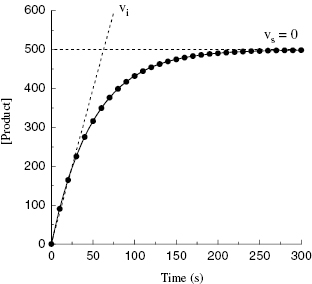
Figure 9.1 Typical enzyme reaction progress curve in the presence of an irreversible enzyme inactivator, highlighting the initial velocity region (vi) and the fact that the terminal velocity (vs) is zero for such compounds.
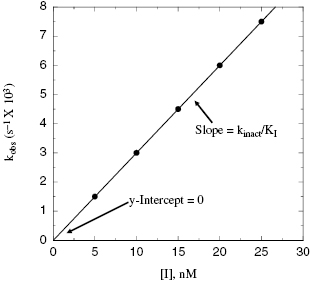
Depending on the mechanism of irreversible reaction, inactivation can appear to proceed through either a single-step or a two-step mechanism (Figure 9.2). In the case of nonspecific affinity labels (see Section 9.2) many amino acid residues on the enzyme molecule, and on other protein molecules in the sample, can be covalently modified by the affinity label. Not every modification event will lead to inactivation; only modification of functionally critical residues will affect enzyme activity. In situations like this, the dependence of kobs on inhibitor concentration can appear nonsaturating. Thus a plot of kobs as a function of [I] (Figure 9.3) will be linear and will pass through the origin because there is no dissociation of inhibitor from the inactivated enzyme complex (see also Chapter 6). The slope of the linear fit of data, as in Figure 9.3, has units of a second-order rate constant (M−1, s−1) and is related to k3 in the single-step inactivation scheme shown in Figure 9.2A. This second-order rate constant is generally considered to be the best measure of relative inactivator potency, or effectiveness. The value of this rate constant is most commonly reported in the literature as kobs/[I] or as kinact/KI; the meanings of the individual terms kinact and KI will be defined next.
Figure 9.2 Mechanisms of irreversible enzyme inactivation. (A) Nonspecific affinity labeling, (B) quiescent affinity labeling, and (C) mechanism-based inactivation.
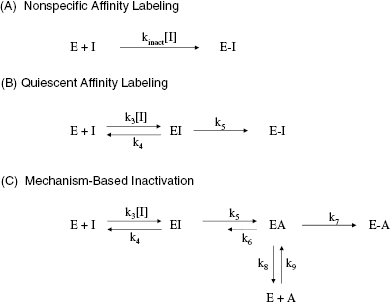
Figure 9.3 Plot of kobs as a function of inactivator concentration for a single-step mechanism of inactivation.
For quiescent affinity labels (as defined in Section 9.2) and for mechanism-based inactivators (Section 9.3), the inactivation follows a two-step mechanism (schemes B and C of Figure 9.2, respectively). The first step for both mechanisms involves reversible binding of the inhibitor to the enzyme, often under rapid equilibrium conditions. For quiescent affinity labels, the second step involves the chemistry of covalent bond formation, while for mechanism-based inactivators, the situation is a bit more complicated, as detailed below. In either case, as with the reversible two-step reactions discussed in Chapter 6, a plot of kobs as a function of [I] will show saturation, reaching a plateau value at high values of [I] (Figure 9.4). Note again that the y-intercept of a plot of kobs vs. [I], for either a saturating or nonsaturating irreversible enzyme inactivator, will be zero, as there is no dissociation of the covalent E–I complex. The data, as in Figure 9.4, can thus be fit to the following equation:
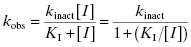
Figure 9.4 Plot of kobs as a function of inactivator concentration for a two-step mechanism of inactivation.
