Adaptive Immune Response to Viral Infections
Thomas J. Braciale
Young S. Hahn
Dennis R. Burton
Introduction
Viruses have been part of human ecology since the human species first evolved on the plains of East Africa. As humans moved out of Africa to populate the continental landmasses,
viruses—already adapted to the human species—were carried along with other microorganisms as part of the human ecological baggage. With the migration of humans around the world, we were exposed to new viruses present in the animal species that humans encountered. Some viruses, such as the herpesviridae family members, Epstein-Barr virus (EBV), and herpes simplex virus (HSV), have co-evolved with humans and have developed complex mechanisms (e.g., latency) to subvert the host defense mechanisms and sustain themselves in the human population. These viruses generally produce mild disease, except where host defenses are absent or weakened (e.g., in immune-deficient or immune-suppressed individuals). Other human viruses (e.g., measles virus) appear to have evolved mechanisms to persist at low levels in some infected individuals and sustain themselves by infecting previously unexposed individuals (typically children) who then go on to develop mild to severe illness and, in turn, serve as reservoirs for further virus propagation. Animal viruses can also jump from their host reservoir to infect humans and produce devastating diseases of potentially pandemic proportions. Notable examples of zoonotic viral diseases are the following: (a) the acquired immunodeficiency syndrome (AIDS) pandemic produced by the human immunodeficiency virus (HIV); (b) the outbreak of severe acute respiratory syndrome (SARS) in 2002 produced by the SARS coronavirus; (c) the 2009 H1N1 swine origin influenza A virus pandemic arising from the genetic reassortment in pigs of viruses with donated genes of human, swine, and avian flu origin; and (d) the periodic outbreaks of highly pathogenic avian influenza infection in poultry since 1997 in Asia and most recently in Africa and Europe, which have resulted in sporadic direct infections of humans with mortality rates approaching 50%. Indeed, the recent reconstruction and analysis of the genome of the influenza virus that caused the 1918 Spanish flu pandemic suggest that this virus may have been an avian influenza virus that spread directly to humans (without modification of virus in an intermediate host or the acquisition of genetic information from then-circulating human influenza strains).288
viruses—already adapted to the human species—were carried along with other microorganisms as part of the human ecological baggage. With the migration of humans around the world, we were exposed to new viruses present in the animal species that humans encountered. Some viruses, such as the herpesviridae family members, Epstein-Barr virus (EBV), and herpes simplex virus (HSV), have co-evolved with humans and have developed complex mechanisms (e.g., latency) to subvert the host defense mechanisms and sustain themselves in the human population. These viruses generally produce mild disease, except where host defenses are absent or weakened (e.g., in immune-deficient or immune-suppressed individuals). Other human viruses (e.g., measles virus) appear to have evolved mechanisms to persist at low levels in some infected individuals and sustain themselves by infecting previously unexposed individuals (typically children) who then go on to develop mild to severe illness and, in turn, serve as reservoirs for further virus propagation. Animal viruses can also jump from their host reservoir to infect humans and produce devastating diseases of potentially pandemic proportions. Notable examples of zoonotic viral diseases are the following: (a) the acquired immunodeficiency syndrome (AIDS) pandemic produced by the human immunodeficiency virus (HIV); (b) the outbreak of severe acute respiratory syndrome (SARS) in 2002 produced by the SARS coronavirus; (c) the 2009 H1N1 swine origin influenza A virus pandemic arising from the genetic reassortment in pigs of viruses with donated genes of human, swine, and avian flu origin; and (d) the periodic outbreaks of highly pathogenic avian influenza infection in poultry since 1997 in Asia and most recently in Africa and Europe, which have resulted in sporadic direct infections of humans with mortality rates approaching 50%. Indeed, the recent reconstruction and analysis of the genome of the influenza virus that caused the 1918 Spanish flu pandemic suggest that this virus may have been an avian influenza virus that spread directly to humans (without modification of virus in an intermediate host or the acquisition of genetic information from then-circulating human influenza strains).288
Virus infection can result in acute or chronic disease in virtually any organ and tissue of the body. Furthermore, infection with human papillomavirus (HPV), human herpesvirus 8 (HHV-8), and hepatitis B virus (HBV) and hepatitis C virus (HCV) have been directly linked to the development of cervical cancer, Kaposi’s sarcoma, and hepatocellular carcinoma, respectively. Given the pandemic outbreak of the swine origin A/California/2009 (H1N1) virus infection, the possibility of another human influenza pandemic from an avian influenza source, as well as the devastating effects of HIV infection all suggest, virus infection represents a continuing threat to human health. We can point to some notable successes in the struggle against viruses (e.g., the eradication of smallpox in the 1970s, and the effectiveness of the live attenuated oral polio vaccine in eliminating paralytic polio in the developing world, raising the prospect of eradicating polio worldwide). Both of these vaccination successes rely on the mobilization of the body’s major defense against virus infection—the adaptive immune system. Our enthusiasm about these successes must be tempered, however. Because of the success of smallpox eradication, mandatory smallpox vaccination was discontinued in the United States in the early 1970s, making a large fraction of the population (i.e., those younger than age 40) now susceptible to smallpox. This fact, along with the advances in poxvirus genetics and genetic engineering, now makes smallpox a major bioterrorism threat. Similarly, administration of live oral polio vaccine to children in underdeveloped countries whose immune function is compromised by malnutrition or HIV infection can result in outbreaks of paralytic polio caused by infection with virulent mutants (revertants) of the live attenuated vaccine strain. This realization has prompted a modification of the guidelines for oral polio vaccine administration in the United States and a reevaluation of the prospect of global polio eradication using current vaccination strategies. To counter the continuing threat posed by viruses, it is essential to understand the body’s primary defense against virus infection in the immune system.
Overview of the Adaptive Immune Response to Virus Infection
Viruses are obligatory intracellular parasites; that is, they replicate within cells of the infected host and use the cell’s biosynthetic machinery for replication and production of progeny virions. Furthermore, most viruses replicate rapidly in infected cells (i.e., within hours) producing progeny virions capable of infecting additional cells, thereby propagating and sustaining infection in the host. These properties of infection by viruses dictate how the immune system must respond to counterinfection. In acute virus infection, the race between the replication of the virus and the host response will result either in the death of the host or in virus clearance and the termination of infection. The tissue injury produced by viruses at the site(s) of infection results both from the cytopathic effects of virus replication and, equally importantly, from the host immune response to infection. In general, therefore, the greater the extent of virus replication in the infected host, the greater the tissue injury and illness severity.
In chronic infection, the time scale of virus replication in the host is not measured in days (as in acute infection) but in weeks, months, or even years, with the host immune response likewise continuing over the same prolonged time scale. Viruses that produce chronic, persistent infection have evolved mechanisms to suppress or alter the immune response (allowing these viruses to persist in the infected host). The tissue injury produced by the immune response to virus infection (termed, immunopathology) occurs with both acute and chronic virus infection. In many chronic virus infections (e.g., HCV infection); however, immune-mediated tissue injury predominates (rather than direct virus-mediated injury).38 Furthermore, the clinical manifestations of virus infection (e.g., fever, headache, myalgia, anorexia) are caused primarily by inflammatory mediators (e.g., cytokines) released by cells of the immune system in response to infection. Undoubtedly, chronic (persistent) virus infection also alters or modifies the immune system and its capacity to respond to microbes and environmental antigens.297 Finally, the triggering of the immune system by virus infection can result in the induction of aberrant immune responses directed both to the virus, and to self-cellular constituents, resulting in autoimmune disease.143
The primary host defenses against virus infection are physical/chemical barriers to infection and the immune system. The immune system can be divided into two components: the innate immune system (see Chapter 8) and the adaptive immune system. This division is based on the properties of
the immune cell types and molecules involved in the response to infection and the tempo of the response (Table 9.1). The response of the innate immune system to virus infection is either immediate (i.e., constitutively active) or rapidly induced (i.e., typically within hours of infection). The innate immune response is triggered in two ways: first, through recognition of viral constituents (e.g., viral nucleic acids) by a limited set of cellular pattern recognition receptors for foreign molecules displayed on innate immune cells; and, second, through the activation of intracellular signaling mechanisms following virus entry into cells and the initiation of virus replication. Importantly, a defining feature of the innate immune response is that repeat exposure to a particular virus generally evokes an identical response from the innate immune system (although a form of that enhanced efficiency of pathogen recognition has recently been reported for natural killer (NK) cells.298 The genes encoding these receptors and associated signaling mechanisms within innate immune cells are encoded within the germline (i.e., fixed in the genome).
the immune cell types and molecules involved in the response to infection and the tempo of the response (Table 9.1). The response of the innate immune system to virus infection is either immediate (i.e., constitutively active) or rapidly induced (i.e., typically within hours of infection). The innate immune response is triggered in two ways: first, through recognition of viral constituents (e.g., viral nucleic acids) by a limited set of cellular pattern recognition receptors for foreign molecules displayed on innate immune cells; and, second, through the activation of intracellular signaling mechanisms following virus entry into cells and the initiation of virus replication. Importantly, a defining feature of the innate immune response is that repeat exposure to a particular virus generally evokes an identical response from the innate immune system (although a form of that enhanced efficiency of pathogen recognition has recently been reported for natural killer (NK) cells.298 The genes encoding these receptors and associated signaling mechanisms within innate immune cells are encoded within the germline (i.e., fixed in the genome).
Table 9.1 Cells of the Immune System | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||
By contrast, the response of the adaptive immune system to a first encounter with a virus takes days to evolve. The response of the cells of the adaptive immune system (i.e., B lymphocytes and T lymphocytes) is triggered by viral constituents, primarily through the engagement of highly specific cell-surface recognition receptors generated by somatic gene rearrangement and displayed by a minuscule fraction of the total repertoire of adaptive immune cells for any given virus. These receptors only recognize the constituents (antigens) of that particular virus. Importantly, this arm of the immune system adapts to repeat exposure to a particular virus or viral protein; this results in a more rapid response of higher magnitude by the specific adaptive immune cells on repeat exposure/infection—a phenomenon called immunological memory, the basis for vaccination.
The constituents of the innate immune response include proteins of the complement system, and so called acute phase proteins (e.g., members of the collectin protein family).251 These molecules are present constitutively in the blood and tissue fluids or are induced rapidly in the liver following infection and can bind directly to certain viruses, thereby inhibiting virus infection. Leukocytes, in particular, neutrophils, blood monocytes, tissue macrophages, dendritic cells, NK cells and NK T cells are the dominant, innate immune cell types responding to virus infection. They are triggered/activated through the engagement of pattern recognition receptors (e.g., Toll-like receptors [TLRs], C-type lectins) expressed on their cell surface, which recognize viral constituents (e.g., viral nucleic acids), and in the case of NK and NK T cells, alterations of the cell surface of infected cells. These cells reside in the tissues, and they respond there or are recruited to sites of infection from the bloodstream through the action of cytokines/chemokines released during infection.
Nonhematopoietic body cells (e.g., epithelial cells, endothelial cells, fibroblasts) that are targets of virus infection, must also be considered part of the innate immune response because, on infection, they are triggered to secrete antiviral proinflammatory mediators such as type 1 interferons and certain cytokines/chemokines (the production of which can initiate and amplify the innate immune response to infection). The induction of the innate immune response and the role of innate immune cells and molecules in controlling virus infection are discussed in detail elsewhere (Chapter 8). One of these innate immune cell types, the dendritic cell (DC), however, bridges the innate and adaptive immune response, and its role in the initiation of the adaptive immune response is discussed in a later section of this chapter.
A hallmark of the adaptive immune response is the exquisite specificity of adaptive immune cells for a particular foreign viral antigen, and the capacity of this system to recognize and respond to a myriad of viruses (or other antigens). This remarkable property of the adaptive immune cells is achieved by the generation of a correspondingly diverse array of cell surface recognition receptors, which are randomly generated within the lymphocytes of every individual and clonally distributed among the adaptive immune cells. As a consequence, in a nonimmune individual, only an extremely small fraction of the adaptive immune cell repertoire (e.g., one cell in a million or less) will have an antigen receptor capable of recognizing a specific virus (viral antigen). Understanding how this diverse array of clonally distributed antigen receptors is generated during lymphocyte development and how that rare adaptive immune cell directed to particular virus finds virus or infected cells in a peripheral site of infection (e.g., the skin or lungs) is critical to the understanding of the adaptive immune response to virus infection.
