Fig. 26.1
Generalized edematous, erythematous rash preceding pustule presentation.
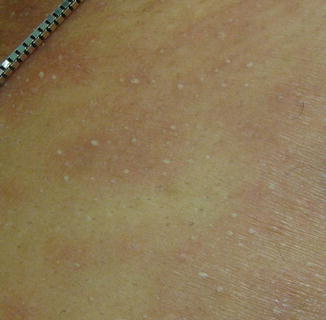
Fig. 26.2
Non-follicular, sterile, pinpoint pustules characteristic of acute generalized exanthematous pustulosis
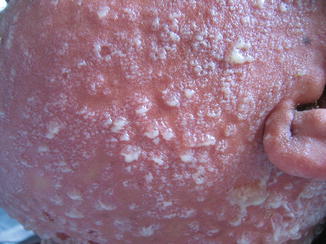
Fig. 26.3
Bullous lesions and lakes of pus
In addition to the cutaneous symptoms, fever greater than 100.4 °F and a neutrophilic (>70 %) leukocytosis (>10,000/mL) is often present. Lymphadenopathy has also been reported in some cases. Possible laboratory abnormalities can include eosinophilia (>700/mL), mild transaminitis (up to twice normal range), and reversible reduction in creatinine clearance. Hypocalcemia may also be present but is often related to hypoalbuminemia. Although Staphylococcus aureus may be present in a few cases, the pustules are most often, and by definition, amicrobial.
AGEP typically occurs anywhere from a few hours to a few days after the administration of the offending drug. In a multinational case–control study of 97 validated cases of AGEP, Sideroff et al. (2007) found that the median time between drug exposure and symptom development was 1 day for antibiotics and 11 days for all other drugs. The pustular eruption typically lasts for 7–10 days and is followed by superficial desquamation lasting several days, characterized at times by collarettes of scale (Fig. 26.3). In most cases, AGEP is self-limited and resolves without treatment 1–2 weeks after removing the offending drug. Courses lasting longer than 2 weeks are rare. AGEP typically has a favorable prognosis, with a reported mortality rate of less than 5 %, usually due to infections in the elderly or immunocompromised, secondary comorbidities, or hemodynamic instability followed by formation of bullae resulting from confluent pustules.
The estimated incidence of AGEP is one to five cases per million per year, most often occurring in adults. It affects both sexes but appears to occur in females more often. One study has found associations with HLA types B51, DR11, and DQ3.
Treatment for AGEP is chiefly symptomatic and supportive. Immediate withdrawal of the presumed causative agent is the mainstay of therapy. Antibiotics are not to be given unless there is a well-documented associated infection. When a patient is taking multiple medications, those frequently associated with AGEP should be stopped. Older or immunocompromised patients with significant fever or widespread eruption may require hospitalization for fluids, electrolyte repletion, and nutritional support. Symptomatic treatment involves moist dressings during the pustular phase to relieve pruritus and prevent superinfection. Emollients may support skin barrier function restoration during the desquamation phase. The use of topical corticosteroids and oral antihistamines has been proposed for symptomatic relief, but their efficacy has not been evaluated in clinical trials. To date, there is limited evidence to support the use of systemic corticosteroids.
Clinical Differential Diagnosis
The clinical differential diagnosis for acute generalized exanthematous pustulosis (AGEP) includes generalized acute pustular psoriasis (von Zumbusch type), subcorneal pustular dermatosis (Sneddon-Wilkinson disease), bullous impetigo, drug reaction with eosinophilia and systemic symptoms (DRESS), Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN), erythema multiforme, Sweet syndrome (neutrophilic dermatosis of the skin), subcorneal immunoglobulin A dermatosis, viral exanthema with secondary pustulation, and infectious folliculitis.
Without additional historical, laboratory, or histologic data, generalized acute pustular psoriasis and AGEP may be difficult to distinguish both clinically and histologically. Factors that support the diagnosis of generalized acute pustular psoriasis include a history of psoriasis, absence of drug exposure, and histologic findings of subcorneal pustules, papillary dermal edema, and dermal eosinophils. Longer duration of symptoms, mainly fever and pustular eruption, also supports this diagnosis. Although pustular psoriasis can be caused by drugs, the spectrum of associated medications (mainly beta-blockers and lithium) is very different from those associated with AGEP. Furthermore, a more abrupt-onset, short duration (2 weeks), association with recently introduced drugs, spontaneous resolution after withdrawal of culprit drugs, and a non-recurrent tendency supports the diagnosis of AGEP.