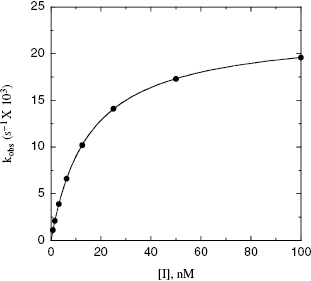
In Equation 9.2, the term kinact defines the maximum rate of inactivation that is achieved at infinite concentration of inactivator, similar to the term kcat in the Michaelis-Menten equation (Chapter 2). At any given concentration of inactivator, one can define the half-life for inactivation as 0.693/kobs (see Appendix 1). Alternatively, one can define a half-life for inactivation at infinite concentration of inactivator as 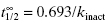 . This latter value is commonly used as a measure of inactivation efficiency. The term KI defines the concentration of inactivator that yields a rate of inactivation that is equal to 1/2 kinact. Again, an analogy with the Michaelis-Menten equation for substrate utilization can be made here, as KI has a meaning for enzyme inactivators that is similar to the meaning of KM for enzyme substrates. As we saw in Chapter 2, it is only under very specific conditions that the kinetic constant KM can be equated with the thermodynamic substrate dissociation constant KS. Likewise the term KI should not be confused with the dissociation constant (Ki = k4/k3) for the initial, reversible encounter complex EI in the two-step reaction schemes of Figure 9.2.
. This latter value is commonly used as a measure of inactivation efficiency. The term KI defines the concentration of inactivator that yields a rate of inactivation that is equal to 1/2 kinact. Again, an analogy with the Michaelis-Menten equation for substrate utilization can be made here, as KI has a meaning for enzyme inactivators that is similar to the meaning of KM for enzyme substrates. As we saw in Chapter 2, it is only under very specific conditions that the kinetic constant KM can be equated with the thermodynamic substrate dissociation constant KS. Likewise the term KI should not be confused with the dissociation constant (Ki = k4/k3) for the initial, reversible encounter complex EI in the two-step reaction schemes of Figure 9.2.
Note that we can divide both sides of Equation (9.2) by [I] to obtain
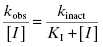
When the concentration of inactivator is far below saturation, such that [I] << KI, the term [I] in the denominator of the right side of Equation (9.3) can be ignored. Under these conditions we obtain
(9.4) 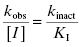
Thus, at concentrations of inactivator well below KI, a plot of kobs as a function of [I] will be linear, and the slope of the line will be equal to kinact/KI. This is exactly the case that we encountered above for nonspecific affinity labeling.
Hence, for any irreversible enzyme inactivator, we can quantify the effectiveness of inactivation using the second-order rate constant kinact/KI. This constant thus becomes the key metric that the medicinal chemist can use in exploring the SAR of enzyme inactivation by a series of compounds. In terms of individual rate constants, the definitions of both kinact and KI depend on the details of the mechanisms of inactivation, as will be described below.
An important point to realize here is that attempts to quantify the relative potency of irreversible enzyme inactivators by more traditional parameters, such as IC50 values, are entirely inappropriate because these values will vary with time, in different ways for different compounds. Hence the SAR derived from IC50 values, determined at a fixed time point in the reaction progress curve, is meaningless and can be misleading in terms of compound optimization. Unfortunately, the literature is rife with examples of this type of inappropriate quantitation of irreversible inactivator potency, making meaningful comparisons with literature data difficult, at best.
9.2 Affinity Labels
An affinity label is a molecule that contains a functionality that is chemically reactive and will therefore form a covalent bond with other molecules containing a complementary functionality. Generally, affinity labels contain electrophilic functionalities that form covalent bonds with protein nucleophiles, leading to protein alkylation or protein acylation. In some cases affinity labels interact selectively with specific amino acid side chains, and this feature of the molecule can make them useful reagents for defining the importance of certain amino acid types in enzyme function. For example, iodoacetate and N-ethyl maleimide are two compounds that selectively modify the sulfur atom of cysteine side chains. These compounds can therefore be used to test the functional importance of cysteine residues for an enzyme’s activity. This topic is covered in more detail below in Section 9.4.