Viruses, because they exist as extracellular virion particles and replicate/assemble within infected cells, pose a unique recognition problem for the adaptive immune system (i.e., how
to recognize the virus [viral proteins or genome] both outside of the cell and within the infected cell). The elegant solution to this problem employed by the adaptive immune system is the partitioning of the adaptive immune system into two distinct cell types: B and T lymphocytes (Table 9.2). B lymphocytes displaying a receptor specific for a particular virus will, on encounter with the virus and activation, release their antigen receptor in a soluble form (i.e., as antibody molecules) capable of binding free (extracellular) virions in a highly specific matter. This interaction typically results in virus neutralization and/or elimination. Antiviral B-lymphocyte responses are exquisitely sensitive to the three-dimensional confirmation of the virus (and its constituents), and they play a dominant role in clearing virus during infection, in preventing or limiting reinfection (after previous virus infection), and in vaccination against a specific virus.
to recognize the virus [viral proteins or genome] both outside of the cell and within the infected cell). The elegant solution to this problem employed by the adaptive immune system is the partitioning of the adaptive immune system into two distinct cell types: B and T lymphocytes (Table 9.2). B lymphocytes displaying a receptor specific for a particular virus will, on encounter with the virus and activation, release their antigen receptor in a soluble form (i.e., as antibody molecules) capable of binding free (extracellular) virions in a highly specific matter. This interaction typically results in virus neutralization and/or elimination. Antiviral B-lymphocyte responses are exquisitely sensitive to the three-dimensional confirmation of the virus (and its constituents), and they play a dominant role in clearing virus during infection, in preventing or limiting reinfection (after previous virus infection), and in vaccination against a specific virus.
Table 9.2 Properties of T and B Lymphocytes | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||
Activated antiviral T lymphocytes retain their antigen-specific receptors on the cell surface and are responsible for recognizing and eliminating virus-infected cells. T lymphocytes, however, do not recognize viral proteins in their native conformation. Rather, the antigen receptors on the T lymphocytes recognize processed peptide fragments of viral proteins displayed on the infected cell surface bound to cell-surface molecular recognition platforms (i.e., the major histocompatibility complex [MHC] locus gene products). Activated virus-specific T lymphocytes halt the spread of infection by killing virus-infected cells through direct cell-to-cell contact and by the release of soluble mediators (e.g., cytokines, such as IFN-γ and tumor necrosis factor α [TNF-α]). By releasing these and other soluble mediators, the activated T lymphocytes recruit and orchestrate the response of the innate immune cells which, in turn, act to clear infection. Understanding how and where B and T lymphocytes activate, the mechanism of antigen presentation to the cells, and the range of effector activities that these cells use to control and eliminate virus is essential for understanding the adaptive immune response to virus infection. These important topics are reviewed in this chapter.
Finally, the importance of the immune system in general and the adaptive immune response in particular in controlling virus clearance and promoting recovery from infection is exemplified by the fact that viruses have evolved a variety of mechanisms to inhibit or alter the host immune response. Whereas the virus immune evasion strategies are best considered in the context of viral pathogenesis (Chapter 10) and are addressed in chapters dealing with individual viruses, this chapter highlights some of the viral evasion strategies directed to the adaptive immune response.
Architecture of the Adaptive Immune System
Generation of T and B Lymphocytes: Primary Lymphoid Organ
Unlike the cells of the innate immune system, T and B lymphocytes possess one unique and profoundly important property. On their surfaces, these cells also display a receptor complex capable of recognizing foreign (nonself) structures—with exquisite specificity. This receptor complex is both randomly generated (by somatic recombination of genetic elements ultimately encoding the mature receptors) and clonally distributed among progeny lymphocytes during their differentiation from progenitor cells. Therefore, of the approximate 109 naive T and B lymphocytes in the human adaptive immune system, each cell may, in principle, display a unique receptor directed to a specific foreign antigenic structure. This common property of the antigen receptor on T and B lymphocytes (i.e., random generation by somatic DNA recombination and clonal receptor display) immediately raises two important questions. First, if these antigen receptors are stochastically generated, how is the generation of lymphocytes with receptors to self-structures (e.g., tissue proteins) avoided/prevented? Second, if the frequency of lymphocytes with a receptor directed to a specific foreign antigen (e.g., the coat proteins of an invading virus) is extremely low (1 in 107 lymphocytes), how can those rare lymphocytes locate the antigen quickly enough to respond before the infection is beyond control? As is discussed below, it is the unique structure of the primary (central) lymphoid organs where lymphocyte progenitors develop into mature T and B lymphocytes, and of the secondary (peripheral) lymphoid organs where mature T and B lymphocytes respond to invading microorganisms that provides the answers to these questions.
Mature T and B lymphocytes are derived from lymphoid progenitors present in the bone marrow, as well as in sites of hematopoiesis in developing fetus (e.g., the liver) through a process of differentiation and selection linked to, and driven by, the antigen receptors displayed by these cells.221,233 As described in Table 9.2, B-lymphocyte lineage development occurs in the bone marrow and follows a well-defined, step-wise program of gene
activation (e.g., expression of the recombination activating gene 1 and 2 [RAG1 and 2], lymphoid DNA-specific recombinases), and sequential rearrangement of immunoglobulin (Ig) VH, DH, JH, and CH heavy (H) chain genetic elements (by random somatic recombination), followed by rearrangement of the Ig K/λ VL-JL-CLK/l light (L) chain locus elements. Then B-lymphocyte progenitors transit from the pro-B, through the pre-B, and to the surface immunoglobulin-positive immature B-lymphocyte stage. It is during this development/differentiation program that the B-lymphocyte receptor (B-cell receptor [BCR]) diversification occurs through random association of one of the 40 VH gene segments with one of the 25 DH and 6 JH and 70 VL with 9 JL to yield (after nucleotide addition and/or excision at the site of gene segment recombination) 107 or more different potential BCR H and L chain combinations. During this development process in the bone marrow, T-Lymphocytes expressing a BCR with reactivity to self-molecules (i.e., proteins, lipids, carbohydrates, nucleic acids) will, as a result of BCR engagement, undergo a process of negative selection. These self-reactive B cells are largely eliminated/deleted through interaction of the BCR on the developing cells, with self-constituents resulting in apoptosis or in activation (“anergy”) of self-reactive B lymphocytes. The immature B lymphocytes that escape negative selection undergo final maturation, migrate from the bone marrow to join the pool of mature B lymphocytes that circulate through the blood, and populate the secondary lymphoid organs. B lymphocytes ultimately express their effector activity by releasing the BCR of the mature B lymphocyte in a secreted form as immunoglobulin (antibody) molecules. This fact has the following two important implications for the recognition of viruses by B lymphocytes: (a) the B-lymphocyte response to viruses will be particularly effective when interacting with free/extracellular virus constituents (e.g., virions); and (b) the BCR and its antibody product will be sensitive to the conformation of viral constituent recognized.
activation (e.g., expression of the recombination activating gene 1 and 2 [RAG1 and 2], lymphoid DNA-specific recombinases), and sequential rearrangement of immunoglobulin (Ig) VH, DH, JH, and CH heavy (H) chain genetic elements (by random somatic recombination), followed by rearrangement of the Ig K/λ VL-JL-CLK/l light (L) chain locus elements. Then B-lymphocyte progenitors transit from the pro-B, through the pre-B, and to the surface immunoglobulin-positive immature B-lymphocyte stage. It is during this development/differentiation program that the B-lymphocyte receptor (B-cell receptor [BCR]) diversification occurs through random association of one of the 40 VH gene segments with one of the 25 DH and 6 JH and 70 VL with 9 JL to yield (after nucleotide addition and/or excision at the site of gene segment recombination) 107 or more different potential BCR H and L chain combinations. During this development process in the bone marrow, T-Lymphocytes expressing a BCR with reactivity to self-molecules (i.e., proteins, lipids, carbohydrates, nucleic acids) will, as a result of BCR engagement, undergo a process of negative selection. These self-reactive B cells are largely eliminated/deleted through interaction of the BCR on the developing cells, with self-constituents resulting in apoptosis or in activation (“anergy”) of self-reactive B lymphocytes. The immature B lymphocytes that escape negative selection undergo final maturation, migrate from the bone marrow to join the pool of mature B lymphocytes that circulate through the blood, and populate the secondary lymphoid organs. B lymphocytes ultimately express their effector activity by releasing the BCR of the mature B lymphocyte in a secreted form as immunoglobulin (antibody) molecules. This fact has the following two important implications for the recognition of viruses by B lymphocytes: (a) the B-lymphocyte response to viruses will be particularly effective when interacting with free/extracellular virus constituents (e.g., virions); and (b) the BCR and its antibody product will be sensitive to the conformation of viral constituent recognized.
T-Lymphocyte lineage development follows a program similar to that of B lymphocytes. The T-cell antigen receptor (TCR) is also a heterodimer consisting of two polypeptide chains, each of which is formed by somatic recombination of variable (V), diversity (D), and joining (J) segments encoding the variable portion of one chain (or V and J joining for the other chain) to a constant (C) gene segment. As in B lymphocytes, the randomly recombined gene segments encoding the V region gene segment joined to the respective C region gene segment for the two TCR chains encodes the TCR on the developing T lymphocyte. Unlike the BCR, however, the TCR always remains cell associated. Therefore, the TCR is normally directed to foreign antigens (or self-structures) that are cell associated. Consequently, T lymphocytes are uniquely suited to recognize cells that have been invaded by microorganisms (e.g., viruses and intracellular bacteria). Unlike the BCR, the TCR does not recognize intact microbial products, but rather recognizes small fragments of molecules—typically, short peptide fragments of microbial or self-proteins bound to protein products of the mammalian MHC genetic locus (e.g., the classically defined major human transplantation antigens). These MHC gene products expressed on cell surfaces serve as molecular platforms, both selecting and displaying the self-peptides involved in TCR selection during T-lymphocyte precursor development, and presenting microbial peptides to mature T lymphocytes responding to uptake/infection of cells by the microbes. The steps in the processing (fragmenting) and presentation of both self and microbial gene products by the MHC locus protein, and the structure of the major classes of MHC locus proteins (i.e., MHC class I and II proteins) are discussed below.
The generation of mature T lymphocyte from committed lymphocyte progenitors (arising in the bone marrow) occurs primarily in the thymus (Fig. 9.1). Here, immature thymocytes undergo the developmental program resulting in the formation of mature T lymphocytes, which then leave the thymus, enter the circulation, and ultimately populate secondary lymphoid organs. Two classes of mature T lymphocytes are produced in the thymus. The major population (∼80% of the peripheral T lymphocyte pool) are the α+β+ T cells (because their heterodimeric TCR express two chains encoded by TCR α and β loci). The second minor population of mature T cells are the γ+δ+ T cells (because they express a related two-chain TCR derived from the TCR γ and δ loci). Much of our current information on T-lymphocyte development comes from analysis of α+β+ T-cell development. These T lymphocytes represent the T-cell subset that responds primarily to viral infection, and the selection process of these T cells in the thymus will be emphasized here. The γ+δ+ T-cell subset likely plays a secondary role in the adaptive response to most virus infections, and the properties/function of this T-cell subset is summarized below.
