Figure 41.1
Schematic representation of two types of gene alterations generated by chromosomal translocations. (a) A protooncogene (B) is activated by the promoter and enhancer elements of a distinct translocation gene partner (A, Regulatory Element). (b) Discrete segments of two different genes (X and Y) are joined as a result of a translocation, creating a fusion gene that encodes a chimeric protein
These genetic alterations have important prognostic implications that can be used to classify precursor lymphoid neoplasms and to guide the selection of therapy [33] (Table 41.1).
Table 41.1
WHO Classification (2008) of acute lymphoblastic leukemia/lymphoma
B lymphoblastic leukemia/lymphoma |
B lymphoblastic leukemia/lymphoma, NOS |
B lymphoblastic leukemia/lymphoma with recurrent genetic abnormalities |
B lymphoblastic leukemia/lymphoma with t(9;22)(q34;q11.2); BCR–ABL1 |
B lymphoblastic leukemia/lymphoma with t(v;11q23); MLL rearranged |
B lymphoblastic leukemia/lymphoma with t(12;21)(p13;q22); ETV6/RUNX1 (TEL–AML1) |
B lymphoblastic leukemia/lymphoma with hyperdiploidy |
B lymphoblastic leukemia/lymphoma with hypodiploidy |
B lymphoblastic leukemia/lymphoma with t(5;14)(q31;q32); IL3–IGH |
B lymphoblastic leukemia/lymphoma with t(1;19)(q23;p13.3); TCF3/PBX1 (E2A–PBX1) |
T lymphoblastic leukemia/lymphoma |
Genetic Abnormalities in B-Lineage ALL
B-lineage ALL is the most frequent acute leukemia in children, corresponding to 80–85 % of cases with about 80 % of these having cytogenetic alterations and characteristic genetic translocations (Table 41.2). The chromosomal translocation t(12;21)(p13;q22) results in the ETV6/RUNX1 (TEL/AML1) fusion gene. This is a cryptic translocation, which cannot be detected by karyotyping. Thus, fluorescence in situ hybridization (FISH) or PCR must be used for detection. It is the most common chromosomal rearrangement in childhood ALL, occurring in about 25 % of cases [34]; by contrast, it is rarely found in adult ALL (1–3 %) [35]. The t(12;21) translocation is almost exclusively associated with a young age (<10 years) and a precursor B-cell phenotype [34]. Both ETV6 and RUNX1 are important regulators of normal hematopoiesis, and are involved in other translocations in both lymphoid and myeloid leukemias, which suggests that the resulting fusion proteins could deregulate normal hematopoietic development leading to leukemia [1, 31]. ETV6-RUNX1 also causes overexpression of the erythropoietin receptor and activation of JAK-STAT signaling [36, 37]. The t(12;21) can be present in blood cells at birth, 5–10 years before the leukemia becomes evident, as demonstrated by studies using neonatal blood spots. The leukemias induced by ETV6-RUNX require a second genetic event for their development. The loss of function of normal TEL protein, through the loss of the normal TEL allele as observed in patients with t(12;21), appears to be this second event [1, 38].
Table 41.2
Main genetic alterations involving B-lineage ALL
Translocation | Involved gene(s) | Frequency |
|---|---|---|
Pre-B cell/early pre-B cell | ||
t(12;21)(p13;q22) | ETV6/RUNX1 | 20–25 % children/1–3 % adults |
t(1;19)(q23;p13) | TCF3/PBX1 | 5 % children |
t(4;11)(q21;q23) | AF4/MLL | 2 % |
t(11;19)(q23;p13) | MLL/ENL | <1 % |
t(9;11)(p21;q23) | AF9/MLL | <1 % |
t(9;22)(q34;q11) | ABL1/BCR | 3–5 % children/25 % adults |
t(17;19)(q22;p13) | HLF/E2A | <1 % |
t(12;v)(p12-p13;V) | ETV6 or KIP1 | 5–10 % |
Dic(9;12)(9p11-p12;p12) | Unknown | 1 % |
Dic(9;20) dic(9;20)(p11-13;q11) | Unknown | 2 % |
t(5;14)(q35;q32) | IL3/IGH | <1 % |
B-cell ALL | ||
t(8;14)(q24;q32) | MYC/IGH | 95 % |
t(8;22)(q24;q11) | MYC/IGL | <5 % |
t(2;8)(p11;q24) | IgK/MYC | <5 % |
New recurrent abnormalities | ||
Del9p | CDKN2A/B | 30–35 % |
Del9p | PAX5 | 30–35 % |
iAMP21 | AML1 | 2 % |
CRLF2 | CRLF2 | 5–7 % (50 % with Down syndrome) |
IKZF1 | IKZF1 | 15 % |
Whether t(12;21) is associated with a good prognosis or with only a marginally better outcome compared to other cytogenetic ALL subgroups remains controversial [34]. ETV6/RUNX1 rearrangement has been observed in about 25 % of relapsing cases treated within European Berlin-Frankfurt-Munster (BFM) group protocols. These patients tend to have a longer duration of remission (>2 years) compared to other childhood ALL cytogenetic groups; however, early relapses have also been observed, illustrating a heterogeneous pattern of clinical behavior in this group of patients [34, 39].