As described here, an affinity label is an inherently reactive molecule and will covalently modify appropriate nucleophiles at any location within the enzyme molecule or external to the enzyme molecule. Because of this general lack of specificity, reactive molecules of this type are referred to as nonspecific affinity labels. Generally, such molecules are not acceptable as drugs, owing to their lack of target specificity. Nevertheless, there are a number of examples of DNA alkylating agents that act as nonspecific affinity labels and that are used clinically in the treatment of some forms of cancer; these include nitrogen mustards, ethylenimines, methanesulfonates, and nitrosoureas.
9.2.1 Quiescent Affinity Labels
The term quiescent affinity label refers to a molecule containing a weak electrophile that does not react generally with nucleophiles in solution at reasonable concentrations. The molecule also contains elements that form reversible interactions with the enzyme active site, so that the compound binds reversibly in the active site with some reasonable affinity. The target enzyme active site must contain a nucleophilic group that is normally involved in catalytic turnover of substrate. In this case the bound compound is sequestered within the solvent-shielded environment of the active site, where the local concentration of the electrophile, and the corresponding active-site nucleophile, are much greater than in general solution. If additionally the reversible binding step orients the weak electrophile appropriately, with respect to the active-site nucelophile, facile reaction can take place. Thus a compound that does not normally undergo nucleophilic attack in solution, can be made to react with a specific nucleophile in the special environment of the enzyme active site. This strategy can impart a good degree of target enzyme selectivity (but generally not specificity) to quiescent affinity labels. For example, aspirin selectively acetylates an active-site serine residue within the substrate binding pockets of COX1 and COX2 to inactivate these enzymes, and thus derive its anti-thrombotic and anti-inflammatory activities. Likewise penicillins, and related β-lactam containing antibiotics, selectively modify the serine hydroxyl group of bacterial peptidoglycan transpeptidases to elicit their antibiotic activity.
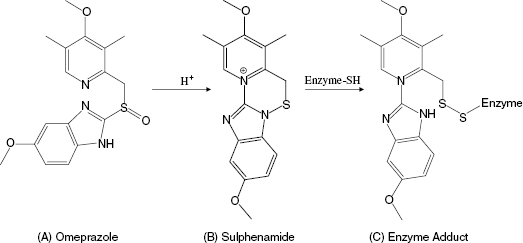
Another interesting, and mechanistically unique, example of quiescent affinity labeling is the H+/K+ ATPase inhibitor omeprazole, which is used to treat peptic diseases, acid reflux disease, and the hypersecretion disease Zollinger-Ellison syndrome. The H+/K+ ATPase is localized to the secretory membranes of parietal cells in the gastric mucosa, where it is responsible for ATP hydrolysis-coupled antiport of potassium ions into the cytosol of the parietal cells and proton transport into the gastric lumen. This cation exchange reaction is the terminal step in gastric acid secretion (Lindberg, 1987). The activity of the gastric H+/K+ ATPase requires the participation of the side chain of a cysteine residue during catalysis. Omeperazole does not inhibit the H+/K+ ATPase in vitro at neutral pH. The compound is weakly basic, so it tends to accumulate in the acid environment of the stomach (Lindberg, 1987). The low pH of the stomach ensures that this is the only location within the body where there is any significant accumulation of omeprazole. Under the acidic conditions of the stomach, omeprazole is quickly (t1/2 ∼ 2 minutes) converted to a sulphenamide that is significantly more reactive with sulphydryl groups, such as cysteine side chains. The high reactivity of the sulphenamide, together with its accumulation in the stomach, result in rapid covalent modification of the catalytically essential cysteine residue of the gastric H+/K+ ATPase (Figure 9.5). The sulphenamide product of omeprazole is cationic, and thus cannot permeate the parietal cell membranes. This minimizes systemic exposure to the compound, enhancing further its in vivo selectivity for covalent modification of the gastric H+/K+ ATPase (Im et al., 1985; Lindberg, 1987). One can reasonably consider omeprazole as an example of a prodrug (see the discussion of enalapril in Chapters 4 and 6).However, the conversion of omeprazole to the active sulphenamide does not result in formation of a reversible enzyme inhibitor, but rather results in in situ formation of a powerful affinity label. Hence we can consider omeprazole to be a unique example of quiescent affinity labeling in which selectivity results from the unique environment of the target enzyme.