T-Lymphocyte development in the thymus proceeds in a stepwise fashion with the sequential generation and expression of one (i.e., TCR-β or TCR-δ), and then the other (i.e., TCR-α or TCR-γ) TCR chain by random somatic recombination as thymocytes mature. A complex of four cell-surface molecules called CD3 (CD3 γ, δ, ε, and ξ), which are noncovalently associated with the TCR and are critical for signaling through the TCR, are also expressed during T-lymphocyte development. Specific antigen recognition by the TCR-α:β (or γ:δ) heterodimer is transduced into the cells by the CD3 complex. Concomitant with the expression of the rearranged TCR chains and the signal transducting CD3 complex, the developing thymocytes simultaneously express two co-receptor molecules, CD4 and CD8. Early in the developmental program, thymocytes do not express CD4 or CD8, and they are classified as immature, double-negative thymocytes. As thymic development/differentiation proceeds, immature thymocytes that express both CD4 and CD8 co-receptors (as well as CD3 complex and the TCR-α and β chains) and, at this point, are classified as are CD4+/CD8+ double-positive thymocytes. CD4 and CD8 molecules recognize structurally conserved domains on the MHC class II and I molecules, respectively. As a result of binding to MHC molecules displaying self-peptides on the surface of thymic stromal cells, CD4 or CD8 also delivers signals to the developing immature thymocytes. The combined signaling by TCR/CD3 complex and the CD4 or CD8 co-receptor directs the development of the immature thymocytes into single-positive CD4+ or CD8+ mature thymocytes (expressing TCR, CD3, and either CD4 or CD8).
Because the TCR does not recognize foreign molecules directly (but rather small fragments of molecules bound to MHC molecules), the TCR generated and expressed on developing thymocytes must recognize both MHC molecules and the peptide fragments bound to these molecules. The source of these peptides bound by MHC class I and class II molecules are self-proteins expressed by several different cell types in the thymus. The selection of the thymocytes that ultimately become mature co-receptor single-positive T lymphocytes (CD4+ or CD8+) and populate the secondary lymphoid organs is a process in the thymus of positive and negative selection.
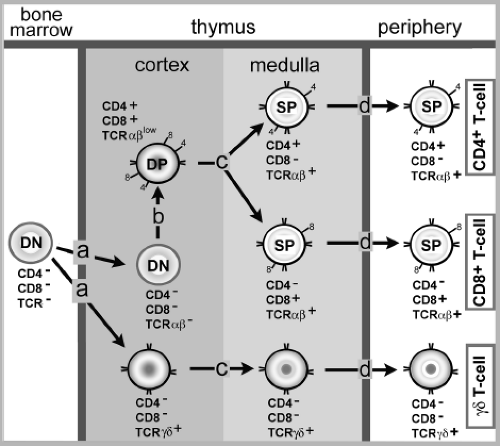 Figure 9.1. T-Lymphocyte development and selection in the thymus. The progenitors of mature peripheral T lymphocytes arise in the bone marrow as mononuclear stem cell progenitors committed to T-cell lineage development, but lacking cell surface receptors characteristic of T-lymphocyte lineage (e.g., T-cell receptors [TCRs] or the co-receptors CD4 and CD8). These TCR- and co-receptor–deficient “double negative” (DN) cells enter the thymic cortex (a) from the bloodstream as CD4/CD8-deficient DN lymphocytes. In the case of conventional TCRαβ T cells, the DN cells also lack antigen receptor. The cells then undergo a multistep differentiation process (b), which results in cells simultaneously expressing both CD4 and CD8 as well as a low level expression of the TCRαβ receptor complex—so-called double-positive (DP) thymocytes. Within the thymic cortex and medulla (c), the DP cells first undergo “positive selection”—that is, selection for those cells with TCRαβ and CD4 or CD8 co-receptor complexes capable of interaction with self-peptide/major histocompatibility complex (MHC) ligands. DP thymocytes that interact with self-peptide/MHC ligand with insufficient avidity die (undergo apoptosis). The surviving DP thymocytes then transition into either CD4+ or CD8+ single-positive (SP) thymocytes, which then undergo a process of “negative selection.” In this case, only those SP thymocytes with an appropriate immediate affinity for self-peptide/MHC complexes survive and go on to traffic through the thymus into the bloodstream to ultimately proceed to the secondary lymphoid organs (d) as mature peripheral CD4+ or CD8+ T cells. The TCRγδ T cells likewise arrive in the thymus (a) as DN progenitors, which eventually upregulate the γ δ TCR (b) and undergo a form of positive and negative selection (c) without upregulation of the canonical co-receptors through a selection process that is not well understood. The TCRγ δ T cells will exit the thymus (d) and populate peripheral sites including mucosal surfaces and secondary lymphoid organs. Natural regulatory T-cells (Treg) cells (not shown) undergo selection for recognition of self-peptide/MHC class II ligands late in thymocyte differentiation by a selection process poorly understood that this time. |
At a specific development stage, immature double-positive thymocytes (expressing the randomly generated α : β TCR, CD3, and both CD4/CD8) encounter, within the thymic cortex, thymic cortical epithelial cells displaying MHC class I and II molecules complexed with self-peptides. Most of these thymocytes (∼80%) have randomly generated TCR that are unable to bind to the self-peptide/MHC molecular complexes. In the absence of a signal delivered through the TCR, these thymocytes will die by apoptosis. Those double-positive thymocytes whose TCR can bind the self-peptide/MHC complexes above a threshold avidity augmented by either a CD4 or a CD8 co-receptor binding to the MHC molecule will undergo positive selection and survive. If the thymocyte TCR can bind peptides bound to MHC class II molecules in association with the CD4 co-receptor, this interaction will result in the silencing of the CD8 co-receptor gene. This signaling event results in the development of a CD4+ single-positive thymocyte whose TCR recognizes (is restricted to) peptides bound to MHC class II molecules. Similarly, the double-positive thymocytes whose TCR and associated CD8 molecule can recognize MHC class I/self-peptide complexes with sufficient avidity to survive (i.e., those that are positively-selected) will give rise to MHC class I–restricted CD8+ single-positive thymocytes by a similar silencing mechanism of the CD4 gene.
After undergoing positive selection, the surviving single-positive thymocytes are all auto-reactive (i.e., directed to self-peptide/MHC complexes). Therefore, a process of negative selection must then ensue to eliminate/reduce self-reactive, mature peripheral T cells. Although less well understood, negative selection of the α : β TCR-expressing single-positive (CD4+ or CD8+) thymocytes probably occurs in both the thymus cortex and medulla. Negative selection is most efficiently mediated by mononuclear cells of hematopoietic origin (e.g., dendritic cells and macrophages) that simultaneously display MHC class I and II molecules expressing self-peptides. The 10% to 20% of thymocytes that survive positive selection, by definition, are auto-reactive. It is likely (but not certain) that the intracellular signal transduction cascade associated with TCR engagement is altered in the cortical and medullary single-positive thymocytes following positive selection. As a result these cortical and medullary thymocytes acquire the TCR-signaling profile of mature T lymphocytes. Consequently, when the thymocytes encounter the self-peptide/MHC complexes displayed by hematopoietic origin cells like DC in the cortex and medulla, those thymocytes whose TCR/CD4 or CD8 co-receptor complex recognizes self-peptide/MHC complexes with a sufficient avidity to activate will be eliminated (i.e., undergo negative selection—death by apoptosis). Thymocytes directed to self- “tissue specific” molecules (e.g., insulin) are deleted through the action of the autoimmune regulator (AIRE) gene. This gene is expressed in thymic medullary stromal cells and triggers the transient low level expression of many extrathymic tissue-specific self-proteins in these cells resulting in negative selection of tissue-reactive single-positive thymocytes. Therefore, only about 2% to 5% of immature thymocytes will survive the positive and negative selection processes. These remaining thymocytes will have the properties of mature CD4+ or CD8+ T lymphocytes (i.e., they have TCR that recognize peptide/MHC complexes, but will only interact with the MHC molecules with sufficient avidity to activate when the appropriate foreign [microbial] peptides are bound to the MHC molecule). Following positive and negative selection, these single-positive α : β T lymphocytes (CD4+ or CD8+) will leave the thymus to populate the secondary lymphoid organs as peripheral T cells (Fig. 9.1). The CD4+ or CD8+ α : β T cells represent 70% to 80% of the circulating extrathymic T-cell population—the primary T-cell population that responds to virus infection.
The second (minor) population of thymus-derived cells that populates peripheral sites is that of the γ : δ T cells, so named because they employ a heterodimeric TCR whose variable (and constant) region is encoded by a distinct set of genetic loci (the γ locus and the δ locus) homologous to the α and β TCR loci103 (Table 9.3). These TCR genes rearrange in the thymus in a manner analogous to that of the
dominant α : β TCR T-cell homolog. The generation of γ : δ T cells in the thymus precedes α : β T-cell development. The γ : δ T cells are generated in two waves during T-cell development in the fetus. The first wave of maturing thymocytes consists of cells with limited TCR V gene diversity (i.e., utilize one or few Vγ and Vδ rearrangement in their TCR). After selection, these γ : δ T cells leave the thymus, traffick through the circulation, and preferentially localize to epithelial surfaces, particularly the skin, gut, and urogenital tract. The second wave of γ : δ T cells, as with α : β T cells, displays a highly diverse TCR array, and these cells localize to the secondary lymphoid tissues. Unlike the α : β TCR, however, the γ : δ TCR does not recognize peptides bound to the MHC class I or II locus products. Rather, γ : δ T cells either directly recognize structures such as heat shock proteins displayed on the surface of the infected cells, or recognize lipid or glyco/phospholipids moieties bound to MHC-like molecules (e.g., nonpolymorphic MHC Ib molecules) on the surface of stressed (infected) cells. The subset of intraepithelial γ : δ T cells, in particular, differs from the α : β T cells, and appears to function like cells of the innate immune system, recognizing not a specific pathogen, but alterations in epithelial cells induced by infection with microbes such as bacteria. At present, no direct role for γ : δ T cells in the control of virus infection has been firmly established although this T-cell subset has been implicated in the control of HIV and herpesvirus infection in humans.5
dominant α : β TCR T-cell homolog. The generation of γ : δ T cells in the thymus precedes α : β T-cell development. The γ : δ T cells are generated in two waves during T-cell development in the fetus. The first wave of maturing thymocytes consists of cells with limited TCR V gene diversity (i.e., utilize one or few Vγ and Vδ rearrangement in their TCR). After selection, these γ : δ T cells leave the thymus, traffick through the circulation, and preferentially localize to epithelial surfaces, particularly the skin, gut, and urogenital tract. The second wave of γ : δ T cells, as with α : β T cells, displays a highly diverse TCR array, and these cells localize to the secondary lymphoid tissues. Unlike the α : β TCR, however, the γ : δ TCR does not recognize peptides bound to the MHC class I or II locus products. Rather, γ : δ T cells either directly recognize structures such as heat shock proteins displayed on the surface of the infected cells, or recognize lipid or glyco/phospholipids moieties bound to MHC-like molecules (e.g., nonpolymorphic MHC Ib molecules) on the surface of stressed (infected) cells. The subset of intraepithelial γ : δ T cells, in particular, differs from the α : β T cells, and appears to function like cells of the innate immune system, recognizing not a specific pathogen, but alterations in epithelial cells induced by infection with microbes such as bacteria. At present, no direct role for γ : δ T cells in the control of virus infection has been firmly established although this T-cell subset has been implicated in the control of HIV and herpesvirus infection in humans.5
Table 9.3 T Lymphocyte Subsets | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
Among the other T-lymphocyte subsets (Table 9.3) generated during development are NK T cells, so-called because these α : β TCR-expressing T cells display cell-surface markers shared with NK cells, most notably the C-type lectin NK 1.1. NK T cells are divided into two subsets CD4+ CD8− or CD4−CD8− based on co-receptor expression. One subset of NK T cells employs a range of α : β V gene combinations in their expressed TCR. The other NK T-cell subset is similar to CD4− and CD8− γ : δ T cells, in that the cells utilize a limited number of TCR α : β V genes in receptor generation and are classified as invariant NK T cells (iNK T cells). The iNK T cells recognize foreign and self-lipid/glycolipid bound to MHC-like cell-surface CD1 molecules. The NK T cells with diverse TCR usage by contrast, directly recognize self- or microbial gene products rather than peptide/MHC complexes. Although not as yet certain, the CD1/lipid complexes and the self-moieties (e.g., heat shock proteins) recognized by NK T cells are likely produced or upregulated by cells in response to virus infection.