The t(1;19)(q23;p13) translocation is one of the most frequently observed translocations in childhood ALL, occurring in approximately 25 % of pre-B cytoplasmic IG-positive cases and in 1 % of early pre-B cytoplasmic IG-negative cases. This translocation most frequently creates a fusion of TCF3 (E2A), a helix-loop-helix (bHLH) protein-coding gene on chromosome 19, with PBX1, a homeobox-containing gene located on chromosome 1 [36, 37, 40]. A few molecular variants of the t(1;19), leading to different species of TCF3/PBX1 mRNAs, have been described [41]. Both genes play a critical role in lymphocyte development and alteration of HOX genes has a clear role in leukemogenesis. Although this translocation has been associated with a poor clinical outcome, the current chemotherapy protocols have improved prognosis for this subgroup of patients [31, 36, 37, 42].
Another rare translocation, found in 1 % of B-precursor ALLs, is t(17;19)(q22;p13.3), which also involves the TCF3 gene but creates a fusion with the HLF gene, resulting in a chimeric transcript and protein that could contribute to leukemogenesis by aberrant regulation in genes that control the fate of early lymphoid progenitors. This translocation has been associated with poor prognosis [3, 36, 37, 43].
Abnormalities of the q23 region of chromosome 11 are seen with relative frequency in childhood ALL (5 %), infant acute leukemia (70 %), and secondary leukemias in patients who received topoisomerase II inhibitors (85 %). Leukemias with translocations involving 11q23 have an early pre-B phenotype and express myeloid antigens. More than 40 different chromosomal loci have been identified as fusion partners in 11q23 translocations. In the majority of cases, the translocation partner is chromosome 4, and less frequently chromosome 1, 10, or 19. At the molecular level, the t(4;11) fuses MLL at 11q23 to AF4 at 4q21, resulting in an MLL/AF4 chimeric gene. MLL-rearranged leukemias exhibit an upregulation of HOX Class I genes. The ability of MLL to regulate expression of the HOX genes suggests a role in hematopoiesis and also in leukemogenesis [36, 37]. This translocation is related to aggressive clinical features such as hyperleukocytosis, organomegaly, frequent central nervous system involvement, and poor outcome [3, 6, 36, 37, 44–49].