Figure 9.5 Chemical transformation of omeprazole to the corresponding sulphenamide under acid conditions and the subsequent modification of an enzyme cysteine residue by the sulphenamide.
More recently attempts to generate highly selective quiescent affinity labels have been made for a number of protease and kinase targets. As examples, inhibitors of the Rhinovirus 3C protease (Mathews et al., 1999) and of the epidermal growth factor receptors (Boschelli, 2002), both incorporating Michael acceptors to covalently inactivate cysteine residues in their target enzymes (Lowry and Richardson, 1981; Figure 9.6), have entered human clinical trials for the treatment of rhinovirus infection and cancer, respectively.
Figure 9.6 A general scheme for reaction of a Michael acceptor with the nucleophilic side chain sulfur of an enzyme cysteine residues.
Rhinoviri are the causal agents of common colds in humans. Viral replication and maturation is dependent on proteolytic processing of a viral polyprotein by a cysteine protease known as 3C protease. The active-site cysteine in 3C protease serves as the attacking nucleophile for catalysis of peptide bond hydrolysis. Covalent modification of this cysteine renders the enzyme irreversibly inactivated.
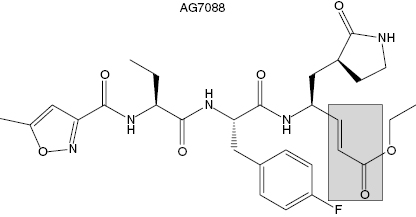
The Agouron (now Pfizer) group solved the crystal structure of the rhinovirus 3C protease and used this structural information to design small peptidic inhibitors of the enzyme. Modest inhibition was observed for a tetrapeptide based on the canonical sequence of the N-terminal side of the polyprotein cleavage sites for 3C protease. When the P1 residue of this inhibitor was appended with an aldehyde group, the active-site cysteine attacked the carbonyl carbon to form a tetrahedral adduct resembling the transition state structure. This compound displayed a Ki of 6 nM for 3C protease. When the aldehyde was reduced to the corresponding alcohol, the peptide showed no significant affinity for the enzyme, indicating that most of the free energy of binding resulted from bond formation with the active-site cysteine. These data implied that it would be very difficult to identify high-affinity inhibitors of the 3C protease based on reversible inhibition modalities. Instead, the Agouron group focused on generating compounds that would irreversibly inactivate the enzyme through modification of the active-site cysteine. They replaced the aldehyde group appended to P1 with an β-unsaturated ethyl ester to impart a Michael acceptor to the inhibitor. Further optimization of the rest of the inhibitor structure led to the final compound, AG7088 (Figure 9.7). The kinetics of AG7088 inactivation of 3C protease was studies in detail (Mathews et al., 1999). The compound was shown to follow a two-step mechanism (scheme B of Figure 9.2) with a second-order rate constant for inactivation (reported as kobs/[I]) of 1.47 × 106 M−1s−1. Crystallization of the AG7088-3C protease binary complex confirmed the expected mode of inactivation. A covalent bond was formed between the β-carbon of the Michael acceptor and Cysteine 147 of the viral protease. Additional favorable binding interactions were observed between the peptidic portion of the inhibitor and the enzyme active site. For example, hydrogen bonds were formed between the prolyl group at P1 in the inhibitor and His161 and Thr142 of the enzyme. Also hydrophobic interactions were observed between the P2 F-phenyl group of the inhibitor and Leu27 and Asn130. Additional interactions also stabilized the final enzyme-inactivator complex (see Mathews et al., 1999, for a more detailed description of these interactions). AG7088 demonstrated good efficacy for inhibiting viral replication in cell culture. The mean IC50 for inhibition of viral replication was 23 nM in HeLa and in MRC-5 cells. No obvious cellular toxicity was seen with the compound at concentrations as high as 100 μM. AG7088 progressed to phase I human clinical trials. The compound was administered intranasally in healthy volunteers either before or after viral challenge. When dosed 5 times per day, starting prior to viral challenge, AG7088 significantly reduced viral shedding. The incidence of colds, total symptom scores, and nasal discharge all showed trends toward reduction in the 5 times per day treated group. AG7088 advanced to phase II trials in first quarter of 2002 (McKinlay, 2001).