One T-lymphocyte subset has recently been recognized as a critical regulator of the adaptive immune responses to both infectious agents and self-molecules: the regulator T lymphocyte (Treg) subset. The TCR on Treg cells utilize rearranged α : β TCR gene products, and appear (at least in experimental animals) to arise late in thymic development (i.e., after the major wave of both γ : δ αvδα : β T-cell generation). As mature thymocytes, Treg cells appear able to recognize self-peptides bound to MHC molecules and directly activate without undergoing negative selection. Therefore, Treg cells that display primarily the CD4 co-receptor exist both in the thymus (as mature thymocytes) and in the periphery (i.e., in secondary lymphoid organs) as activated CD4+ T cells. The encounter with self and the resulting activation of Treg cells, however, does not result in autoimmunity. Rather, recent evidence suggests that these T cells suppress/downregulate the activity of conventional CD4+ and CD8+ T cells responding to viral infection. Although both the role of Treg cells in adaptive immune responses and their mechanism of action are not yet fully understood, these cells may prevent the development of excessive injury during T-cell responses to infection, and suppress the development of autoimmune responses during viral infection. Recent emerging information on the role of Treg cells in virus infection is discussed in a separate section below.
Secondary Lymphoid Organs: Structure and Function
The mature naïve (resting) B and T lymphocytes that egress from their development site (bone marrow and thymus, respectively) enter the blood, and then migrate to secondary (peripheral) lymphoid organs (SLOs). The SLOs play a critical role in the initiation of the adaptive (T- and B-cell) immune response to viruses and other microbes. The major SLOs are the lymph nodes (LNs), the spleen, and the so-called mucosal-associated lymphoid tissue (MALT). LNs are highly organized structures located at vessel convergence points in the lymphatic system, a system that collects fluids and cells from the tissues of most internal organs (e.g., liver, stomach) and body surfaces (e.g., skin, mucosa of the gastrointestinal [GI], genitourinary [GU], and respiratory tracts). The capacity of lymphatic vessels to collect antigen (e.g., virus, viral constituents) from the body surfaces/tissues, and deliver them to the LNs (where antigen concentrates) is essential for the efficient triggering of the adaptive immune response to microbes. The spleen plays a role similar to that of the LN network; however, the spleen is uniquely suited to collect antigen (virus) directly from the bloodstream, because of its unique architecture and extensive blood circulation. The third class of SLOs, the MALT, is composed of specialized lymphoid aggregates associated with major mucosal surfaces and sites of microbial invasions (e.g., the GI, GU, and respiratory tracts). The gut-associated MALT (also called GALT) includes tonsils, adenoids, appendix, and Peyer’s patches in the small intestine. In the respiratory tract are similar, but more diffuse, aggregates of lymphoid tissues associated with the epithelium of small/medium airways (i.e., bronchial-associated lymphoid tissue [BALT]). As with the other SLOs, the MALTs are sites of antigen localization where the initiation/induction of the adaptive immune response can occur.
The common feature of SLOs is the localization of adaptive immune cells (i.e., T and B cells) to distinct regions of the SLOs. For example, most B cells are localized in distinct follicles within the LN cortex. Activation of B cells specific for a pathogen/antigen leads to their proliferation and formation of a germinal center within the LN cortex. T cells within the LN localize to, and are diffusely distributed within, the LN paracortical areas. Other cell types are also distributed within the outer cortex and central medulla of the normal LN. These include stromal cells and cells of macrophage and DC lineage. These cells (particularly DC) play a crucial role in the induction of the adaptive immune response, and serve to direct T-cell/B-cell localization to distinct LN regions through the constitutive release of small (low molecular weight) chemotactic proteins—chemokines. A similar segregation of B cells/T cells to distinct regions occurs within the white pulp of the spleen and the MALT (likewise, under control of chemokines elaborated by SLO-resident cells).
Induction of Adaptive Immune Responses in the Secondary Lymphoid Organs
The body’s complement of B and T cells consists of more than 109 cells, each of which may display a unique antigen receptor directed to a specific epitope. Therefore, the frequency of B or T cells directed to a specific pathogen is extremely low (e.g., a frequency of 1 of 104 to 106 naïve B or T cells). The problem faced by the adaptive immune systems is that these rare, antigen-specific cells are distributed randomly throughout the body, and must be rapidly mobilized to respond to invading organism. These rare antigen-specific cells must first contact antigen, then activate and generate effector cells capable of eliminating the pathogen. The solution to this problem is achieved through the process of lymphocyte recirculation through the SLO.57,273
Naïve (primary) T cells and most naïve B cells do not permanently reside in the SLOs. Rather, the T and B cells are constantly migrating from the blood through the SLOs and back to the bloodstream. Lymphocyte recirculation is controlled by the expression of distinct homing receptors (adhesion molecules) whose ligands are expressed on cells within the SLO. Naïve CD4+ and CD8+ T cells within the bloodstream express on their surface the adhesion/homing receptor, CD62L (L-selectin). T cells entering the blood vessels encounter the CD62L ligands—the mucin-like moieties CD34 and GlyCam-1 that are constitutively displayed in the endothelium of specialized high endothelial venules (HEVs) within the LN cortex. The interaction of CD62L (on the T cell) with its endothelial cell ligands arrests the T cell’s migration through the blood vessel, which results in its binding to the HEV endothelial cell. Other adhesion receptors displayed on endothelial cells that recognize carbohydrate ligands displayed on the surface of lymphocytes (as well as the αLβ2 integrin, lymphocyte function-associated antigen 1 [LFA-1] displayed on lymphocytes) also contribute to the arrest of T-cell migration. The naïve circulating T cells also constitutively express a chemokine receptor (CCR7) that recognizes specific chemokines (CCL19, CCL21) displayed on the surface of the HEV endothelial cells. These chemokines are produced by the HEV cells and by cell types within the LN (e.g., DC and stromal cells). The chemokines produced by the latter cell types diffuse into the HEV and are bound to/displayed on, the surface of the HEV endothelial cell. The interaction between the CCR7 receptor on the arrested T cells and its chemokine ligand triggers the migration of the naïve T cells through the HEV and into the LN cortex (parafollicular T-cell region). If the T cells do not encounter antigen, they traffic through the LN cortex to the LN medulla, and then exit the LN though the efferent lymphatic vessels, and ultimately return to the bloodstream where the process of recirculation though the SLO begins again. The egress of naïve T cells from the LN is as noted above, controlled by a chemotactic receptor S1P1R for the lipid sphingosine-1-P (S1P). S1P is present at higher concentrations in lymph and blood than in the LN. S1P1R engagement attracts naïve T cells away from the LN and back into the circulation. Antigen recognition by T cells transiently downregulates S1P1R retaining the antigen detecting T cells in the LN.
Naïve B cells traffic through the LN by similar mechanisms and transiently localize to the lymphoid follicles along a chemokine gradient mediated by a specific chemokine receptor, CXCR5, displayed on the surface of circulating naïve B cells. Lymphocyte trafficking through the spleen and MALT are likewise regulated by the interplay of adhesion receptor/ligand interactions and chemokines constitutively expressed in these SLOs. In the case of MALT, a set of unique receptor/ligand interactions (restricted to these sites) guides the trafficking of adaptive immune cells to these specialized mucosal sites.
Afferent lymphatic vessels are localized in all body surfaces/tissues, and they drain into the LN. Virus infection of a body surface/tissue triggers the innate immune system—the initial defense against infection. Innate immune cells located at the body surfaces/tissues (specifically, DC and monocyte/macrophages) encounter pathogen, take up virus/viral antigen, and then activate/migrate through the afferent lymphatics to the draining LN. In the draining LN, these migrating cells (along with DC and, to a lesser extent, monocyte/macrophages present in the LN cortex) serve as antigen-bearing presenting cells (APCs) to the naïve B and T lymphocytes trafficking through the LN. As a result of encounter with antigen displayed on the APCs, the lymphocytes with antigen receptors specific for the antigen or pathogen will no longer traffic through the LN, but rather will be retained in the LN (due to S1P1R downregulation) and initiate the process of activation and differentiation, leading to the generation of specific effector B and T cells directed to the pathogen. Therefore, the SLO concentrate antigen in a specific site where the rare antigen-specific, naïve B and T cells circulating through the body can encounter and respond to the pathogen. Afferent lymphatics can also carry large particulates (e.g., viruses as well as “soluble” viral proteins) directly to the LN where the particulates can be captured by macrophages lining subcapsular lymph node sinuses and subsequently delivered to DC or presented to naïve T cells, as well as directed along with soluble antigens through fibroreticular conduits to B cells and a lymphoid follicles.95,125
If infection by a virus is localized to the body surface (e.g., papillomavirus infection of the skin or influenza virus infection of the respiratory tract), then the adaptive immune response to that pathogen will be induced primarily in the LN draining these sites. Virus infections that result in systemic spread (i.e., viremia) will also induce adaptive immune responses in the spleen. Viruses that infect sites where MALT is prominent (e.g., rotavirus infection in the Peyer’s patches of the small intestine) will trigger the local induction of the adaptive immune responses at the site of MALT localization.196
Antigen Presenting Cells and the Induction of Adaptive Immune Responses
As with most microorganisms, viruses enter the body surfaces where the initial encounter with the immune system occurs. These body surfaces (e.g., skin and mucosa of the GI, GU, and respiratory tracts) contain epithelial lining cells, which can serve as the initial cells supporting virus replication. The epithelial surfaces also contain DC, specialized cells of bone marrow origin, which serve as both infection sentinels and APCs, which migrate to the SLOs and deliver antigen to the adaptive immune cells that are trafficking from the blood to the SLOs. A large body of evidence now suggests that DCs are critical APCs for the induction of the primary adaptive immune (T cell) response.184
For our purposes, two classes or types of DCs, conventional DCs (cDCs) and plasmacytoid DCs (pDCs) can be distinguished. cDCs are localized to the body surfaces and SLOs where they exist as immature (inactive) DCs prior to infection and carry out the aforementioned sentinel and APC function. As discussed below, a subset of cDCs identified by expression of CD8α in the SLO and αE integrin CD103 in mucosal tissue has the capacity to efficiently take up and present cellular material—for example, apoptotic-infected cells, particulates like viruses, and soluble proteins—to naïve CD8+ T cells without direct infection of the DC. pDCs, so-called because of their plasma cell–like morphology are derived from blood monocyte precursors and recruited to sites of inflammation/infection where, following activation, they are major producers of type 1α-interferons.80 In most circumstances these important innate immune effectors cells have limited capacity to act as APC for naïve T cells.
cDCs localize to the epithelium within the epithelial cell layer (e.g., Langerhans’ cells in the skin) at the junction of the epithelial basement membrane in GI and respiratory tracts, as well as in the mucosa and submucosa underlying the epithelial surface. The cDCs at body surfaces are derived from circulating monocytic progenitors originating in the bone marrow, which are present in the blood and migrate to the body surfaces and differentiate into peripheral (tissue) DCs. These tissue DCs exist as immature (inactive) DCs at these sites, turn over at a low variable rate, and are replenished by the circulating blood monocyte progenitors that enter the tissues.