The t(9;22) translocation is identified in 3–5 % of childhood ALL and approximately 25 % of adult ALL [47, 48, 50]. This translocation creates a novel chromosome (the Philadelphia chromosome, Ph+) and a fusion between the protooncogene ABL1 on chromosome 9 and the “breakpoint cluster region” gene (BCR) on chromosome 22. Whereas the ABL1 breakpoint on chromosome 9 is consistently between exons a1 and a2, the BCR breakpoints on chromosome 22 can occur in two different gene regions. The “minor” breakpoint cluster region (m-bcr), between exons e1 and e2, is present in approximately 90 % of childhood Ph+ ALL; by contrast, a “major” breakpoint cluster region (M-bcr), between exons 13 and 14 or 14 and 15 (formerly called b2 and b3 or b3 and b4, respectively), is a usual finding in chronic myelogenous leukemia [50] and occasionally present in Ph+ ALL. The m-bcr generates a fusion protein of 190 kDa (p190), whereas the M-bcr results in a fusion protein of 210 kDa (p210) [47, 48, 50–53]. The presence of t(9;22) is associated with a high risk of treatment failure in children and adults with ALL. However, Ph+ childhood ALL is a heterogeneous disease with regard to treatment response. High leukocyte count, old age, and poor response to prephase treatment with prednisone and intrathecal methotrexate influence treatment outcome. Patients with a good prednisone response have a significantly lower risk of treatment failure compared to those with a poor prednisone response (PPR), when treated with intensive BFM protocol chemotherapy, whether associated with bone marrow transplantation (BMT) or not. These PPR children have a clinical response and prognosis as poor as that of Ph+ ALL adults [47, 48, 54]. For the treatment of these patients, s elective tyrosine kinase inhibitors have been used in association with chemotherapy and/or BMT. The use of the ABL tyrosine kinase inhibitor imatinib, and derivatives of second generation (e.g., nilotinib, dasatinib) has transformed the treatment and outcome of ALL patients with the BCR–ABL1 fusion gene [55, 56].
More recently, a group of childhood B-lineage ALLs termed “BCR-ABL1-like” has been described. These ALLs exhibit a similar gene expression profile as that observed in BCR–ABL1 positive ALL patients. This subtype represents up to 15 % of all B-lineage cases and is frequently associated with deletion or mutation of IKZF1. In 50 % of the cases, the CRLF2 gene is rearranged, which confers a very poor prognosis [6, 36, 37, 57, 58].
The t(8;14)(q24;q32), involving the MYC gene (8q24) and the IGH locus (14q32), occurs in 85–90 % of cases of surface immunoglobulin positive B-cell ALL. The dysregulation of MYC expression appears be responsible for the B-cell proliferation. Two other variants, t(2;8)(p11;q24) and t(8;22)(q24;q11), involving the MYC gene and IGK and IGL, respectively, are less commonly observed [3, 29, 59, 60]. The t(5;14)(q31;q32) is an uncommon translocation, observed in less than 1 % of ALL patients, and results in the fusion of the IGH gene with the interleukin-3 (IL3) gene. The overexpression of IL3 appears be involved in the pathogenesis of the leukemia and hypereosinophilia in these patients [3].
Subgenetic alterations involving genes that regulate normal B lymphoid development are identified in 40 % of B-lineage ALL. The most commonly involved gene is PAX5, required for B-lineage maturation. Other regulating B-lymphoid development genes frequently involved include the IKAROS family of transcription factors IKZF1, IKZF2, IKZF3, as well as EBF1, TCF3, LEF1, RAG1/2, BLNK, and VPREB1 [30, 57, 61]. Deletions or sequence alterations of the lymphoid development associated IKZF1 gene and tumor suppressor gene CDKN2A/B have been associated with relapse risk, confirming that genetic alterations detected at diagnosis can predict the risk of relapse [6, 36, 37, 61]. Overexpression of CRLF2, associated or not with corresponding genomic lesions (IGH–CRLF2, P2RY8–CRLF2 or the p.Phe232Cys mutation) occurs in 5–7 % of childhood B-lineage precursor ALL and in 50 % of individuals with Down syndrome and ALL. These alterations commonly are associated with JAK1/2 mutations and, in patients who do not have Down syndrome, with IKZF1 mutations and poor prognosis [6, 36, 37].
Genetic Abnormalities in T-Lineage ALL
T-lineage ALL accounts for approximately 15 % of all childhood ALL and in general is considered a high-risk category. This leukemia characteristically presents with chromosomal rearrangements involving T-cell receptor genes, proto-oncogenes, or transcription factor genes, or alterations of key genes or pathways related to T-cell lymphogenesis (Table 41.3).