Figure 9.7 Chemical structure of the Rhinovirus 3C protease inactivator AG7088. The shaded box highlights the Michael acceptor group within the compound.
Epidermal growth factor receptors (EGFR) represent a family of receptor-based protein kinases. The activity of two members of this protein family, EGFR and ErbB2, has been associated with certain forms of human cancer, particularly with breast cancer where some tumors show augmented expression of these enzymes. A variety of strategies have been put forth to block the activity of these kinases, including antibody-based therapies, receptor antagonists, reversible small molecule kinase inhibitors, and irreversible kinase inactivators. Groups at Parke-Davis and Wyeth (both now Pfizer) have independently incorporated a Michael acceptor into compounds that bind to the ATP binding pocket of EGFR to covalently associate with Cys 773 within this pocket. The Wyeth compound, EKB-569 (Figure 9.8A), irreversibly inhibits EGFR in vitro and in cells. No information on the kinetics of irreversible inactivation were reported, but the apparent IC50 for inhibiting the autophosphorylation of EGFR and ErbB2 was reported to be 83 nM and 1.23 μM, respectively (Wissner et al., 2003). In human A431 tumor cells, which overexpress EGFR, cell proliferation was also inhibited by EKB-569 with an IC50 of around 80 nM. The compound has also been tested in a nude mouse xenograft model in which A431 human tumor cells were implanted on day zero. Tumors began to grow and the mice were treated for 10 days with an oral dose of EKB-569 of 10 mg/kg. Efficacy was quantified by measuring the median tumor mass in the treated animals and in the control animals. The ratio of these median values, multiplied by 100 yields a percentage value referred to as the T/C ratio. At the end of the dosing interval, the T/C ratio for EKB-569 treatment was 14%. When the dose was increased to 40 mg/kg for 10 days, efficacy could be extended well beyond the dosing interval. At this dose a T/C ratio of 20% was realized 18 days after stopping drug treatment (Tsou et al., 2001; Boschelli, 2002). This compound is thought to be selective for EGFR and ErbB2; however, it has been demonstrated that the compound forms covalent adducts with the reduced form of glutathione. Hence the potential for off-target effects of EKB-569 cannot be discounted (Tsou et al., 2001). Nevertheless, the compound has entered human clinical trials for the treatment of cancer.
Figure 9.8 Chemical structures of the EGRF protein kinase inactivators EKB-569 (A) and CI-1033 (B). The shaded boxes highlight the Michael acceptor group within each compound.
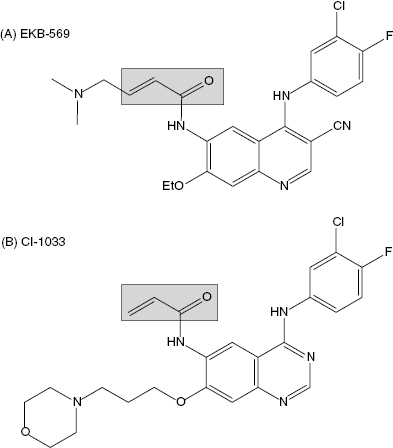
The Pfizer compound CI-1033 (Figure 9.8B) uses an essentially identical strategy to inactivate EGFR and ErbB2. Again, no report of kinetics of inactivation has been published for this compound, but the reported IC50 for inhibition of EGFR in solution is 1.5 nM. Cellular assays of EGFR and ErbB2 autophosphorylation demonstrate inhibition by CI-1033 with IC50 values of 7.4 and 9.0 nM, respectively. This compound was also tested in a nude mouse A431 human tumor xenograft model. When dosed orally at 5 mg/kg on days 10 to 24 (after tumor implantation) the compounds demonstrated a T/C ratio of 4%. The difference in time for tumors to reach a mass of 750 mg was delayed by 53.2 days in the CI-1033 treated animals, relative to the control animals (Small et al., 2000). No information on off-target covalent adduct formation has been reported for CI-1033. This compound has also entered human clinical trials for the treatment of cancer (Bonomi, 2003). In phase I studies CI-1033 was shown to have an acceptable side effect profile. Evidence of antitumor activity was observed in some patients, and biomarker studies also indicated that inhibition of the target (EGFR) was being achieved in patients (Allen et al., 2003). The compound has now advanced to phase II studies.