Immature DCs are sessile cells that have a particular set of phenotypic markers or characteristics. They express low to intermediate levels of cell-surface MHC class I and class II molecules; the latter (class II) molecules are found predominately not on the DC surface, but within specialized endocytic vesicles. Immature DCs also express low levels of cell-surface molecules, which serve as co-stimulatory ligands for naïve lymphocytes (e.g., CD80, CD86, CD40, CD83).272 These molecules play a critical accessory role in the triggering of naïve antigen-specific (e.g., virus-specific) T cells, ultimately leading to the generation of effector (i.e., antiviral) T and B cells, and the formation of memory T and B cells directed to the pathogen. In addition, immature DCs have the ability to take up particulate and fluid-phase antigen into pinocytic/phagocytic vesicles with high efficiency. The enhanced capacity of these DCs to take up material (including the remains of infected apoptotic cells) likely accounts for their unique capacity to serve as APCs.185
The initiation of an inflammatory response at the body surface (as produced by virus infection) triggers the DC activation (maturation) process. Activation of the tissue-resident DC results in the de novo expression/upregulation of chemokine receptors (notably, CCR7), which renders the activated DCs capable of migrating from the peripheral tissue, primarily through lymphatic vessels along a gradient of constitutively expressed CCR7 ligands, the CCL19/CCL21 chemokines produced by lymphatic endothelial cells. Activation of the tissue DCs is also accompanied by upregulated expression of MHC class I/II molecules, and co-stimulatory ligands resulting in the maturation of the DCs, which, on activation/maturation, characteristically express high cell-surface levels of MHC class I/II molecules and the co-stimulatory ligands, CD80/86, CD40, and CD83. Mature DCs migrate to the SLOs where they act as the principal APCs for the induction of the adaptive immune response. Therefore, DCs (and to a lesser extent macrophages) serve the dual role of delivery (concentration) of antigen to the SLOs and acting as APCs to retain/activate the rare antigen-specific adaptive immune cell entering the SLOs from the circulation.
Immature DCs can be triggered to activate by virus infection through at least three mechanisms:
Direct infection of immature DCs. The stimulus for DC activation/maturation is provided primarily by the interaction of viral nucleic acids with (and signaling through) microbial pattern-recognizing Toll-like receptors (TLRs) displayed by DC,129 and/or activation of intracellular antiviral signaling mechanisms mediated by the RNA-activated protein kinase (PKR kinase),240 the retinoic-acid–inducible protein I (RIG-I) helicase systems,130 and for some viruses the caspase/IL-1β–associated inflammasome.289
Uptake of virus and soluble antigen (or more likely the remnant of infected epithelial cells that have undergone apoptosis in response to infection) with activation of immature DC through binding to one or more scavenger or C-type lectin receptor(s), for example, dendritic and epithelial cells, 205 kDa (DEC-205); dendritic cell-specific intercellular adhesion molecule-3 grabbing non-integrin (DC-SIGN) displayed on DC with specificity for carbohydrates on viral glycoproteins/glycolipids274 or through the process of phagocytic uptake.
Dendritic cell activation/maturation induced by inflammatory mediators (i.e., cytokines, such as TNF-α) released by infected epithelial cells and tissue macrophages in response to TLR and/or intracellular antiviral signaling.
The first two types of virus encounters with immature DC will result in the initiation of the adaptive immune response by delivery of virus/viral antigen to the SLO presented by the activated/mature DC. The significance of inflammatory mediator–induced DC maturation in the induction of the antiviral immune response is still not clear, because the activated, mature DC would not directly deliver viral antigen and would not serve as an APC.
Direct infection of immature DCs and the subsequent migration of the activated/mature DCs to the SLOs would appear to be the most likely mechanism to ensure the induction of an effective antiviral adaptive immune response. Whereas this mechanism of viral antigen delivery to T and B lymphocytes in the SLO occurs,36 it raises the important question: How do viruses with a very narrow cell tropism, which are unable to either infect or express viral genes in hematopoietic cells like DC (e.g., papillomaviruses) initiate an immune response?
The capacity of certain immature cDCs to take up and internalize cellular debris such as apoptotic remnants of infected epithelial cells provides the most likely mechanism for triggering adaptive immune T-cell responses. This process of the uptake of infected cell material with presentation to T cells by cDCs is called cross-presentation259 and is carried out by CD8α+ cDCs in SLO and by CD103+ cDCs at mucosal surfaces. Indeed, the speculation based on evidence from experimental viral infection is that endocytic uptake of virions or material from virus-infected cells may be the primary/sole mechanism of viral antigen presentation—at least to virus-specific naïve CD8+ T cells.230 The relative contribution of direct presentation (virus infection of DC), and cross-presentation of viral antigen remains controversial296 and will differ for different viruses.
Naïve (primary) T lymphocytes have stringent requirements for activation that include both TCR antigen receptor engagement by the appropriate processed antigenic peptide/MHC complexes and engagement of co-stimulatory receptors (e.g., CD28) displayed on naïve T cells by their corresponding ligands (e.g., CD80/86) along with peptide/MHC complexes by APC. Mature, antigen-bearing DCs are uniquely suited to carry out this process. The inflammatory response triggered by virus infection triggers DC maturation/migration. Virus infection also results in the infection of and/or antigen uptake by tissue macrophages. However, the migration of infected DC and macrophages along with infectious virions (either cell-associated or free) from the body surface to the SLO, not only initiates an immune response in the SLO, but also can in many instances spread infection to other sites in the body. One classic example of this is the dissemination of poxvirus from a peripheral infection site (body surface) to the SLOs, where free (infectious) virions and infected cells carried from the inoculation site to the SLOs can result in additional virus replication there, followed by virus entry into the bloodstream.78 Migrant macrophages are probably not as efficient as mature DCs in triggering primary immune response, but they may also serve as a potential APC in the induction of recall memory (secondary) immune responses where the requirements for memory T- and B-lymphocyte activation are less stringent.71 In this regard, it should also be noted that the mature migrant DCs that have entered the SLOs in response to virus infection may not be the primary APCs for T-cell activation. Several lines of evidence suggest the LN-resident DCs (particularly, CD8α expressing cDCs) can capture viral antigen delivered by migrant DCs (or macrophages), and present that antigen to naïve (primary) T lymphocytes in the SLOs—not by direct infection, but by the transfer of viral antigen from migrant antigen-bearing DCs to SLO-resident DCs through cross-presentation.296 Intact virions and/or soluble (free, intact) viral antigen, however, undoubtedly contribute to the induction of effective antiviral antibody response by B lymphocytes (as discussed in a later section of this chapter).
Studies examining the induction of adaptive immune responses using confocal and intravital multiphoton microscopy in intact viable LN have provided new insight into the early interaction events between DC-APCs and antigen-specific B and T lymphocytes.27,255 Some studies indicate that, on entering the draining LN, activated (mature) DCs migrate under a chemokine gradient stimulus to the paracortical T-cell regions in the vicinity of the HEV.35 Therefore, antigen-bearing DCs would be in the precise location to interact with naïve virus-specific B and T lymphocytes that have entered the draining LN from the circulation and, thereby, initiate the adaptive immune response. However, soluble antigens (viral proteins) and particulates such as virions that entered the LN via lymphatics may be presented to T cells, and particularly B cells, by mechanisms not involving DC migration from the site of infection.94
Activation of T and B Lymphocytes in the Secondary Lymphoid Organs
The precise localization of T cells, B cells, DCs, and macrophages within the SLOs is controlled by chemokines, which are largely produced by stromal cells of the SLOs, and by bone marrow–derived cells (e.g., SLO-resident DCs). In draining LN, naïve (primary) T cells that are present in the circulation will enter the LN through the specialized HEV, where they encounter mature DCs (and macrophages) displaying processed antigen bound to MHC molecules, which are localized to sites in the parafollicular cortex of the LN facilitate interaction with T cells. Most naïve CD8+ T cell and CD4+ T cells entering the LN will not recognize the antigen (i.e., they have TCRs that fail to recognize the processed antigen with sufficient avidity to activate the cells). These T cells will ignore the DCs and migrate from the LN cortex to the medulla, then enter the efferent lymphatics, and ultimately return to the bloodstream to continue their perpetual search for relevant antigen.273 A similar process of lymphocyte egress through the HEV occurs in the MALT; for example, Peyer’s patches of the small intestine where antigen uptake by mucosal DCs and direct viral antigen transfer to the gut MALT through specialized intestinal epithelial cells, called M cells, result in antigen delivery to these mucosal lymphoid aggregates.150 Viruses that enter the bloodstream (i.e., produce viremia) as well as any antigen-bearing tissue DCs that, on maturation, bypass the LN and gain access to the circulation will localize to the splenic white pulp. Here, circulating T and B lymphocytes enter the splenic white pulp by transiting through splenic arterioles where viral antigen-bearing DCs can encounter lymphocytes. The lymphocytes that do not recognize antigen will exit the splenic white pulp by entering trabecular veins, and re-enter the circulation.
The rare naïve CD8+ T cells and CD4+ T cells that recognize processed antigenic peptides displayed on the APC surface in the SLO do so through TCR engagement. The activation of naïve T cells leading to the induction of the adaptive immune response is a multistep process (at both the molecular and cellular level).112 For induction of antigen-specific (virus-specific) CD8+ T-cell responses, the activation process requires both interaction of the antigen-specific TCR α : β chain complex with processed antigen fragments bound to MHC class I molecules on the APC surface, with sufficient avidity and interaction of the CD8 molecule on the T cell with a region of the MHC class I molecule distant from the site of peptide binding. To fully activate the naïve CD8+ (or CD4+) T cells, additional signaling events must occur, including binding of the co-stimulatory receptor, the CD28 molecule, displayed on naïve T lymphocytes with its ligands, CD80/86, which are displayed at high levels on mature (activated), antigen-bearing DCs. The sequence of events resulting in the activation of antigen-specific naïve CD4+ T cells parallels the steps in CD8+ T-cell activation, except that the antigen-recognizing TCR α : β chain complex must bind (with sufficient avidity) the antigenic peptides bound to MHC class II molecules displayed by APC; and the CD4 co-receptor on CD4+ T cells, in turn, must interact with a distinct site (outside of the region of peptide binding) on the MHC class II molecule. One important early consequence of CD4+ T-cell activation by antigen is the upregulation of the expression of the CD154 molecule on activated CD4+ T cells. CD154 (also called CD40 ligand or CD40L) is a member of the TNF gene family, and, as its name implies, is a ligand for CD40 (a TNF receptor gene family member). CD40 is expressed on both activated/mature DCs and antigen-activated B cells. Engagement of CD40 on DCs and B cells by CD154 (CD40L) on activated CD4+ T cells has important consequences for the function of these cells. Engagement of CD40 on DCs results in increased synthesis of certain cytokines (notably IL-12) by the antigen-bearing DCs in the SLO, which promotes CD4+ and CD8+
T-cell differentiation into effectors.142 For B cells, the interaction between CD40-activated B cells and CD40L expressed on activated CD4+ T cells is critical for B-cell differentiation into antibody-secreting effector B cells (plasma cells).182 Strategies to modulate the CD40/CD40L interaction in vivo may have important consequences in vaccine development.
T-cell differentiation into effectors.142 For B cells, the interaction between CD40-activated B cells and CD40L expressed on activated CD4+ T cells is critical for B-cell differentiation into antibody-secreting effector B cells (plasma cells).182 Strategies to modulate the CD40/CD40L interaction in vivo may have important consequences in vaccine development.