Histopathology and Histologic Differential Diagnosis
Roujeau et al. (1991) described the main histologic findings of AGEP to be subcorneal and/or intraepidermal pustules (66 %), papillary dermal edema (61 %), a polymorphous perivascular infiltrate with eosinophils (34 %), necrotic keratinocytes (25 %), and leukocytoclastic vasculitis with fibrinoid necrosis (20 %). In most cases, the epidermis was uninvolved or exhibited spongiosis without psoriasiform hyperplasia (61 %). Additional findings include pustular or dermal eosinophilic exocytosis, leukocytoclastic vasculitis with fibrinoid deposits, and erythrocyte extravasation. Hyperplastic epidermal changes, such as acanthosis and papillomatosis, as well as follicular pustules, are rare.
The histologic differential diagnosis of AGEP includes pustular psoriasis, subcorneal pustular dermatosis, pustular contact dermatitis, bullous leukocytoclastic vasculitis, drug hypersensitivity syndrome, and IgA pemphigus. The main differential diagnosis is pustular psoriasis. Features that are more characteristic of AGEP include the presence of papillary dermal edema, necrotic keratinocytes, and dermal eosinophils (Fig. 26.4a–c). Subcorneal pustular dermatosis (Sneddon-Wilkinson disease) exhibits only subcorneal pustules; whereas intraepidermal pustules are often noted in AGEP. A few cases of pustular contact dermatitis exhibiting subcorneal pustules have been reported in the literature and, thus, can be difficult to distinguish from AGEP. While pustular lesions may arise in some cases of leukocytoclastic vasculitis, vasculitis is an uncommon feature of AGEP. Drug hypersensitivity syndrome, or DRESS (drug rash with eosinophilia and systemic syndrome), may present with pustules, but these typically are less pronounced than those seen in AGEP. In addition, patients with drug hypersensitivity syndrome often have lymphadenopathy, eosinophilia, mononucleosis, and significant visceral involvement, such as hepatitis, nephritis, pneumonitis, and/or myocarditis. Mild acanthosis would usually be noted in cases of IgA pemphigus. In addition, direct immunofluorescence would demonstrate intercellular IgA deposition.
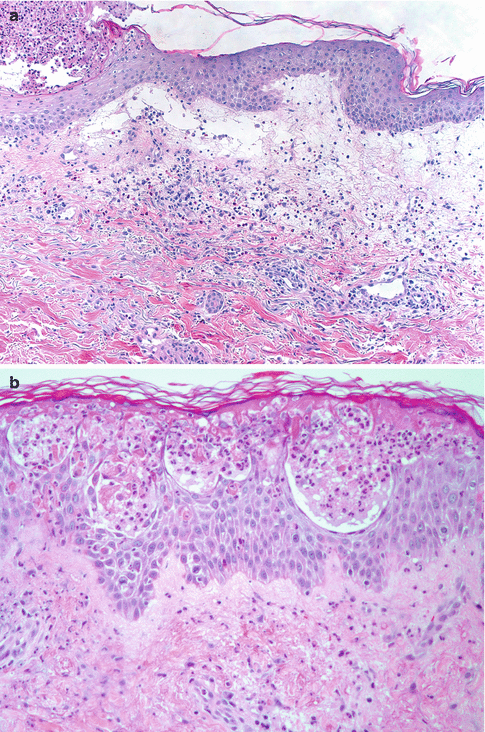
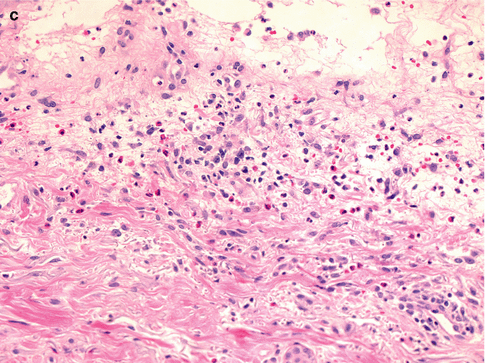
Fig. 26.4
Salient histologic features of acute generalized exanthematous pustulosis include (a) papillary dermal edema, (b) necrotic keratinocytes, and (c) a prominent dermal infiltrate of eosinophils
Etiology
It appears that greater than 90 % of all reported cases of AGEP are drug-related, with a wide variety of medications suspected to cause this reaction pattern. In 2001, Sidoroff and colleagues put forth a comprehensive list of medications that had been published in case reports and larger series of AGEP. Among the medications, antibacterials of the β-Lactam, macrolide, and cephalosporin drug classes were most commonly implicated. In addition, antimycotics, calcium channel blockers, hydroxychloroquine, antalgics and antipyretics, antiparasitics, antiarrhythmics, tricyclic antidepressants, anxiolytics, and others have been found to cause AGEP. Of note, there have been cases reported of AGEP caused by aspirin. According to a EuroSCAR study, pristinamycin, aminopenicillins, quinolones, (hydroxy) chloroquine, sulfonamides, terbinafine, and diltiazem were the drugs that conferred the highest risk. Lower risk etiologic medications include corticosteroids, macrolides, non-steroidal anti-inflammatory drugs, and antiepileptic drugs. Others include terazosin, omeprazole, sennoside, and anti-retroviral protease inhibitors. Topical medications, including bufexamac and other mercury products, have also been linked to AGEP through contact sensitivity. A more recent review of the literature by Speeckaert et al. (2010) identified case reports of AGEP caused by anticonvulsants such as phenytoin, low-molecular weight heparin, and many others.
