmacologic intervention.
Reuptake of catecholamine into the neuronal cytoplasm is mediated by a selective catecholamine transporter (e.g., norepinephrine transporter, or NET) that is also known as Uptake 1 (Fig. 11-2). Approximately 90% of the released norepinephrine is taken up by this process (recycled); the remainder is either metabolized locally or diffuses into the blood. Uptake 1 is a symporter that uses the inward Na+ gradient to concentrate catecholamines in the cytoplasm of sympathetic nerve endings, thus limiting the postsynaptic response and allowing neurons to recycle the transmitter for subsequent release. Inside the nerve terminal, catecholamines can be further concentrated in synaptic vesicles via VMAT, the same transporter used to transport dopamine into the vesicle for catecholamine synthesis. Thus, the pool of catecholamines available for release comes from two sources: molecules that are synthesized de novo and molecules that are recycled via neuronal reuptake.
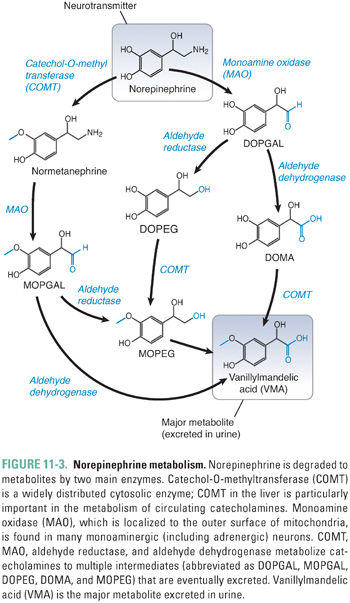
Catecholamine metabolism involves the two enzymes MAO and catechol-O-methyltransferase (COMT) (Fig. 11-3). MAO is a mitochondrial outer-membrane enzyme that is expressed in most neurons. It exists in two isoforms: MAO-A and MAO-B. The two isoforms have some degree of ligand specificity: MAO-A preferentially degrades serotonin, norepinephrine, and dopamine, while MAO-B degrades dopamine more rapidly than serotonin and norepinephrine. As indicated in the clinical case, MAO inhibitors are efficacious in the treatment of depression. The MAO-A isoform is responsible for detoxifying substances in cheese and wine before they reach the general circulation. COMT is a cytosolic enzyme that is relatively nonspecific and is expressed primarily in the liver.
Adrenergic receptors (also called adrenoceptors) are selective for norepinephrine and epinephrine. Supraphysiologic concentrations of dopamine can also activate some adrenoceptors. These receptors are divided into three main classes, termed α1, α2, and β (Table 11-1). Each of these major classes has three subtypes: α1A, α1B, and α1D; α2A, α2B, and α2C; and β1, β2, and β3. Each of the adrenergic receptor subtypes is a member of the G protein-coupled receptor (GPCR) superfamily (also known as seven-transmembrane helix receptors). GPCRs regulate complex intracellular signaling networks through intermediate transducing molecules, which are called G proteins because of their GTP binding and hydrolysis activity. G proteins are heterotrimeric, with α, β, and γ subunits. In the resting (inactive) state, Gα binds guanosine 5′-diphosphate (GDP) and is associated with Gβγ. Binding of agonist to the GPCR triggers the dissociation of GDP and the binding of guanosine 5′-triphosphate (GTP) to the Gα subunit. GTP binding initiates a conformational change that leads to the dissociation of Gβγ and to the activation of Gα. Both Gα and Gβγ can activate downstream effectors. In mammals, at least 27 Gα, 5 Gβ, and 13 Gγ subtypes are present, and downstream GPCR signaling depends on the specific Gαβγ combination. On the basis of the primary sequence of the Gα subunit, G proteins can be divided into four major families—Gs, Gi, Gq/11, and G12—and each family of Gα subunit activates specific downstream signaling pathways (see Chapter 1, Drug–Receptor Interactions).