The engagement of the TCR/co-receptor complex, and the CD28 co-stimulatory receptor on T cells in the SLO are the first two critical steps in the induction of a T-cell response to a foreign antigen. This initial antigen encounter by the TCR on specific T cells is called signal 1 in the lexicon of lymphocyte activation/differentiation. The engagement of the CD28 co-stimulatory receptor is designated as signal 2. As a consequence of their interaction with antigen and co-stimulatory ligand displaying DC APC, the naïve T cells halt their transit through the SLO (i.e., downregulation of S1P1R, and are retained there). The APC/T cell encounter leading to signal 1 and 2 transmission in the T cells appears to be of short duration (from 6 to 24 hours), after which the initial programming for T-cell activation/differentiation is independent of additional interaction with antigen.174,293 Delivery of signals 1 and 2, although necessary, is not sufficient in itself, however, to ensure full T-cell activation/differentiation and the generation of effector and memory T cells to the antigen. A third signal, signal 3, is required to complete the T-cell activation/differentiation program in the SLO. Signal 3 is delivered by soluble mediators (i.e., cytokines and chemokines) produced primarily by cells of the innate immune system (e.g., pDCs mature/activated in cDCs, NK cells, NK T cells), which enter the SLO in response to infection/antigen deposition in the SLO. IL-12 produced by mature (activated) DC in the SLO is a particularly critical type of signal 2 in T-cell responses to virus, because engagement of the IL-12 receptor on activated CD8+ T cell and CD4+ T cells promotes their differentiation into effector T cells with potent antiviral activity (e.g., the generation of cytolytic T cells).199
Other cytokines produced during T-cell activation (e.g., type 1 and type II/III IFNs [IFN-α, β, IFN-γ, IFN-l, respectively], IL-1, IL-4, IL-6, IL-10 and so on) by innate immune cells responding in the SLOs to virus infection will dramatically modulate the magnitude and type of both the effector and memory T-cell responses. These and other soluble mediators modify the course of T-cell activation/differentiation during the T cell’s residence in the SLO.142
Similarly, in addition to CD28, other co-stimulatory receptors belonging (like CD28) to the Ig receptor gene family (e.g., Cytolytic T-Lymphocyte antigen-4 [CTL A-4]; inducible T-cell co-stimulator [ICOS]; programmed death-1 [PD-1]) and the TNF receptor gene family (e.g., CD27, OX40, 4-1BB, CD40L) whose expression on activated T cells is temporarily regulated following T-cell activation in the SLO, can either positively (ICOS, CD27) or negatively (PD-1, CTLA-4) regulate the tempo and magnitude of T-cell activation and differentiation into effector cells in the SLO or in peripheral sites of virus infection following interaction with their perspective ligands on APC.248,252
The temporal sequence of signal 1, 2, and 3 delivery to T lymphocytes in the SLO is reflected in the morphologic change of the naïve (primary) T cells from small, resting cells to activated lymphoblasts. This is followed by proliferative expansion of the antigen-specific T-cell clones in the SLO, and their differentiation into both effector T cells and an expanded population of memory CD8+ T cell and CD4+ T cells. T-cell proliferation is extremely rapid (cell cycle time of ≤6 hours and in one report ≤2 hours,317 with estimates of individual naïve T-cell precursors undergoing 8 to 20 divisions during this programmed proliferation.137 During their proliferative expansion and differentiation in the SLO, the responding T cells lose cell-surface adhesion molecules (e.g., CD62L) and receptors (e.g., CCR7) necessary for naïve T-cell circulation, and they upregulate the expression or express de novo adhesion molecules (e.g., the β2 integrin, LFA-1, and β1 integrin, very late antigen 4 [VLA-4], respectively), and chemokine receptors (e.g., CCR5, CXCR3) essential for effector T cells to localize to sites of inflammation.165 At the end of the proliferation/differentiation sequence, most activated antigen-specific effector T cells will upregulate S1P1R and exit the SLO (through the efferent lymphatics in the case of T cells responding in the draining LN), enter the circulation, and traffic to the site of infection/inflammation. The homing of activated T cells is controlled by chemokines released by innate immune cells (as well as by epithelial and endothelial cells and fibroblasts) at the site of inflammation/infection (e.g., CXCL9 [Mig/CXCL9 (Monocline Induced by Gamma interferon)], CXCL10 [IP-10 (Interferon gamma induced Protein-10)], and CXCL11 [I-TAC (Interferon-inducible T cell Alpha Chemoattractant)] for CXCR3; and CCL3 [macrophage inflammatory protein-1α; MIP-1α] and CCL4 [MIP-1β] for CCR5). This homing is also controlled by the inflammation-induced upregulation of integrin ligands (e.g., intercellular adhesion molecule-1 [ICAM-1] for LFA-1, and vascular adhesion molecule [VCAM] for VLA-4) and adhesion receptors (P-selectin, E-selectin) on endothelial/epithelial cells at these sites.
The generation of effector T cells in the SLOs responding to infection can occur in as few as 5 to 7 days in both experimental models and human viral infection.68 Given the rapid replication of most viruses, it is obviously essential to get specific T cells activated and mobilized as soon as possible through the concentration of antigen and the accumulation of the rare antigen-specific T cells in the SLO, followed by rapid proliferative expansion/differentiation of these cells into effectors in the SLO. The rapid induction of an adaptive immune response to virus infection is not a foregone conclusion, however. The process of T- and B-cell activation can be delayed for weeks to months in the case of infection with viruses such as HIV and HCV, which induce chronic infection.111,187 In these instances, the virus has the capacity to suppress/delay the induction of the adaptive immune response.
The process of naïve B-cell activation in the SLOs is similar to that of naïve T-cell activation. In the case of B-cell responses to viruses and other T cell–dependent antigens, however, antigen-specific CD4+ T cells play a critical role in driving B-cell activation/differentiation. As with naïve T cells, naïve B cells enter the LN though the HEV; unlike T cells, however, the B cells migrate from the paracortical (T-cell rich) area of the LN and into the B cell–enriched LN cortical follicle. This migration is controlled by the chemokine CXCL13, produced by a stromal cell type resident in the follicle (called a follicular DC because of its dendritic morphology, but not of hematopoietic origin). CXCL13 is the ligand for CXCR5 chemokine receptor that is constitutively expressed by naïve B cells. If the B cell does not encounter antigen in the follicle, it will ultimately migrate out of the cortical follicle to the medulla, then enter the efferent lymphatics, and rejoin the circulating pool of B cells in the bloodstream.
The B cells that display a BCR antigen receptor (i.e., the cell-surface membrane-bound form of the immunoglobulin molecule that recognizes antigen) for an antigen present in the LN would encounter this antigen immediately after migrating out of the HEV in the parafollicular (T-cell rich area) of the LN. In the case of viral antigen, the relative contribution of free virus, soluble viral proteins, and viral proteins transported by migrant DC/macrophages from the infection site to the reservoir of antigen present in the SLO and available to specific B cells is not certain. However, it has been recently appreciated that structures (i.e., conduits) within the SLO (in the case of LN) direct particulates like viruses and soluble proteins directly to regions of naïve B-cell accumulation in the SLOs.125 Binding of antigen to the BCR (i.e., cross-linking cell surface immunoglobulin) provides the initial stimulus (signal 1) for specific B-cell activation. Signal 1 delivery halts the constitutive migration of the B cell from the paracortical T-cell zone to the LN follicle, and activates the chemokine-dependent migration of the B cells to the junction between the T-cell zone and the follicle. The strength of signal 1 can be greatly enhanced if the BCR co-receptor complex201 is ligated simultaneously with the BCR by interaction with complement components bound to antigen or antigen–antibody complexes. In addition, cross-linking of the immunoglobulin receptor induces internalization of the antigen–immunoglobulin complex into an endosomal compartment where the antigen is fragmented and associates with MHC class II molecules that are constitutively expressed by B cells. The antigen/MHC class II complexes are then cycled to the B-cell surface, where they are available for recognition by antigen-specific CD4+ T cells. Another consequence of signal 1 delivery (by antigen to B cells) is CD40 upregulation. The CD4+ T cells, which interact with antigen-stimulated B cells, are themselves activated by prior encounter with antigen/MHC class II complexes on APC (i.e., mature, antigen-bearing DC), and have upregulated CD154 (CD40L) expression. The activated antigen-specific CD4+ T cells recognize the antigen/MHC complexes on the antigen-stimulated B cells, and deliver signal 2 to the B cells in two forms: interaction of CD40L (on T cells) with CD40 (on B cells), and the release of T-cell–derived cytokines (notably, IL-4, IL-21, and/or IFN-γ) whose synthesis is triggered in the CD4+ T-cell TCR recognition of antigenic peptides displayed by the B cells. Antigen-activated CD4+ T cells in LN may support B cell responses by differentiating into so-called T follicular helper (TFH) cells. These activated CD4+ T cells appear to represent a distinct CXCR 5+/PD-1+ CD4+ T-cell subset, the generation of which is in part regulated by IL-6 produced within the responding LN. The cells secrete IL-21 in response to TCR engagement and also express CD154 (CD40L). The interaction of the TFH with the naïve parafollicular B cells through CD40 engagement (on B cells) by CD40L on the T cells, and IL-21 signaling through its receptor on naïve T cells helps support the formation of B-cell germinal centers.
The fully activated (signals 1+2+) B-cell lymphoblasts undergo programmed proliferation after migration to the follicles to produce germinal center, which are follicular structures consisting primarily of responding/proliferating B-cell lymphoblasts. The proliferation/differentiation of the germinal center B-cell lymphoblasts results in the formation of antibody-secreting plasma cells, which migrate from the LN to the sites of infection/inflammation, and more importantly to the bone marrow where the plasma cells serve as a depot of long-lived antibody-secreting cells.167 B cells that encounter antigen without CD4+ T-cell help (i.e., receive signal 1, but not signal 2) are capable of producing specific secreted antibodies; however, these antibodies will be of the immunoglobulin M (IgM) isotype and of low affinity. The isotype switch of antibody production from IgM to the IgG, IgA, and so on isotypes, as well as, the process of antibody affinity maturation requires T-cell help (signal 2).167
Viral Antigen Recognition By B Cells
Viral antigens are found on virions, on virus-infected cells, as soluble molecules produced by virally infected cells, and as breakdown products from virions and infected cells. These antigens can be recognized by antibodies in soluble form in plasma or tissues and by antibodies on the surface of B cells (i.e., BCR). If the individual has had previous exposure to viral antigen(s) through infection or vaccination, then specific soluble high-affinity antibodies will likely be present in the plasma. A minority of these antibodies will be reactive with viral antigens presented on virions and virally infected cells and able to function antivirally. Many will be directed to a range of nonsurface viral antigens; that is, they will be functionally inert, but may be useful in the diagnosis of viral infection. In the case of prior infection or vaccination, memory B cells that express specific antibody will be activated on contact with viral antigen, leading to the production of soluble, specific high-affinity antibody and the increase of specific serum antibody levels. In the absence of a previous encounter with viral antigen, antibodies on the surface of naïve B cells will bind antigen with relatively low affinity, setting in motion antibody affinity maturation and class-switching processes, which are discussed below.
Nature of B-Cell Epitopes
Most of what is known about how antibodies recognize antigens has been determined from studies on the interaction of soluble antibodies or antibody fragments with antigen.61,208,308 Given that B-cell surface antibody is essentially identical to soluble monomeric antibody, with an extra segment ensuring membrane localization, the conclusions should apply broadly to cell surface antibody. Antibodies recognize molecular shapes, termed epitopes, on the surface of antigens. The antibody-combining site makes multiple contacts with the surface of the antigen to form complementary surfaces. The more complementary these surfaces are to one another—in terms of geometry and chemical character—the more favorable interactions will be formed between the antibody and antigen and the higher will be the affinity of the antibody for antigen. The affinity of the antibody for the antigen is one of the most important factors in determining the efficacy of the antibody in vivo.
The antibody-combining site is made up of residues contributed primarily from six highly variable segments or loops referred to as hypervariable loops or complementary determining regions (CDRs). The site can vary greatly in shape and character depending on the length and characteristics of the CDRs. Generally, most or all of the CDRs contribute to antigen binding, but their relative contributions vary. The heavy chain CDRs (particularly the third heavy chain CDR [CDR H3]) tend to contribute disproportionately to antigen binding. The CDR H3 in human antibodies can be very long, with
distinctive shapes such as fingers and hammerheads that contact viral epitopes. The combining site of antibodies against smaller molecules such as carbohydrates and organic groups (haptens) are often more obviously grooves or pockets, rather than the extended surfaces typically found in antiprotein antibodies.
distinctive shapes such as fingers and hammerheads that contact viral epitopes. The combining site of antibodies against smaller molecules such as carbohydrates and organic groups (haptens) are often more obviously grooves or pockets, rather than the extended surfaces typically found in antiprotein antibodies.