AGEP has also been associated with viral infections, such as with Adenovirus, Coxsackie B4 virus, Cytomegalovirus, E. coli, Echovirus, Enterovirus, Epstein-Barr virus, Hepatitis B virus, and Human Parvovirus B19. Case reports of AGEP associated with bacterial and parasitic infections, such as with Mycoplasma pneumoniae, Chlamydia pneumoniae, and Echinococcosis, have also been documented. Exposure to mercury was a suspected cause for 8 of 63 patients reported by Roujeau et al. (1991). Vaccinations, illicit drug use, herbal medications, spider bites, intravenous contrast media, lacquer chicken, and progesterone have also been associated with AGEP. However, given the preponderance of evidence behind medications and other supplements as causing AGEP and the rather limited, weaker evidence supporting infectious causes, such cases are best considered as parainfectious causes of AGEP until proven otherwise.
Pathophysiology
Our understanding of the pathogenic mechanisms underlying AGEP is incomplete. However, immunohistologic investigation of patch test studies as well as direct immunologic study of immune cells generated from these patch tests and from the peripheral blood of patients with drug-induced AGEP have demonstrated the crucial role of CXCL (chemokine (C-X-C motif) ligand)-8 producing, drug-specific T cells in orchestrating the neutrophil response.
Based on these studies, Britschgi and Pichler (2002) have proposed a three-phase model for the regulation of T-cell/keratinocyte-orchestrated neutrophil-rich inflammation in AGEP. Phase 1 involves the activation and expansion of drug-specific T cells with subsequent migration to the skin. After exposure to the offending drug, professional antigen-presenting cells activate drug-specific T cells by presenting the drug on major histocompatibility complex (MHC) class I (for CD8+) and class II (for CD4+) in the lymph nodes. These drug-specific T-cells then expand and subsequently migrate into the dermis and epidermis.
Phase 2 is characterized by the functional, orchestrating activity of drug-specific T-cells in the skin as well as drug presentation by Langerhans’ cells and keratinocytes. The principal role of infiltrating CD4+ T-cells is the massive secretion of the neutrophil recruiting factors CXCL8 and granulocyte macrophage colony-stimulating factor (GM-CSF), as well as of other factors (interferon-γ, interleukin-4, interleukin-5, and RANTES (regulated on activation, normal T expressed and secreted). On the other hand, CD8+ T-cells produce interferon-γ and destroy tissue/keratinocytes through cytotoxic mechanisms including perforin/granzyme B and the Fas/Fas-L apoptotic system. These T-cells are further stimulated by drug-presenting keratinocytes (MHC class I) and by Langerhans’ cells (MHC class I and II). It is thought that the release of inflammatory cytokines such as interferon-γ may stimulate the keratinocytes to secrete CXCL8. At this point, CD4+ and CD8+ cells are scattered throughout the epidermis, but the subcorneal vesicles are populated mainly by CD4+ cells. Very few neutrophils and eosinophils are present at this stage.
In Phase 3, neutrophils are recruited to the skin in increasing numbers by attachment to the site of inflammation via adhesion molecules (e.g., intercellular adhesion molecule-1), expressed on activated endothelial cells. They migrate through the dermis into the epidermis along an increasing CXCL8 gradient, and eventually fill the vesicles, transforming them into pustules. At this point, T-cells are mainly gathered in the dermis (CD4+ more so than CD8+) and around blood vessels (where CD4+ and CD8+ are more similar in number).
This final phase may proceed as long as the drug is present. Resident antigen-presenting cells and keratinocytes may continue to present the drug to and stimulate T-cells, which will continue to orchestrate the inflammation by activation and destruction of the tissue and by recruitment of more neutrophils to the skin. The release of interleukin-5 and RANTES may contribute to the eosinophilia seen in some patients.
Conclusions
AGEP is a rare, dramatic drug reaction. It mimics generalized pustular psoriasis, and some authors consider it a variant of that disease. It is usually self-limited but the offending medication must be identified and discontinued. Clinicians need to be alert to this illness so early recognition can lead to discontinuation of the offending medication as quickly as possible.
Suggested Reading
Albanesi C, Cavani A, Girolomoni G. IL-17 is produced by nickel-specific T lymphocytes and regulates ICAM-1 expression and chemokine production in human keratinocytes: synergistic or antagonistic effects with IFN-gamma and TNF-alpha. J Immunol. 1999;162(1):494–502.PubMed
Baker H, Ryan TJ. Generalized pustular psoriasis: a clinical and epidemiological study of 104 cases. Br J Dermatol. 1968;80(12):771–93.CrossRefPubMed
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