Table 41.3
Main genetic alterations involving T-cell ALL
Involved gene(s) | Frequency | |
|---|---|---|
Translocation involving T-cell receptor genes | ||
t(7;10)(q34;q24) and t(10;14)(q24;q11) | TLX1 (HOX11) | 7 % children; 31 % adults |
t(5;14)(q35;q32) | TLX3 (HOX11L2) | 20 % children; 13 % adults |
inv(7)(p15q34), t(7;7) | HOXA genes | 5 % |
t(1;14)(p32;q11) and t(1;7)(p32;q34) | TAL1 | 3 % |
t(7;9)(q34;q32) | TAL2 | <1 % |
t(7;19)(q34;p13) | LYL1 | <1 % |
t(14;21)(q11.2;q22) | BHLHB1 | <1 % |
t(11;14)(p15;q11) | LMO1 | 2 % |
t(11;14)(p13;q11) and t(7;11)(q35;p13) | LMO2 | 3 % |
t(1;7)(p34;q34) | LCK | <1 % |
t (7;9)(q34;q34.3), | NOTCH1 | <1 % |
t(7;12)(q34;p13) and t(12;14)(p13;q11) | CCND2 | <1 % |
Translocation involving oncogenes | ||
1p32 deletion | SIL–TAL1 | 9–30 % |
t(10;11)(p13;q14) | CALM–AF10 | 10 % |
11q23 | MLL | 8 % |
t(9;9)(q34;q34) | NUP214–ABL1 | 5 % |
t(9;14)(q34;q32) | EML1–ABL1 | <1 % |
t(9;22)(q34;q11) | BCR–ABL1 | <1 % |
t(9;12)(p24;p13) | ETV6–JAK2 | <1 % |
Cryptic deletions | ||
9p21 | P16 | 65 % children; 15 % adults |
Del(6q) | Unknown | 20–30 % |
Mutations | ||
Notch1 | NOTCH1 | 50 % |
PTEN | PTEN | 17 % |
IL7R | IL7R | 7–9 % |
Developmental genes abnormalities have been found in T-lineage ALL (T-ALL) comprising the basic helix-loop-helix (bHLH) gene family (MYC, TAL1, LYL1), the homeobox gene family (HOX) including the HOXA cluster, and the LIM gene family (LMO1 and LMO2). When rearranged near enhancers with the loci 14q11.2, 7q34-q35 or 7p15, that contain the TRA/TRD, TRB, and TRG genes, respectively, these regulatory genes become active and lead to dysregulated expression of transcription factor genes. The TAL1 gene can be involved due to t(1;14)(p32,q11), which occurs in 5 % of childhood T-ALL, or can be affected by a submicroscopic interstitial deletion between the SIL and TAL1(SCL) genes at 1p32. This “TAL deletion” occurs in 20–25 % of childhood T-ALL [3, 29–31, 60].
Different T-ALL signatures, based on gene expression profiles, indicate that the leukemic arrest in specific genetic subgroups (LYL1, TLX1, TAL1) occurs at particular stages of thymocyte development (i.e., at the pro-T, early cortical thymocytes, or late cortical thymocyte stage) [62–65] and seems to comprise at least four distinct molecular-cytogenetic subgroups: TAL/MLO, TLX3/HOX11L2, TLX1/HOX11, and HOXA subgroups [66, 64, 65, 67, 68]. A fifth subtype of T-ALL termed “early T precursor” (ETP) comprises approximately 15 % of T-ALL and is associated with a high risk of relapse. Inactivating alterations that disrupt hematopoietic development and cause histone-modification of genes are possible in ETP, as well as somatic activation of regulating cytokine receptors and RAS signaling [67, 69].
The TAL deletion leads to expression of a SIL–TAL chimeric transcript, resulting from the fusion of the 5′ part of SIL to the 5′ region of TAL1. In addition, high levels of TAL1 have been observed in 40–60 % of T-ALL. The aberrant expression of TAL1 may activate a specific set of target genes that are normally quiescent in T-cell progenitors or could exert a negative effect through inhibition of E2A or E2A-HEB heterodimers, leading to a leukemogenic effect. The LYL1 gene is involved in the t(7;19)(q34;p13) leading to constitutive overexpression, usually in a more immature T-lineage phenotype. The MYC gene can be translocated to a TCR locus [t(8;14)(q24;q21)] in some cases of T-ALL [3, 29–31, 62].
In addition to genes encoding bHLH proteins, gene rearrangements of the LIM gene family (encoded by LMO1 and LMO2) and the TCR loci have been described in T-ALL. The t(11;14)(p13;q11) (LMO1/TCRD) occurs in 7–13 % of T-ALL and has been associated with males, high WBC count and extramedullary disease. The t(11;14)(p15;q11) (LMO2/TCRD) is infrequent and found in <1 % of the cases [3, 29, 70, 71].