On the antigen, epitopes come in as many different shapes and sizes as do antibody-combining sites. The area of antigen that contacts antibody, referred to as a footprint, is typically between about 400 and 1,000 Å2. All antibodies recognize a topographic surface of a protein antigen. Most usually, key residues in the epitope will arise from widely different positions in the linear amino acid sequence of the protein because of the manner in which proteins are folded. The linear sequence typically traverses from one side of the protein to the other a number of times. Such epitopes are described as discontinuous. Occasionally, key residues arise from a linear amino acid sequence. In such cases, the antibody may bind with relatively high affinity to a peptide incorporating the appropriate linear sequence from the antigen. Furthermore, the peptide may inhibit the antigen binding to the antibody. The epitope in such cases is described as continuous. An example of a continuous epitope would be a loop on the surface of the protein for which an antibody recognized successive residues in the loop. It should be noted, however, that an antibody that recognizes a continuous epitope does not bind a random or disordered structure. Rather, it recognizes a defined structure that is found in the complete protein but can be readily adopted by the shorter peptide.
Antigenicity and Immunogenicity
A clear distinction is seen between the ability of an epitope to be recognized by antibody (antigenicity) and its ability to stimulate an antibody response when presented to a host antibody system (immunogenicity). A number of factors appear to be involved in how well a given epitope elicits an antibody response. Perhaps one of the most important factors is the accessibility of the epitope on the protein surface. Loops that protrude from the surface of the folded protein tend to elicit particularly good antibody responses. Figure 9.2 shows epitopes on the hemagglutinin (HA) protein from the surface of the influenza virus, with those at the “top” of the molecule distant from the viral membrane and most accessible to antibody. Natural infection and classical vaccination strategies typically induce antibodies to the top of the HA and mostly to variable regions that are therefore strain specific. Changes within these immunodominant variable regions allow neutralization escape and dictate repeated annual influenza vaccinations to provide robust protection to large populations. More conserved regions in the stem region of the HA appear to induce weaker antibody responses, but if these could be presented in a more immunodominant format they might induce broadly neutralizing antibodies and thereby provide a universal influenza vaccine that would replace the current annual vaccines. HIV is another virus in which highly exposed variable regions on the viral surface glycoprotein are immunogenic.313 Following primary infection, it takes some time (weeks) for neutralizing antibodies to reach a level where they interfere with virus replication.229,303 These antibodies are typically elicited to exposed variable loops on the virus. While these antibodies are being elicited, the virus has diversified (i.e., it has become a swarm of related viruses through the errors associated with reverse transcription of this RNA retrovirus). Among this swarm is a virus that has sequence changes in the epitopes targeted by the neutralizing antibody response that allows it to escape from the response. This new virus becomes predominant. Eventually, a response is mounted to this virus and a second new virus emerges and so on. The antibody response chases the virus over many years but never appears to gain control.
 Figure 9.2. Neutralizing epitopes on influenza hemagglutinin (HA). The epitopes were determined from crystal structures of neutralizing Fab fragments in complex with HA. The viral membrane is at the bottom of the figure. Most of the epitopes toward the top of the HA molecule are relatively variable in different influenza strains. The epitopes defined by the antibodies CR6261 and CR8020 in the stem of the HA molecule are conserved and might be targeted by a universal influenza vaccine. (Courtesy of Damian Ekiert and Ian Wilson.) |
An important consideration in relation to viral surface antigens and immunogenicity is the spacing of the antigens on the virion surface.12 It is well established that haptenated polymers, in which the haptens are spaced at 5 to 10 nm, can induce antibody responses in the absence of T-cell help. For vesicular stomatitis virus (VSV), in which the G protein is found in a highly organized quasicrystalline state on the virion surface, a strong immunodominant T-cell–independent neutralizing antibody response is induced by infection. A poorly organized recombinant form of VSV-G expressed in micelles induces a weaker B-cell response that is partially T-cell dependent. Soluble VSV-G alone, in the absence of adjuvant, fails to induce a B-cell response.
Generation of the Primary B-Cell Repertoire
The humoral immune system first senses the presence of novel viral antigens through the interaction of B-cell surface
antibody (monomeric IgM and/or IgD) with antigen. Individual B cells possess multiple BCRs of identical specificity, which when occupied by antigen, are activated to divide and differentiate. The final results include the production of large amounts of specific, soluble high-affinity antibody and the generation of memory B cells that can be readily triggered to produce high-affinity antibody on repeated encounter with viral antigen.226 The first encounter of viral antigen is with the primary B-cell repertoire of the individual. This repertoire is large, allowing recognition with low affinity of essentially any molecular shape and, therefore, any viral variant. It is produced in B cells by recombination of a limited set of germline segments.121,134,169,175 Hence, an individual’s primary repertoire is unique to that individual and is in a constant state of flux. Briefly, the variable regions of antibody heavy and light chains are produced by V(D)J recombination in which numerous unique immunoglobulin genes can be made by joining different combinations of the V, D, and J segments at the heavy, and V and J segments at the light, chain loci. In humans, the potential heavy chain repertoire is approximately 50 VH × 27 DH × 6 JH = 8 × 103 different combinations. Similarly, approximately 165 (33 Vλ × 5 Jλ) and 200 (40 Vκ × 5 Jκ) different combinations exist, for a total of 365 light chain (λ and κ) combinations. If we consider that each heavy chain could potentially pair with each light chain, then the diversity of the immunoglobulin repertoire is of the order of 106 possible combinations. Further diversity (junctional diversity) is generated because the joining of the V, D, and J gene segments is imprecise.
antibody (monomeric IgM and/or IgD) with antigen. Individual B cells possess multiple BCRs of identical specificity, which when occupied by antigen, are activated to divide and differentiate. The final results include the production of large amounts of specific, soluble high-affinity antibody and the generation of memory B cells that can be readily triggered to produce high-affinity antibody on repeated encounter with viral antigen.226 The first encounter of viral antigen is with the primary B-cell repertoire of the individual. This repertoire is large, allowing recognition with low affinity of essentially any molecular shape and, therefore, any viral variant. It is produced in B cells by recombination of a limited set of germline segments.121,134,169,175 Hence, an individual’s primary repertoire is unique to that individual and is in a constant state of flux. Briefly, the variable regions of antibody heavy and light chains are produced by V(D)J recombination in which numerous unique immunoglobulin genes can be made by joining different combinations of the V, D, and J segments at the heavy, and V and J segments at the light, chain loci. In humans, the potential heavy chain repertoire is approximately 50 VH × 27 DH × 6 JH = 8 × 103 different combinations. Similarly, approximately 165 (33 Vλ × 5 Jλ) and 200 (40 Vκ × 5 Jκ) different combinations exist, for a total of 365 light chain (λ and κ) combinations. If we consider that each heavy chain could potentially pair with each light chain, then the diversity of the immunoglobulin repertoire is of the order of 106 possible combinations. Further diversity (junctional diversity) is generated because the joining of the V, D, and J gene segments is imprecise.
Although mice and humans use combinatorial and junctional diversity as a mechanism to generate a diverse repertoire, in many species, including birds, cattle, swine, sheep, horses, and rabbits, V(D)J recombination results in assembly and expression of a single functional gene.169,179 Repertoire diversification is then achieved by gene conversion, a process in which pseudo-V genes are used as templates to be copied into the assembled variable region exon. For example in the chicken, during B-cell development in the bursa of Fabricius, rapidly proliferating B cells undergo gene conversion to diversify the antibody repertoire. Stretches of sequences from germline variable region pseudogenes, located upstream of the functional V genes, are introduced into the VL and VH regions. This process takes place in the ileal Peyer’s patches of cattle, swine, and horses, and in the appendix of rabbits.
B-Cell Activation by Viral Antigens
B cells can recognize and respond to both soluble and membrane-bound viral antigens, although it is likely that, in vivo, the most relevant interaction is with antigen on the surface of APCs.14,15 The encounter of viral antigens with BCR has two consequences. First, a signal is transmitted into the B cell that is amplified via intracellular pathways and results in the activation of transcription factors such as nuclear factor-kappaB (NF-κB) and AP-1. These factors act to induce specific gene transcription promoting B-cell proliferation and differentiation. Second, the BCR delivers viral antigens to intracellular sites where they are proteolytically cleaved and some viral peptides are bound to MHC class II molecules. The peptide/MHC II complexes are returned to the B-cell surface where they can be recognized by viral-specific TCR on specialized CD4+ T cells (T helper cells), known as TFH or T follicular helper cells.52,200 These TFH cells are then stimulated to produce cytokines and provide surface costimulatory molecules that promote B-cell proliferation and act on later generations of these B cells to promote differentiation to antibody-producing and memory B cells. Some viral antigens, as described above, can activate B cells directly in the absence of T-cell help (so-called T-independent antigens).
Cell signaling from the BCR occurs when multiple antigen contacts lead to clustering of cell surface IgM molecules.219 In the BCRs, IgM monomer is associated with a heterodimer of membrane-bound Igα and Igβ molecules, which bear immunoreceptor tyrosine activation motifs (ITAM) on their cytoplasmic domains. Following BCR clustering, tyrosines in the ITAM become phosphorylated to trigger a series of events that eventually lead to activation of transcription factors. Signaling by the BCR is modulated by co-receptors, most notably Fc receptor IIB for IgG (FcRγ IIB) and CD19. FcRγ IIB is a potent inhibitor of B-cell signaling197 by reversing protein kinase activities and by inhibiting the formation of BCR microclusters. CD19 is an activating coreceptor64 that functions to recruit activating molecules to the BCR. In addition, CD19 occurs on the surface of B cells alone and is linked to CD21, a receptor for the complement fragment C3d. If, following complement activation, C3d has been linked to viral antigen or to antibody–virus complex, the co-receptor complex can be linked to the BCR and BCR activation enhanced. The involvement of complement can be crucial in the development of normal B-cell memory responses (see below). Mice with deficiency in C3, C4, or CD21/CD35 and infected with HSV show a reduction in specific IgG and germinal centers compared to normal mice.59
The interaction of B cells with viral-specific TFH cells in lymphoid tissue leads to B-cell activation and proliferation and differentiation. It is important to note that the B-cell surface antibody and the TCR must recognize epitopes from the same molecular complex for the help signal to be delivered and B-cell activation to occur. Therefore, a B cell that is expressing surface antibody to the surface protein of a virus could be activated by a T helper cell expressing a TCR to an internal viral protein if antigen challenge involved whole virions.241 A subunit vaccine consisting only of the envelope protein could not obtain help from such a T helper cell. The process is as follows. The B cell binds a virion through antibody recognizing surface protein. The whole virion is internalized and degraded, and peptides displayed on MHC class II molecules are carried to the B cell surface. Specific TFH cells that have previously been primed by DCs recognize the peptide-MHC class II complex on B cells through specific TCRs.44 Antigen recognition results in T-cell adhesion to the B cell and the provision of “help” to the B cell via a range of transmembrane costimulatory receptors and cytokines. TFHs are highly enriched for selective expression of an array of cell surface molecules, reflecting their specialized role for cell–cell interaction with B cells.52 The most critical cell–cell interaction is CD40–CD40L. CD40 on B cells engages CD40 ligand (denoted CD40L and also known as CD154) on TFH cells, driving B-cell proliferation. The combination of B- and T-cell interactions and locally released cytokines can further enhance B-cell proliferation or direct differentiation, depending on the signals provided by
the TFH cell and the cell surface molecules expressed by the B cell. Of interest, the process of shared CD4 T-cell help to B cells specific for a different viral protein may occur only for small viruses, as the B cell and antibody responses to infection with vaccinia virus (a large poxvirus used as the smallpox vaccine) are strictly dependent on matched CD4 T-cell help.254 This likely reflects a limit on the pathogen or particle size that a B cell can endocytose whole.
the TFH cell and the cell surface molecules expressed by the B cell. Of interest, the process of shared CD4 T-cell help to B cells specific for a different viral protein may occur only for small viruses, as the B cell and antibody responses to infection with vaccinia virus (a large poxvirus used as the smallpox vaccine) are strictly dependent on matched CD4 T-cell help.254 This likely reflects a limit on the pathogen or particle size that a B cell can endocytose whole.
There are two pathways for B cells that have contacted specific T cells. In the first, after several rounds of proliferation, B cells differentiate into plasma cells in extrafollicular foci168 and secrete relatively large amounts of soluble antibody to the virion surface protein of our example. This antibody is encoded by the unmutated germline repertoire and of the IgM isotype or class-switched to IgG or IgA. The plasma cells are short-lived with a lifespan of a few days. This early antibody is believed to be important as a first-line humoral response against viral infection.