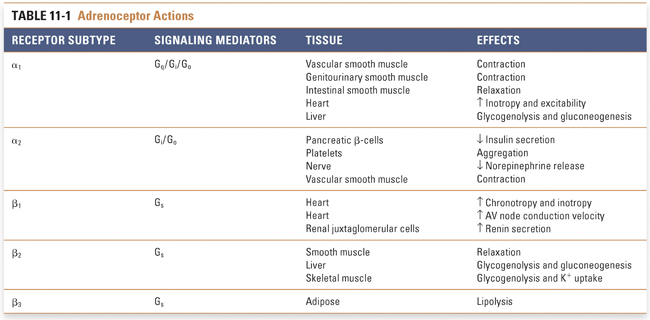
α1-Receptors are expressed in vascular smooth muscle, genitourinary tract smooth muscle, intestinal smooth muscle, prostate, brain, heart, liver, and other cell types. The prototypical signaling mechanism of α1-receptors involves Gq/11, which is generally a stimulatory protein that activates various effectors including phospholipase C, phospholipase D, phospholipase A2, Ca2+ channels, K+ channels, Na+/H+ exchangers, several members of the mitogen-activated protein (MAP) kinase pathways, and a variety of other kinases including phosphatidylinositol 3-kinase. Phospholipase C cleaves phosphatidylinositol-4,5-bisphosphate, generating the two second messengers inositol trisphosphate (IP3) and diacylglycerol (DAG). IP3 acts to increase intracellular [Ca2+] via both release of endogenous Ca2+ stores and influx of Ca2+ from extracellular fluid. Increased intracellular [Ca2+] activates various regulatory proteins that mediate physiologic responses in various tissues. DAG activates protein kinase C, which further activates a variety of protein substrates including ion channels such as Na+/H+ exchangers, Ca2+ channels, and K+ channels. Phospholipase D catalyses the hydrolysis of phosphatidylcholine to phosphatidic acid and choline. Phosphatidic acid may act directly as a signaling molecule or be further metabolized to DAG by phosphatidic acid hydrolase. Gq/11-stimulated activation of phospholipase A2 is mediated by increased intracellular [Ca2+] or by activation of protein kinase C and MAP kinase pathways. The various α1-receptor subtypes likely differ in their tissue-specific localization and their capacity to activate downstream signaling pathways.
The downstream signaling pathways activated by α1-receptors can be complex. Stimulation of α1-receptors in vascular smooth muscle cells increases intracellular [Ca2+], leading to activation of calmodulin, phosphorylation of myosin light chain, increased actin–myosin interaction, and muscle contraction (see Chapter 22, Pharmacology of Vascular Tone). Therefore, α1-receptors are important in mediating increases in peripheral vascular resistance, which can increase blood pressure and redistribute blood flow. While α1-receptor antagonists would seem to be attractive in the therapy of hypertension, their clinical efficacy in preventing the complications of hypertension is uncertain. α1-Receptor activation also causes contraction of genitourinary smooth muscle, and α1-receptor antagonists are clinically efficacious in the symptomatic treatment of benign prostatic hyperplasia (BPH) (see below).
α2-Adrenoceptors activate Gi, an inhibitory G protein. Gi has multiple signaling actions, including inhibition of adenylyl cyclase (thus decreasing cAMP levels), activation of G protein-coupled inward rectifier K+ channels (causing membrane hyperpolarization), and inhibition of neuronal Ca2+ channels. These effects tend to decrease neurotransmitter release from the target neuron. α2-Receptors are found on both presynaptic neurons and postsynaptic cells. Presynaptic α2-receptors function as autoreceptors to mediate feedback inhibition of sympathetic transmission. α2-Receptors are also expressed on platelets and pancreatic β-cells, where they mediate platelet aggregation and inhibit insulin release, respectively. The latter observations have led to the development of agents that are selective inhibitors of α2-receptors. The main pharmacologic approach to α2-receptors, however, has been in the treatment of hypertension. α2-Receptor agonists act at CNS sites to decrease sympathetic outflow to the periphery, resulting in decreased norepinephrine release at sympathetic nerve terminals and, therefore, decreased vascular smooth muscle contraction.