9.2.2 Potential Liabilities of Affinity Labels as Drugs
The lack of target enzyme specificity is a critical liability for the use of affinity labels as drugs. The inherent chemical reactivity of these compounds almost ensures that one will see reaction with proteins and peptides other than the target enzyme, and these off-target relativities will often translate into acute or cumulative toxicities. There is no question that nonspecific affinity labels should generally be avoided for human clinical use, with the possible exception of acute cancer chemotherapy (see above). Much of past cancer chemotherapy has been focused on acute treatments, where moderate adverse events could be accepted. Today, however, the goal of most research efforts in cancer therapeutics is to improve efficacy, minimize adverse events, and prevent disease progression, so that patients can tolerate longer durations of drug treatment. Hence the use of nonspecific affinity labels in cancer treatment is likely to diminish significantly.
TABLE 9.1 Some Examples of Quiescent Affinity Labels of Clinical Interest
| Compound or Compound Class | Enzyme Target | Clinical Indication |
|---|---|---|
| Aspirin | COX1 and COX2 | Anti-thrombosis, anti-inflammatory |
| Penicillins | Bacterial peptidoglycan transpeptidases | Antibiotic |
| Cephalosporins | Bacterial peptidoglycan transpeptidases | Antibiotic |
| Penems | Bacterial peptidoglycan transpeptidases | Antibiotics |
| CI-1033 | ErbB1 and ErbB2 | Cancer |
| EKB-569 | ErbB1 and ErbB2 | Cancer |
| AG7088 | Rhinovirus 3C protease | Common colds |
We have seen in Section 9.2.1 that greater target selectivity can be achieved with quiescent affinity labels, and there are a number of examples of the use of such agents in human medicine. These precedences notwithstanding, drug design strategies based on quiescent affinity labeling nevertheless bring added potential safety risks. Quiescent affinity labels remain chemically reactive species, even though their reactivity is attenuated relative to nonspecific affinity labels. Hence careful testing often reveals off-target relativities for these compounds. Often the rate of covalent modification is much faster for the target enzyme than for off-target proteins, and this can give the researcher a false sense of therapeutic index. Especially for drugs that are intended for long-term, chronic therapies, seemingly minor side reactions can lead to cumulative effects that can significantly erode the utility of a drug. It must also be recognized that one cannot screen compounds for reactivity against all proteins and peptides that are likely to be encountered in vivo. Hence one only knows the selectivity with respect to those proteins that the compound is tested against. Given the already high rate of compound attrition in drug development, it is, in my opinion, seldom worth the risk of discovering a safety issue late in compound development, due to incorporation of chemically reactive functionalities into a drug molecule.
