Although enzymes are essential for life, dysregulated enzyme activity can also lead to disease states. In some cases mutations in genes encoding enzymes can lead to abnormally high concentrations of the enzyme within a cell (overexpression). Alternatively, point mutations can lead to an enhancement of the specific activity (i.e., catalytic efficiency) of the enzyme because of structural changes in the catalytically critical amino acid residues. By either of these mechanisms, aberrant levels of the reaction product’s formation can result, leading to specific pathologies. Hence human enzymes are also commonly targeted for pharmacological intervention in many diseases.
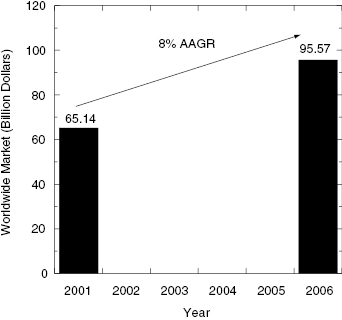
Enzymes, then, are attractive targets for drug therapy because of their essential roles in life processes and in pathophysiology. Indeed, a survey reported in 2000 found that close to 30% of all drugs in clinical use derive their therapeutic efficacy through enzyme inhibition (Drews, 2000). More recently Hopkins and Groom (2002) updated this survey to include newly launched drugs and found that nearly half (47%) of all marketed small molecule drugs inhibit enzymes as their molecular target (Figure 1.1). Worldwide sales of small molecule drugs that function as enzyme inhibitors exceeded 65 billion dollars in 2001, and this market was expected to grow to more than 95 billion dollars by 2006 (see Figure 1.2). Some contraction of the worldwide market has occurred due to withdrawal of several products since 2005. Revised forecasts suggest that the worldwide market will now grow at a rate of about 6.7% as of 2005 (Business Communications Company, Inc., 2006, “Enzyme Inhibitors with Broad Therapeutic Application”).
Figure 1.1 Distribution of marketed drugs by biochemical target class. GPCRs = G-Protein coupled receptors.
Source: Redrawn from Hopkins and Groom (2002).
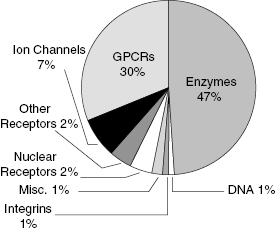
Figure 1.2 Worldwide market for small molecule drugs that function as enzyme inhibitors in 2001 and projected for 2006. AAGR = average annual growth rate.
Source: Business Communications Company, Inc. Report RC-202R: New Developments in Therapeutic Enzyme Inhibitors and Receptor Blockers, www.bccresearch.com.
The attractiveness of enzymes as drug targets results not only from the essentiality of their catalytic activity but also from the fact that enzymes, by their very nature, are highly amenable to inhibition by small molecular weight, drug-like molecules. Because of this susceptibility to inhibition by small molecule drugs, enzymes are commonly the target of new drug discovery and design efforts at major pharmaceutical and biotechnology companies today; my own informal survey suggests that between 50 and 75% of all new drug-seeking efforts at several major pharmaceutical companies in the United States are focused on enzymes as primary targets.
While the initial excitement generated by the completion of the Human Genome Project was in part due to the promise of a bounty of new targets for drug therapy, it is now apparent that only a portion of the some 30,000 proteins encoded for by the human genome are likely to be amenable to small molecule drug intervention. A recent study suggested that the size of the human “druggable genome” (e.g., human genes encoding proteins that are expected to contain functionally necessary binding pockets with appropriate structures for interactions with drug-like molecules) is more on the order of 3000 target proteins (i.e., about 10% of the genome), a significant portion of these being enzymes (Hopkins and Groom, 2002). As pointed out by Hopkins and Groom, just because a protein contains a druggable binding pocket does not necessarily make it a good target for drug discovery; there must be some expectation that the protein plays some pathogenic role in disease so that inhibition of the protein will lead to a disease modification. Furthermore the same study estimates that of the nearly 30,000 proteins encoded by the human genome, only about 10% (3000) can be classified as “disease-modifying genes” (e.g., genes that, when knocked out in mice, effect a disease-related phenotype). The intersection of the druggable genome and the disease-modifying genome thus defines the number of bona fide drug targets of greatest interest to pharmaceutical scientists. This intersection, according to Hopkins and Groom (2002), contains only between 600 and 1500 genes, again with a large proportion of these genes encoding for enzyme targets.