β-Adrenoceptors are divided into three subclasses, termed β1, β2, and β3 (Table 11-1). All three subclasses activate a stimulatory G protein, Gs. Gs activates adenylyl cyclase, which catalyzes the formation of intracellular cAMP from adenosine triphosphate (ATP). Increased intracellular cAMP activates protein kinases, especially protein kinase A (PKA), by binding to the regulatory subunit of the enzyme. This results in the release and activation of the catalytic subunit of PKA, which phosphorylates and activates a variety of intracellular proteins including ion channels and transcription factors. The nature of the signaling differences among the β-adrenoceptor subtypes is unclear since they all appear to couple efficiently to Gs, and stimulation of β1– and β2-adrenoceptors causes increased intracellular cAMP. It has been suggested that specificity may be conferred by differences in the composition of the G protein subunits associated with the two receptors. β1-Adrenoceptors couple exclusively to Gs, but β2-adrenoceptors can also activate effectors via coupling to Gi. Thus, β2-adrenoceptors can limit and spatially restrict cAMP production by switching between Gs– and Gi-mediated signaling, which, in turn, affects PKA-dependent regulation of target proteins.
Pharmacologic selectivity among the β-adrenoceptors appears to reside in the tissue-selective distribution of each β-adrenoceptor subtype. β1-Adrenoceptors are localized primarily in the kidney and heart. In the kidney, they are present mainly on renal juxtaglomerular cells, where receptor activation causes renin release (see Chapter 21, Pharmacology of Volume Regulation). Stimulation of cardiac β1-receptors (which represent 70–80% of all cardiac β-adrenergic receptors) causes an increase in both inotropy (force of contraction) and chronotropy (heart rate). The inotropic effect is mediated by increased phosphorylation of Ca2+ channels by protein kinase A, including calcium channels in the sarcolemma and phospholamban in the sarcoplasmic reticulum, and by phosphorylation of troponin I and troponin C, which reduces myofilament sensitivity to Ca2+ (see Chapter 25, Pharmacology of Cardiac Contractility). The increased chronotropy results from a β1-mediated increase in the rate of phase 4 depolarization of sinoatrial node pacemaker cells. Both effects contribute to increased cardiac output (recall that cardiac output = heart rate × stroke volume). Activation of β1-receptors also increases conduction velocity in the atrioventricular (AV) node because the β1-stimulated increase in Ca2+ entry increases the rate of depolarization of AV node cells.
β2-Adrenoceptors are expressed in smooth muscle (including bronchial smooth muscle), liver, skeletal muscle, and heart. In smooth muscle, receptor activation stimulates Gs, adenylyl cyclase, cAMP, and protein kinase A. Protein kinase A phosphorylates several contractile proteins, especially myosin light chain kinase. Phosphorylation of myosin light chain kinase reduces its affinity for calcium-calmodulin, leading to relaxation of the contractile apparatus. β2-Adrenoceptor activation may also relax bronchial smooth muscle by Gs-independent activation of K+ channels. Increased K+ efflux leads to bronchial smooth muscle cell hyperpolarization and, therefore, opposes the depolarization necessary to elicit contraction. In hepatocytes, activation of the Gs signaling cascade initiates a series of intracellular phosphorylation events that result in glycogen phosphorylase activation and glycogen catabolism. The result of β2-adrenoceptor stimulation of hepatocytes is, therefore, an increase in plasma glucose. In skeletal muscle, activation of these same signaling pathways stimulates glycogenolysis and promotes K+ uptake. Recent studies in cardiac myocytes suggest that β2-adrenoceptor-mediated activation of the Gβγ subunit of Gi leads to activation of phosphatidylinositide-3 kinase γ, which, in turn, activates the protein kinase B (also known as Akt) pathway that confers anti-apoptotic activity.
β3-Adrenoceptors are expressed in adipose tissue and in the gastrointestinal tract. Stimulation of β3-receptors leads to an increase in lipolysis and thermogenesis in adipocytes and to a decrease in gastrointestinal tract motility. These physiologic actions have led to speculation that β3-agonists may be useful in the treatment of obesity, noninsulin-dependent diabetes mellitus, and other potential indications, but such selective pharmacologic agents remain to be developed for clinical use.