One of the more difficult to manage aspects of compound reactivity is the potential for idiosyncratic immunological reactions to covalent protein-compound complexes. Normally the immune system does not respond to xenobiotics of molecular weight less than 1000 Daltons. When, however, a drug is covalently linked to a protein, the immune system may now recognize the modified protein as foreign and therefore mount an immunological response. Immune responses to protein–drug conjugates can range from acute anaphylaxis to hemolytic anemia to less acute systemic and organ-specific tissue damage (Naisbitt et al., 2000, 2001). This can be an issue even for some nonreactive drug molecules, where metabolic bioactivation can produce reactive species that go on to covalently modify proteins and subsequently elicit an immune response. In most patients, bioactivation and detoxification mechanisms (see Chapter 10) are in good balance, so that a buildup of protein–drug conjugates and the subsequent immune response are not seen. In a small fraction of patients, however, severe reactions can be seen. Most disturbingly, it is very difficult to predict whether or not a nonreactive drug molecule will elicit such an idiosyncratic immune response, and if so, in what fraction of the patient population. However, some progress has been made in identifying specific chemical functionalities that appear to be problematic in this regard (Uetrecht, 2003). When the parent compound is itself chemically reactive, the problem of protein–drug conjugate formation is significantly exacerbated. For example, antibiotics that contain β-lactams, such as the penicillins, are known to form protein conjugates with human serum albumin and other proteins. These protein conjugates are thought to be the progenitors of the range of immune-based adverse reactions that are seen in some patients after treatment with these drugs. Thus the uncertainty of formation of protein–drug conjugates with quiescent affinity labels, which may subsequently elicit an immunological reaction, generally makes these compounds far less desirable than noncovalent, tight binding inhibitors for clinical use.
9.3 Mechanism-Based Inactivators
Mechanism-based inactivators are generally defined as compounds that are transformed, by the catalytic machinery of the enzyme, into a species that acts as (1) an affinity label, (2) a transition state analogue, or (3) a very tight binding reversible inhibitor, prior to release from the enzyme active site (see Szewczuk et al., 2004, for an interesting example of another form of mechanism-based inactivation, the redox-mediated “hit-and-run” inactivation of COX1 by resveratol). Thus the chemical entity added to the enzyme sample is not inherently reactive and is also not, per se, an inhibitor of the enzyme. Rather, the molecule is recognized by the enzyme as an alternative substrate that is acted upon by groups within the active site to catalytically generate the inhibitory species. Because mechanism-based inactivators rely on the chemistry of enzyme turnover, they must bind within the active site of the enzyme. Hence all mechanism-based inactivators are competitive with the normal substrate of the enzyme. For these reasons mechanism-based inactivators are also commonly referred to as suicide substrates (Abels and Maycock, 1976; Walsh, 1978; Silverman, 1992).
Owing to their reliance on enzyme catalysis to generate the inhibitory species, mechanism-based inactivators can be very specific for the target enzyme, or at the very least, highly selective for a family of enzymes that catalyze a common reaction. Hence molecules that act through this type of inactivation mechanism can be quite useful in human medicine.
A kinetic scheme for mechanism-based inactivation is illustrated in Figure 9.2C, which specifically illustrates the more common case of mechanism-based inactivation leading to formation of an affinity label and subsequent covalent modification of the target enzyme. In this mechanism the parent compound binds in a reversible fashion to the enzyme to form an initial encounter complex, EI. This is exactly analogous to formation of ES in the normal catalytic reaction of the enzyme. The bound compound is then chemically transformed by the catalytic machinery of the enzyme to form the inhibitory species A (for Affinity label), still within the context of a binary complex, EA. The rate constant k5 then is the forward rate constant for the catalytic conversion of I to A within the enzyme active site. As with normal substrates of an enzyme, each reaction step displays microreversibility; hence there is some possibility of the reverse reaction, going from EA to EI. This reverse reaction is governed by the rate constant k6, which is almost always exceedingly small under laboratory and most physiological conditions. Once the binary EA complex has been formed, there are two potential fates for the complex. The affinity label can stay bound to the enzyme active site and react with an enzyme nucleophile to form a covalent species E–A that is irreversibly inactivated. Alternatively, the newly formed species A can dissociate from the enzyme to reform the free enzyme and the free affinity label, A. The released species A can then rebind to the enzyme to inactivate it, but this would not be considered mechanism-based inactivation, as part of the definition of mechanism-based inactivation requires inactivation prior to compound release from the enzyme (see above). Thus the efficiency of a compound as a mechanism-based inactivator depends on the relative rates of covalent modification and inactivator dissociation subsequent to formation of the EA complex. The ratio of A released to inactivation is defined by the ratio of first-order rate constants k8/k7. This ratio is given the symbol r, and is referred to as the partition ratio. The partition ratio is used as a quantitative measure of efficiency for mechanism-based inhibitor. When the EA complex goes on to inactivate the enzyme with 100% efficiency (i.e., there is no release of A from the complex), r = 0. The closer the value of r is to zero, the more efficient the mechanism-based inactivator is in irreversibly modifying the enzyme. There are some reported cases of compounds for which r = 0, so that every turnover event leading to EA formation also leads to inactivation (e.g., see Silverman and Invergo, 1986). Experimental methods for determining the partition ratio are described later in this chapter.