Translocations involving the HOX genes TLX1 (HOX11) and TLX3 (HOX11L2), complete the list of developmental genes that are inappropriately placed under control of the TCR loci. Activation of expression of the HOX11 gene by chromosomal translocations t(10;14)(q24;q11) and t(7;10)(q34;q24) interferes with normal T-cell development and promotes malignant transformation. HOX11 expression is associated with a favorable prognosis in childhood T-ALL. HOX11L2 is activated by t(5;14)(q35;q32) and (5;14)(q35;q11). Despite the fact that this finding has been associated with a poor prognosis, this does not seem to apply in children receiving more intensive chemotherapy [3, 29–31, 60].
Recurrent translocations involving the HOXA cluster have been described. This subgroup of T-ALL is characterized by elevated expression of the HOXA genes and includes MLL rearrangements, inv(7)(p15q34), t(10;11)(p13;q23), and CALM–AF10 rearrangements as well as the cryptic del(9)(q34.11q34.13) deletion that results in a SET–NUP214 fusion product. MLL fusions are found in 4–8 % of T-ALL. The t(11;19)(q23;p13) that involves MLL/ENL is the most frequent. Other identified translocations include t(6;11)(q27;q23) (MLL/AF6), t(10;11)(p13;q23) (MLL/AF10), t(X;11)(q13;q23) (MLL/AFX1), and t(4;11)(q12;q23) (MLL/AF4). T-ALL with an MLL fusion is characterized by a specific expression profile with differentiation arrest in early stage thymocyte development [29–31, 60, 61, 72].
Chromosomal translocations involving NOTCH1, a gene that regulates the normal embryologic development of T-cells and other tissues, are rare in T-ALL. In contrast, activating NOTCH1 mutations are present in more than 50 % of T-cell ALLs. NOTCH1 regulates oncogenes including the MYC and RAS, and this is probably the mechanism by which aberrant NOTCH signaling causes T-ALL. This oncogenic mechanism is supported by experimental models, in which mutations of the NOTCH1 gene can induce T-cell ALL [29–31]. Small molecule inhibitors of the NOTCH pathway thus have the potential to induce remission in T-cell ALL [31].
Cryptic deletions are common in T-ALL and may be concomitant with other changes. The most common cryptic deletion causes loss of the INK4/ARF locus at 9p21, important to cell cycle control. Del(6q) also has been observed, but the involved gene is unknown [29, 31]. Inactivating gene mutations in PTEN have been described in approximately 17 % of pediatric T-ALL and are associated with poor prognosis [73]. Somatic-gain mutations in IL7R have been observed in 7–9 % of pediatric T-ALL [74]. The main genetic alterations in T-ALL are summarized in Table 41.3.
Indications for Testing
Many genetic alterations have important prognostic implications that can guide the selection of therapy. Treatment of the acute leukemias has progressed from uniform strategies devised for large groups of patients to more refined protocols tailored to the risk of relapse in discrete subgroups. Although routinely recorded features such as blast cell immunophenotype and presenting WBC count provide useful criteria for risk assessment, molecular genetic changes are the most sensitive markers of potential leukemia aggressiveness. Hence, overall these are the best suited to guide treatment. IG and TCR gene rearrangement studies are useful for the diagnosis of childhood ALL, for detection of cerebrospinal involvement [24, 25, 51–53, 75], and for MRD assessment because they occur in the vast majority of ALL patients [16, 45, 46, 76–80].
Molecular detection of chromosomal abnormalities in blast cells of leukemia patients has important prognostic implications that can guide staging and selection of treatment. In addition, detection of specific translocations is used to MRD monitoring [81].
The term “minimal residual disease” has been used to define the lowest level of disease detected by conventional methods of analysis in patients who are in complete continuous remission. At clinical presentation, the number of leukemic cells is approximately 1011–1012; if the patient is not treated, the clone continues to expand, and death occurs with approximately 1013 leukemic cells. With cytotoxic treatment, the number of neoplastic cells is diminished and when less than 5 % of leukemic blasts are identified in the bone marrow (BM) by conventional cytology, the patient is considered to be in complete remission. Thus, patients in complete remission have from zero to 1010 leukemic cells. The detection of MRD, either after clinical remission was achieved or during treatment, can help direct adjustments to therapeutic strategies and identify patients who are at higher risk of relapse [16, 45, 46, 76–80]. The presence of MRD at the time of transplantation as well as in the post transplant period are also powerful, independent predictors of adverse outcome [82–87].