The “druggability” of enzymes as targets reflects the evolution of enzyme structure to efficiently perform catalysis of chemical reactions, as discussed in the following section.
1.2 Enzyme Structure and Catalysis
From more than a thousand years of folk remedies and more recent systematic pharmacology, it is well known that compounds that work most effectively as drugs generally conform to certain physicochemical criteria (Table 1.2). To be effective in vivo, molecules must be absorbed and distributed, usually permeate cell membranes to reach their molecular targets, and be retained in systemic circulation for a reasonable period of time (i.e., pharmacokinetic residence time). These and other necessary biological features of small molecule drugs are dictated by the physicochemical nature of the drug molecules. Over the years there have been a number of published surveys that relate specific physicochemical properties of small molecules to their utility as therapeutic agents (Ajay et al., 1998; Lipinski et al., 2001; Veber et al., 2002; Vieth et al., 2004; Keller et al., 2006). With respect to orally administered small molecule drugs, a specific set of physicochemical features is commonly articulated as important for success; these are summarized in Table 1.2. Generally, drug molecules need to be relatively small, with molecular weights less than 1000 Da and preferably less than or equal to 500 Da. Drug molecules are generally hydrophobic, but very often contain polarizable groups at precise locations within the molecule. Hence, drug molecules typically contain a number of specifically oriented heteroatoms and hydrogen-bond donors (for more details on chemical features of drug-like molecules, see Ajay et al., 1998; Lipinski et al., 1997; Veber et al., 2002). Note that these “rules” of chemical structure for drug molecules are significantly relaxed, and sometimes altered completely in the case of natural products (Clardy and Walsh, 2004). Nevertheless, even in the case of natural products, target binding affinity and in vivo delivery are dictated largely by specific physicochemical properties of the drug molecule.
TABLE 1.2 Some Physicochemical Properties of Drug-like Molecules
Sources: Data from Lipinski et al. (2001), Veber et al. (2002), and Keller et al. (2006).
| Molecular Property | Typical Value |
|---|---|
| Molecular weight | ≤500 Da |
| cLog(P) | ≤5 |
| Number of H-bond donors | ≤5 |
| Sum of N and O atoms | ≤10 |
| Polar surface area | ≤140 Å2 |
| Rotatable bonds | ≤10 |
Accepting the premise that drug molecules conform to specific stereochemical, electrostatic, hydrophobic, and other physiochemical properties, it follows that drug targets must contain binding pockets for these molecules that demonstrate structural and electronic complementarity to the small molecule drugs. Thus a “druggable target” is one that contains a “druggable binding pocket” as part of its three-dimensional structure, and a druggable binding pocket conforms to specific structural and chemical requirements.
The features that make a binding pocket on a protein “druggable” have been reviewed by several authors (Liang et al., 1998; Hajduk et al., 2005). Generally, drug binding pockets are cavities or clefts along the protein surface, with small molecular volumes (relative to that of the entire protein) of around 1000 Å3 (Liang et al., 1998). Estimates of the volume relationship between a ligand binding pocket and the overall protein have suggested that the ligand binding pocket constitutes around 1–5% of the total volume of the protein molecule (Liang et al., 1998). Drug binding pockets tend to display a large surface area to volume ratio, a factor referred to as surface roughness (Pettit and Bowie, 1999) and which reflects the stereochemical uniqueness of the binding pockets; by having a large surface area to volume ratio, the potential for favorable van der Waals interactions between the pocket and ligand is enhanced.
Ligand binding pockets are usually designed to exclude bulk solvent, and are generally composed of hydrophobic amino acids. Nevertheless, the pockets may contain highly ordered water molecules, incorporated as part of a specific architectural motif to participate in ligand interactions (see, for example, Figure 1.5). This exclusion of bulk water favors the formation of stronger hydrogen bonds and other electrostatic interactions between the protein and the ligand. Complementary to the drug molecules themselves, these pockets also often contain specific loci for hydrogen bonding, salt bridge formation, and other noncovalent, electrostatic interactions between the binding partners. The combination of electrostatic determinants of binding, the general hydrophobicity of the pockets, and surface roughness make for significant surface complexity in drug binding pockets (Hajduk et al., 2005).