The value of kobs for this type of mechanism is a saturable function of [I], as was the case for quiescent affinity labels (vide supra). For this mechanism, kinact (as defined above) is a complex mixture of rate constants:
(9.5) 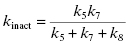
Only when k5 is rate-limiting can we equate kinact with k5. Likewise KI has a complex form for mechanism-based inactivation:
(9.6) 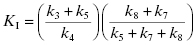
Hence, as we saw with quiescent affinity labels, we must treat KI as a kinetic constant, not an equilibrium constant. Only in the situation that both k3 and k4 are very large (i.e., rapid equilibrium) and k5 is rate-limiting, can we equate KI with Ki. If, for example, k7 is even partially rate-limiting, KI > Ki, hence the two constants have different meanings.
Despite the mechanistic differences in the definitions of kinact and KI between quiescent affinity labels and mechanism-based inactivators, the dependence of kobs on [I] is the same for both mechanism. Hence we cannot determine whether or not a compound is acting as a mechanism-based inhibitor, based merely on this two-step kinetic behavior. However, there is a set of distinguishing features of mechanism-based inactivation that are experimentally testable. Compounds that display all of these features can be safely defined as mechanism-based inactivators.
9.3.1 Distinguishing Features of Mechanism-Based Inactivation
Several authors have discussed specific criteria for designating a compound as a mechanism-based inactivator. Abels and Maycock (1976) and Walsh (1978) were among the first to set out specific experimental tests for mechanism-based inactivation. More recently Silverman (1988, 1992, 1995) has described a comprehensive set of seven distinguishing features that mechanism-based inactivators must display. These are described here.
Inhibition Must Be Time Dependent
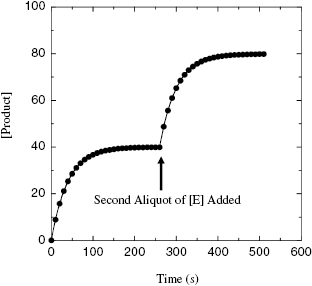
This criterion should be obvious from our previous discussions. Nevertheless, it is critical that one experimentally verify that there is a slow onset of inhibiton for the compound, using the experimental methods described in Chapter 6. Related to this, one must also demonstrate that the observed curvature in the progress curves is due to inactivation of the enzyme, and not due to substrate depletion or other artifacts of the experimental design. One simple test that can be used in this regard is to allow the progress curve, in the presence of a large excess of inhibitor over enzyme concentration, to go to a plateau (as in Figure 9.1) and to then add another aliquot of enzyme to the reaction mixture. If the plateau in the original progress curve was attained due to depletion of active enzyme through inactivation, then addition of the new aliquot of enzyme should reinitiate the reaction, and further product formation should be observed (Figure 9.9). If the inactivation is mechanism based, this new aliquot of enzyme should be inactivated at the same rate as was the first aliquot, as described later in this section.
Figure 9.9 Reaction progress curve in the presence of a mechanism-based inactivator when a second aliquot of enzyme is added to the reaction solution. The reaction is allowed to reach a plateau before a second, equal concentration aliquot of enzyme is added at the indicated time point. Note that the rate of inactivation for this second aliquot of enzyme is the same as that seen in the initial progress curve.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