Available Assays
Detection of Clonality by PCR
Monoclonality in ALL is detected by PCR amplification of the IG and/or TCR V(D)J region, typically using consensus primers to conserved sequences within the framework regions of the V region and the D and/or J regions (Fig. 41.2). The V(D)J region varies in size and sequence across a population of B or T cells and allows for detection of a monoclonal leukemic cell population by PCR amplification of a unique PCR product specific to the leukemic clone. For a population of nonlymphoid cells, where the IG and TCR genes are in the germline configuration, amplification will not occur, due to the great distance between the closest V and J regions [8, 9, 11, 15, 17].
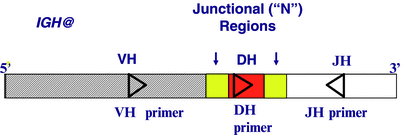
Figure 41.2
Schematic representation of an IGH VDJ recombination region and the strategy used to amplify the N regions. Probes for the N regions can be used as patient-specific probes (boxes with vertical arrows above them indicate N regions)
RT-PCR for Leukemia-Specific Translocations
PCR analysis of fusion genes is based on the design of oligonucleotide primers within exons at the opposite sides of the breakpoint fusion regions, so that the PCR product contains the tumor-specific fusion sequence. The exact breakpoint at the DNA level may be different from patient to patient and therefore difficult to determine. In the majority of ALL translocations, the breakpoint regions span greater than 2 kb, exceeding the conventional range for PCR. However, because the breakpoints are mostly intronic and spliced out at the messenger RNA (mRNA) level, the preferred target for detection of translocations is the chimeric mRNA [26].
Reverse transcription-PCR (RT-PCR) requires extraction of total RNA or mRNA from mononuclear cells, reverse transcription of the RNA into complementary DNA (cDNA) and then PCR, followed by a method to detect the RT-PCR product, such as electrophoresis. The sensitivity of the method is specific for each target and can be assessed by amplification of serial dilutions of RNA from diagnostic specimens or cell lines containing the specific translocation into RNA from healthy individuals who do not have the translocation. The presence of a very small number of abnormal cells, in the range of 1 in 105 to 1 in 106, has been consistently detected using appropriate conditions [88]. This sensitivity is approximately 1000–10,000 times greater than with Southern blot analysis. A PCR test that is sufficiently sensitive to detect one leukemia cell in 102–103 normal cells is acceptable for diagnostic testing. A higher sensitivity of one leukemia cell in 104–105 normal cells is required for MRD assessment during follow-up testing [16]. More than one primer set may need to be used to detect all fusion transcripts when the translocation breakpoints can be in different introns of one or both of the fusion genes (Fig. 41.3).
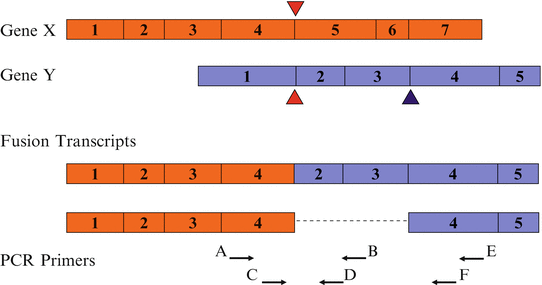
Figure 41.3
RT-PCR detection of different fusion transcripts generated by the same translocation. The possibility of different fusion transcripts requires the use of multiple primer pairs to detect fusion transcripts formed by breakpoints in different introns of the gene(s), depending on the size of the transcript
MRD Quantification of IG and TCR Gene Rearrangements
Real time quantitative PCR (RT-qPCR) of IG and TCR gene rearrangements can be used to quantify MRD by using allele-specific oligonucleotide (ASO) probes. Sensitivities of one in 103–105 (Fig. 41.4) are achievable with this strategy [89–94]. Although initial assays used an ASO fluorescent probe to the junctional region, a more useful approach is to use a fluorescent probe complementary to the germline IGH and TCR gene segments, in combination with an ASO primer complementary to the junctional region [95, 96]. The ASO primer approach theoretically results in more sensitive MRD detection compared with the use of germline primers, because no competition can occur with the amplification of similar rearrangements in normal cells. Although specific amplification can be easily distinguished from incidental nonspecific amplification, conditions with higher stringency of amplification may need to be used to overcome nonspecific amplification while maintaining the efficiency of the method.