Druggable binding pockets on protein surfaces have largely evolved to bind physiologically relevant small molecular weight ligands, such as nucleotide analogs (e.g., ATP, GTP, NADH), amino acids, steroid hormones, metabolites, peptides, cofactors (e.g., flavins, hemes), and the like. The interactions of these natural ligands with the protein binding site typically effects a change in the biological activity of the target protein. For example, binding of the physiologic agonist (a ligand that stimulates the biological activity of a receptor) to a G-protein coupled receptor (GPCR) on the surface of a cell elicits a conformational transition of the receptor, often leading to post-translational modification of cytosolic domains of the receptor protein. These post-translational modifications lead to recruitment and/or activation of various proteins, thus initiating cellular signal transduction cascades that are critical for a number of cellular activities, such as cell proliferation, mobility, and programmed cell death.
In the organism, the extent and duration of signal transduction—hence the interactions between the receptor and ligand—need to be responsive to the changing needs and environment of the cell. This need for facile responsiveness at the receptor level is facilitated by three characteristics of protein interactions with physiologic ligands:
All of these properties are dicated by equilibrium binding between the protein receptor and the ligand, as discussed in more detail in Chapters 2 and 3 and Appendix 2. Hence, the elements of molecular recognition between proteins and their physiologic ligands are largely mediated through the cumulative effects of multiple, weak, reversible chemical forces, such as hydrogen bonds, salt bridges, van der Waals forces, and hydrophobic forces (Copeland, 2000). This is exemplified in Figure 1.5 where we illustrate the collective interactions between the enzymatic active site of dihydrofolate reductase and its substrate dihydrofolate. These same weak, noncovalent chemical forces typically also form the structural determinants of interaction between protein binding sites and drug molecules; this is also exemplified in Figure 1.5 where we see the same types of chemical interactions forming between the enzymatic active site of dihydrofolate reductase and the drug methotrexate.
Thus, the best molecular targets for drug intervention are those containing a relatively small volume, largely hydrophobic binding pocket that is polarized by specifically oriented loci for hydrogen bonding and other electrostatic interactions and that is critical for the biological function of the target (Liang et al., 1998). These criteria are well met by the structures of enzyme active sites and additional regulatory allosteric binding sites on enzyme molecules.
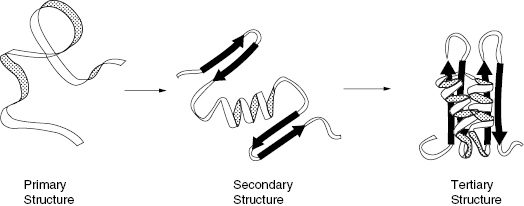
The vast majority of biological catalysis is performed by enzymes, which are proteins composed of polypeptide chains of amino acids (natural peptide synthesis at the ribosome, and a small number of other biochemical reactions are catalyzed by RNA molecules, though the bulk of biochemical reactions are catalyzed by protein-based enzymes). These polypeptide chains fold into regular, repeating structural motifs of secondary (alpha helices, beta pleated sheets, hairpin turns, etc.) and tertiary structures (see Figure 1.3). The overall folding pattern, or tertiary structure of the enzyme, provides a structural scaffolding that presents catalytically essential amino acids and cofactors in a specific spacial orientation to facilitate catalysis. As an example, consider the enzyme dihydrofolate reductase (DHFR), a key enzyme in the biosynthesis of deoxythymidine and the target of the antiproliferative drug methotrexate and the antibacterial drug trimethoprim (Klebe, 1994; Copeland, 2000). The bacterial enzyme has a molecular weight of around 180,000 (162 amino acid residues) and folds into a compact globular structure composed of 10 strands of beta pleated sheet, 7 alpha helices, and assorted turns and hairpin structures (Bolin et al., 1982). Figure 1.4 shows the overall size and shape of the enzyme molecule and illustrates the dimensions of the catalytic active site with the inhibitor methotrexate bound to it. We can immediately see that the site of chemical reactions—that is, the enzyme active site—constitutes a relatively small fraction of the overall volume of the protein molecule (Liang et al., 1998). Again, the bulk of the protein structure is used as scaffolding to create the required architecture of the active site. A more detailed view of the structure of the active site of DHFR is shown in Figure 1.5, which illustrates the specific interactions of active site components with the substrate dihydrofolate and with the inhibitor methotrexate. We see from Figure 1.5 that the active site of DHFR is relatively hydrophobic, but contains ordered water molecules and charged amino acid side chains (e.g., Asp 27) that form specific hydrogen bonding interactions with both the substrate and inhibitor molecules.
Figure 1.3 Folding of a polypeptide chain illustrating the hierarchy of protein structure from primary structure through secondary structure and tertiary structure.
Source: From Copeland (2000).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