In the second pathway involving germinal centers (GCs) in lymphoid tissues (see Section 2), the quality and versatility of the antibody to the virion surface protein can be improved by affinity maturation and isotype switching.40,73,154,166,172,182,188,195 In GCs, B cells divide very rapidly, every 6 to 8 hours. CD4 T cells are required for the formation and maintenance of GCs. GC TFH cells provide signals that regulate maintenance of GC B cells and GC B-cell differentiation into plasma cells and memory B cells.52 GC TFH cells critically stimulate somatic hypermutation and selection of high affinity clones. Somatic hypermutation177 involves the introduction of nontemplated point mutations into V regions of the rapidly proliferating B cells at a rate on the order of 1 × 10−3 mutations per base pair per generation, which is approximately 106 times higher than the mutation rate of cellular housekeeping genes. The enzyme activation–induced cytidine deaminase (AID) has been demonstrated to be essential for somatic hypermutation. AID is a cytidine deaminase capable of carrying out targeted deamination of C to U, and shows strong homology with the RNA-editing enzyme APOBEC-1. It seems that AID directly deaminates DNA to produce U:G mismatches that ultimately can result in sequence changes in the V regions. Selection from the array of B cells expressing slightly different mutated forms of cell surface antibody was thought to occur based directly on the ability of higher affinity clones to compete for antigen and receive stronger BCR signals and survive. More recently however,56,294a somewhat different view has emerged in which B cells gather antigen as they move over the network of follicular DCs in GCs. Cells with the highest affinity for antigen acquire the most antigen, thereby leading to greater presentation of peptide/MHC II complexes to T cells, which in turn provide the requisite cytokine signals for B-cell survival and selection.
Isotype switching, in which the constant region of IgM is replaced by that of another isotype, also occurs in germinal centers. This might lead for example to a switch from IgM to IgG or IgA antibodies against a virion surface protein. Switching occurs through deletional DNA recombination events and involves repetitive DNA sequences or switch regions. The enzyme AID, described above, appears to be critically involved in the process. Cytokines such as IFN-γ and IL-17, which are expressed by germinal center TFH cells in a context-dependent manner, control appropriate isotype switching to different pathogens.
The B cells surviving the germinal center differentiate into plasma cells or memory B cells.97,171,182,304 Plasma cells accumulate in the spleen or draining lymph nodes during the early stages of acute infection, but as the immune responses subside, the majority of plasma cells are found in the bone marrow.11,124,267,318 These cells no longer express surface antibody but are committed to the secretion of soluble antibody—up to 10,000 antibody molecules per second.113,118 Because the half-life of IgG is on the order of 1 to 3 weeks, but specific antibody can be present for many years after antigen exposure, it appears that plasma cells maintain serum antibody levels. A favored hypothesis is that plasma cells are very long-lived and continue secreting antibody long after antigen has disappeared (see below). The half-lives of antibody responses in humans to measles and mumps viruses for example are estimated at more than 200 years.6 Memory B cells can also last for many decades.54,319 Memory B cells maintain surface antibody but secrete little if any antibody, unless activated. They divide slowly if at all. B-cell memory is considered in greater detail below.
Viral Antigen Recognition By T Cells
The T-cell response to viruses is largely mediated by T cells that utilize the TCR α : β chain heterodimer to recognize a foreign antigen. T cells, which employ the γ : δ chain complex (the gamma/delta cells), as well as certain rare TCR α : β expressing T cells with restricted variable α (or β) gene segment usage in TCR generation (e.g., NK T cells) have a less clearly defined role in the adaptive immune response to viruses (and will be briefly discussed below). The conventional TCR α : β T cells, which have left the thymus and populated the SLOs and bloodstream, express, along with their TCRs, either the CD4 or the CD8 co-receptor molecules. One defining feature of the response of these cells is that α : β antigen receptors recognize nonnative peptide fragments of foreign antigen (including viral polypeptides). Therefore, in contrast to the immunoglobulin receptor (BCR), which is sensitive to antigen conformation, T-cell recognition of antigen is insensitive to three-dimensional antigen structure. Furthermore, T-cell recognition of antigens (e.g., viruses) is limited primarily to the polypeptide constituents of the virus or, more precisely, to viral gene products expressed in infected cells (although some rare exceptions exist to this general rule, which are noted later in this chapter).
A second defining feature of the α : β TCR is that the antigen receptor recognizes these antigenic peptides bound to MHC molecules displayed on the surface of the antigen-containing (e.g., virus-infected) cells. Therefore, CD4+ and CD8+ T cells are restricted in their recognition of foreign antigen to cells displaying the foreign peptide complexed to the MHC molecule to which that T cell was selected during development in the thymus. This is the phenomenon of MHC restriction324 that defines antigen recognition by T cells. CD8+ T cells will recognize cells displaying the foreign antigen peptide bound to one of several distinct MHC class I molecules expressed on the cell surface. CD4+ T cells, likewise, are restricted in foreign peptide recognition by the cell surface MHC class II molecule to which the peptide fragment is bound.
The implications of MHC-restricted recognition of processed peptide fragments by T cells for virus recognition are immediate and profound. Conventional TCR α : β T cells
responding to virus infection will recognize infected cells and not free virions or soluble viral proteins. Therefore, the activation of naïve T cells and the expression of the effector activity of activated T cells responding to virus infection will be primarily mediated through cell-to-cell contact (i.e., contact between naïve T cells and APCs or the infected cell targets of effector T cells, such as epithelial cells). In addition, T cells have the potential to recognize any viral gene product (structural or nonstructural) that can be fragmented into peptides, and bound to/displayed by MHC molecules on the surface of infected cells. Consequently, CD8+ T cells will only recognize virus-infected cells that display peptide fragments of viral protein bound to cell surface MHC class I molecules, whereas CD4+ T cells will recognize only those cells in which display viral peptide/MHC class II complexes. The differences in the cell types that display MHC class I and II molecules, and the distinct mechanisms by which foreign polypeptides are processed (fragmented) and bound to/presented by MHC class I and II molecules determine how CD8+ and CD4+ T cells respond to virus infection.
responding to virus infection will recognize infected cells and not free virions or soluble viral proteins. Therefore, the activation of naïve T cells and the expression of the effector activity of activated T cells responding to virus infection will be primarily mediated through cell-to-cell contact (i.e., contact between naïve T cells and APCs or the infected cell targets of effector T cells, such as epithelial cells). In addition, T cells have the potential to recognize any viral gene product (structural or nonstructural) that can be fragmented into peptides, and bound to/displayed by MHC molecules on the surface of infected cells. Consequently, CD8+ T cells will only recognize virus-infected cells that display peptide fragments of viral protein bound to cell surface MHC class I molecules, whereas CD4+ T cells will recognize only those cells in which display viral peptide/MHC class II complexes. The differences in the cell types that display MHC class I and II molecules, and the distinct mechanisms by which foreign polypeptides are processed (fragmented) and bound to/presented by MHC class I and II molecules determine how CD8+ and CD4+ T cells respond to virus infection.
The function of the MHC molecules is to bind peptide fragments from pathogens, and to display them on the cells harboring the pathogen for recognition by T cells expressing the appropriate (i.e., specific) TCR. The consequences of this MHC-TCR–dependent cell–cell interaction are almost always deleterious to the pathogen: virus-infected cells are killed; macrophages are activated to destroy intracellular bacteria living within their intracellular vesicles; and B cells are activated to produce antibodies that eliminate or neutralize extracellular forms of pathogens. The MHC locus is located on chromosome 17 in the human (chromosome 4 in the mouse), and extends over approximately 4 centimorgans of DNA (∼4 × 106 base pairs). Two properties of the MHC locus product make it difficult for pathogens to evade the adaptive immune response. First, the MHC locus is polygenic.258 It encodes several different MHC class I and II gene products, and these differences in nucleotide/amino acid sequence control the range of peptide fragments that an individual MHC molecule can bind, so that every individual possesses a set of MHC molecules with a different range of peptide-binding specificity. Second, the MHC locus is highly polymorphic. Multiple versions (alleles) exist of each MHC gene product expressed in the population as a whole. In fact, the MHC genes are the most polymorphic genes in the human genome.258
The MHC locus contains three major classes of genes: class I, II, and III. The MHC class I and II genes (and their products) were first identified because of their critical role as targets in graft rejection. Hence, the name for the human MHC locus, HLA, (for human leukocyte antigen locus), and H-2 (histocompatability-2 locus) for the corresponding genes in the mouse. The MHC class I genes (or more precisely MHC class Ia genes) consist of eight exons and encode a 45-kD protein containing three external domains (α1, α2, α3), along with a membrane-spanning segment and a short cytoplasmic tail. The α1 and α2 domains fold to form a molecular platform on the upper, outer surface of the molecule, with a groove or cleft to accommodate peptide fragments from pathogens.83 Amino acid differences in the α1 and α2 domains among several class I molecules encoded within the MHC locus genes of an individual and between the numerous alleles of the gene encoding a given MHC class I gene product among humans dictate the types of peptides that can be accommodated in the peptide-binding groove. The α3 domain contains the site for the binding CD8 molecules to the MHC class I. The MHC class I molecule is a heterodimer consisting of the MHC encoded 45-kD heavy (H) chain complexed noncovalently with a non-MHC locus-encoded small molecule, β-2 microglobulin (β-2M), which interacts with the H chain α3 domain. Stable cell surface expression of the MHC class I molecule usually requires β-2M association. Importantly, MHC class I molecules are expressed at varying levels on most cells of the body, with neurons being a notable exception.218 The expression levels of class I molecules on the cell surfaces can be upregulated by exposure of the cell to inflammatory mediators (e.g., type I and II IFN, TLR agonist, and so on).85,141 Therefore, the T cells that recognize peptide/MHC class I complexes (i.e., the CD8+ T cells) have the capacity to survey most body cells and interact with/destroy cells displaying the appropriate peptide/MHC class I complex. Three class I genes (more precisely, three class Ia genes) exist in the human: HLA-A, HLA-B, and HLA-C (H-2K, H-2D, and H-2L in the mouse), and each gene is polymorphic (i.e., displays multiple different alleles) within the human population.
The MHC class II molecules are also heterodimers, consisting of two chains (α and β), each encoded by a separate gene. These genes have a similar exon–intron structure, with the class II α chain genes encoding two extracellular domains (α1 and α2), and the class II β chain gene likewise encoding two extracellular domains (β1 and β2). The α1 and β1 domains of the class II molecules interact to form a molecular platform with a groove to accommodate the foreign peptide, which is like the α1 and α2 domain of the class I molecule on the upper outer surface of the molecule and, therefore, available for recognition by the TCR on CD4+ T cells. The CD4 molecule on the T cell interacts with the surface of the β2 domain, distal to (away from) the TCR binding site. The class II heterodimer is anchored to the cell membrane by transmembrane segments of both chains, and each chain contains a short cytoplasmic tail. In contrast to class I molecules, the expression of class II molecules is largely restricted to immune system cells: B cells, macrophages, DCs, and activated T cells in the human (and to a lesser extent, for activated at T cells in the mouse), as well as specialized epithelial cells in the thymus and type II alveolar epithelial cells in the lungs. The expression of MHC class II molecules on these cells can be upregulated by cytokines (in particular, IFN-γ)64; evidence indicates that certain somatic (nonimmune) cells can be induced to express MHC class II molecules in response to inflammation.85 Three pairs of MHC class II α and β chain genes exist, which are called HLA-DR, HLA-DP, and HLA-DQ (two pairs in the mouse: H-2I–A and H-2I–E). In some instances, the DR cluster contains the genes for two β chains (DRβ1, DRβ2), each of which can pair with the DRα chain. Again, multiple alleles exist for each gene pair. The amino acid differences among the alleles are localized primarily to the α1 and β1 domains, where these residue differences control the range of foreign peptide bound by the various MHC class II allelic gene products.66
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