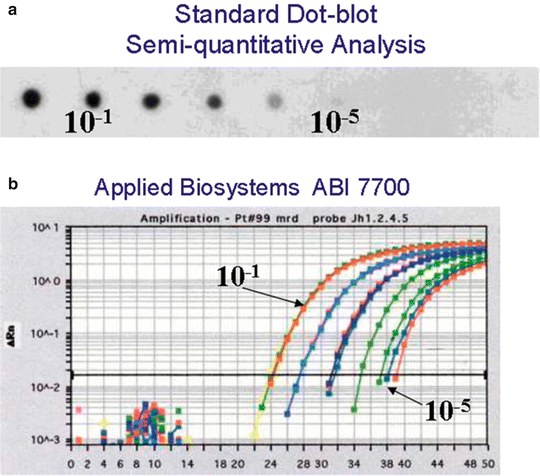
Figure 41.4
Assessement of assay sensitivity by two different methods of a patient-specific IgH clonotypic marker. (a) semiquantitative dot-blot analysis; (b) real-time quantitative PCR using Applied Biosystems ABI 7700
Quantification of Leukemia-Specific Translocations for MRD Assessment
Numerous publications have demonstrated the feasibility of the RT-qPCR method to quantify chimeric transcripts resulting from chromosomal translocations that occur in ALL [81, 91]. Although the principles of RT-qPCR are the same whether DNA or RNA is being analyzed, the RT step for RNA represents a major assay variable for accuracy of quantification and sensitivity. In fact, assays must be designed to correct for variations linked to differences in RNA input amount and, more importantly, in efficiency (or inhibition) during RT. For this reason, the number of target gene copies has to be normalized using a ubiquitously and constantly expressed housekeeping gene as a reference (e.g., ABL1, B2M, and PBGD) [97]. The number of chimeric transcripts are expressed in relation to the number of copies of the reference gene transcript.
Interpretation of Test Results
PCR for IG and TCR Gene Rearrangements
Leukemia-specific IG and TCR gene rearrangements identified at diagnosis can be used for MRD assessment of BM or peripheral blood samples during and following treatment. However, similar IG and TCR gene rearrangements from normal lymphocytes in these specimens also are amplified. To discriminate between the leukemia-derived PCR products and PCR products of normal cells with rearrangements resulting in comparable size PCR products, the amplified bands are subjected either to “fingerprint” [98–100] or homo-heteroduplex analysis [18, 101, 102]. What was traditionally called fingerprint analysis consists of PCR amplification with a fluorescent primer and analysis by capillary electrophoresis. Clonal amplification results in a single peak, often in a background of polyclonal, constitutional amplification products. The homo-heteroduplex analysis takes advantage of the different migration properties in a polyacrylamide gel of V-J rearrangements containing a few mismatches (heteroduplex) compared with the fully matched V-J junctions (homoduplex) typical of a clone.
A variety of IG and TCR gene rearrangements can be present at diagnosis in childhood B-precursor ALL. Oligoclonal IGH rearrangements have been observed in 30–40 % of cases and IGK rearrangements in 5–10 % and are due to VH replacements, VH-DJH junctions, or de novo IGH gene rearrangements [18, 103–106]. Oligoclonality for TRB and TRG gene rearrangements is thought to be rare in B-lineage ALL [107–109, 111]. One exception was reported by Szczepanski et al., who found TRG oligoclonality in 38 % of B-lineage ALLs [17]. Oligoclonality of TRD is frequently observed in incomplete rearrangements, such as VD2DD3 or DD2DD3 s[109, 109, 110]. In T-lineage ALL, oligoclonality is rarely seen at diagnosis [107, 111, 24, 25].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