Regulation of Receptor Response
The ability of receptor agonists to initiate downstream signaling is related to the number of receptors activated, and changes in the density of receptors on the cell surface often alter the apparent efficacy of an agonist. Thus, both short-term (desensitization) and long-term (down-regulation) changes in the number of functional adrenoceptors are important in regulating tissue response (see Fig. 1-10).
When an agonist activates an adrenoceptor, the dissociation of its associated heterotrimeric G protein subunits leads not only to downstream signaling, as discussed above, but also to a negative feedback mechanism that limits tissue responses. Activation of β-adrenoceptors recruits GPCR-specific protein kinases (GRKs), which phosphorylate serine and threonine residues in the carboxyl-terminal tail of the receptor. Protein kinase A and protein kinase C can also phosphorylate G protein-coupled receptors. The phosphorylated state of the receptor promotes the translocation to the membrane of a cytosolic protein called β-arrestin, which binds to the intracellular domain of the receptor and sterically inhibits interaction between the receptor and the G protein. This effectively silences receptor signaling. β-Arrestins also recruit clathrin and the clathrin adaptor protein AP2 to the phosphorylated receptor, and this complex targets the adrenoceptors to clathrin-coated pits. These pits are pinched off from the membrane with the help of the large GTPase dynamin, and the internalized receptors are then either rapidly recycled, targeted to endosomes and recycled more slowly, or degraded in lysosomes. Each of these processes is important in regulating tissue responsiveness on a short- or long-term basis. Over the last decade, evidence has suggested that β-arrestins can turn on (rather than off) novel signaling pathways by serving as scaffold proteins for signaling complexes that promote G protein-independent pathways involving the activation of Erk1/2, Src, and small GTP-binding proteins. A newly discovered aspect of GPCR signaling is that some antagonists that block G protein signaling pathways may also function as agonists in alternative signaling pathways such as β-arrestin signaling. In this way, both the desensitization/down-regulation and the signaling roles for β-arrestins may be involved in physiologic and pathologic situations.
Physiologic and Pharmacologic Effects of Endogenous Catecholamines
The endogenous catecholamines epinephrine and norepinephrine act as agonists at both α- and β-adrenoceptors. At supraphysiologic concentrations, dopamine can also act as an agonist at α- and β-receptors. The overall effect of each catecholamine is complex and depends on the concentration of the agent and on tissue-specific receptor expression.
Epinephrine is an agonist at both α- and β-adrenoceptors. At low concentrations, epinephrine has predominantly β1 and β2 effects, while at higher concentrations, its α1 effects become more pronounced. Acting at β1-receptors, epinephrine increases cardiac contractile force and cardiac output, with consequent increases in cardiac oxygen consumption and systolic blood pressure. Vasodilation mediated by β2-receptors causes a decrease in peripheral resistance and a decrease in diastolic blood pressure. Stimulation of β2-receptors also increases blood flow to skeletal muscle, relaxes bronchial smooth muscle, promotes glycogenolysis, and increases the concentrations of glucose and free fatty acids in the blood. Recent studies suggest that β1-receptors are responsible for vasodilation in large arteries such as the femoral and pulmonary arteries, while β2-receptors have a predominant role in vasodilation of the arterioles that contribute to peripheral vascular resistance. These β1 and β2 effects are all components of the “fight-or-flight” response.
Epinephrine was used to treat acute asthmatic attacks shortly after its discovery more than 100 years ago; other drugs with higher selectivity for β2-receptors, and which are delivered directly to pulmonary β2-receptors by inhalation, are now more often used in the treatment of asthma, chronic obstructive pulmonary disease, and other pulmonary conditions. Epinephrine remains a drug of choice for the treatment of anaphylaxis. Locally injected epinephrine causes vasoconstriction and prolongs the action of local anesthetics; for example, it is often used in combination with a local anesthetic in dentistry. It is ineffective orally due to extensive first-pass metabolism. Epinephrine has a rapid onset and a brief duration of action when injected intravenously. Adverse consequences of rapid intravenous infusions include increased cardiac excitability that may lead to cardiac arrhythmias and excessive increases in blood pressure.
Norepinephrine is an agonist at α1– and β1-receptors but has relatively little effect at β2-receptors. Because of the lack of action at β2-receptors, systemic administration of norepinephrine increases not only systolic blood pressure (β1 effect) but also diastolic blood pressure and total peripheral resistance. Norepinephrine is used in the pharmacologic treatment of hypotension in patients with distributive shock, most frequently due to sepsis.
Although dopamine is a prominent CNS neurotransmitter, systemic administration has few CNS effects because it does not readily cross the blood–brain barrier. Dopamine activates one or more subtypes of catecholamine receptor in peripheral tissues, and the predominant effect is dependent on the local concentration of the compound. At low doses (<2 μg/kg per min), a continuous intravenous infusion of dopamine acts predominantly on D1 dopaminergic receptors in renal, mesenteric, and coronary vascular beds. D1 dopaminergic receptors activate adenylyl cyclase in vascular smooth muscle cells, leading to increased cAMP levels and vasodilation. At higher rates of infusion (2–10 μg/kg per min), dopamine is a positive inotrope via its activation of β1-adrenergic receptors. At still higher rates of infusion (>10 μg/kg per min), dopamine acts on vascular α1-adrenergic receptors to cause vasoconstriction.
Dopamine is used in the treatment of shock, particularly in states of shock caused by low cardiac output and accompanied by compromised renal function leading to oliguria. However, efficacy in protecting the kidneys has not been clearly demonstrated.
 PHARMACOLOGIC CLASSES AND AGENTS
PHARMACOLOGIC CLASSES AND AGENTS
Pharmacologic intervention is possible at each of the major steps in catecholamine synthesis, storage, reuptake, metabolism, and receptor activation. The following discussion pre-sents the various classes of agents in the order of their action on adrenergic pathways, from neurotransmitter synthesis to receptor activation.
Inhibitors of Catecholamine Synthesis
Inhibitors of catecholamine synthesis have limited clinical utility because such agents nonspecifically inhibit the formation of all catecholamines (see Fig. 11-1). α-Methyltyrosine is a structural analogue of tyrosine that is transported into nerve terminals, where it inhibits tyrosine hydroxylase, the first enzyme in the catecholamine biosynthesis pathway. This agent is used occasionally in the treatment of hypertension associated with pheochromocytoma (a tumor of the enterochromaffin cells of the adrenal medulla that produces norepinephrine and epinephrine). Its clinical use is limited, however, because it causes significant orthostatic hypotension and sedation, and many other antihypertensive drugs with fewer adverse effects are available for this indication.
Inhibitors of Catecholamine Storage
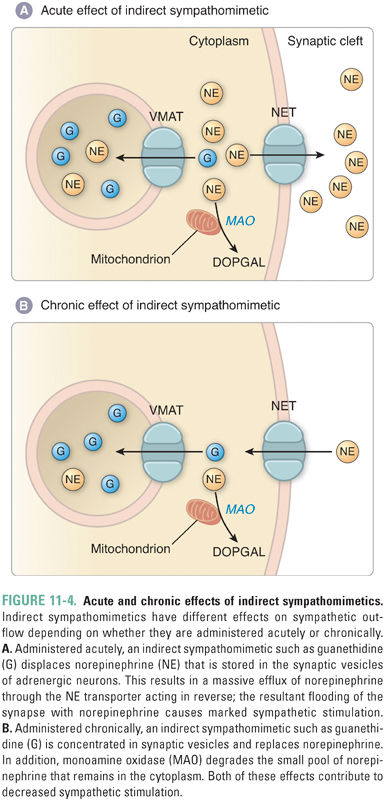
Catecholamines originate from two pools—de novo synthesis and recycled transmitter. An agent that inhibits catecholamine storage in vesicles generally has two sequential effects. In the short term, the agent increases the net release of catecholamine from the synaptic terminal, and thus mimics sympathetic stimulation (sympathomimetic). Over a longer time period, however, the agent depletes the pool of available catecholamine and thus acts as a sympatholytic (inhibitor of sympathetic activity) (Fig. 11-4).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


