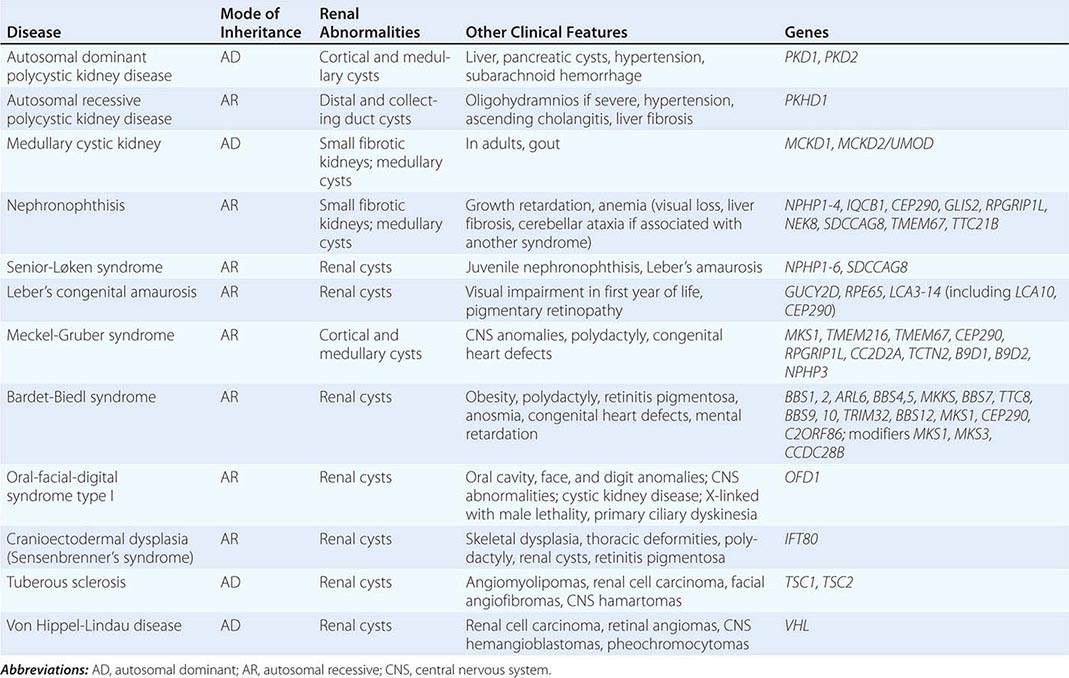
339 | Polycystic Kidney Disease and Other Inherited Disorders of Tubule Growth and Development |
The polycystic kidney diseases are a group of genetically heterogeneous disorders and a leading cause of kidney failure. The autosomal dominant form of polycystic kidney disease (ADPKD) is the most common life-threatening monogenic disease, affecting 12 million people worldwide. The autosomal recessive form of polycystic kidney disease (ARPKD) is rarer but affects the pediatric population. Kidney cysts are often seen in a wide range of syndromic diseases. Recent studies have shown that defects in the structure or function of the primary cilia may underlie this group of genetic diseases collectively termed ciliopathies (Table 339-1).
INHERITED DISEASES COMMONLY ASSOCIATED WITH A CYSTIC PHENOTYPE |
AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE
Etiology and Pathogenesis (Fig. 339-1) ADPKD is characterized by progressive formation of epithelial-lined cysts in the kidney. Although cysts only occur in 5% of the tubules in the kidney, the enormous growth of these cysts ultimately leads to the loss of normal surrounding tissues and loss of renal function. The cellular defects in ADPKD that have been known for a long time are increased cell proliferation and fluid secretion, decreased cell differentiation, and abnormal extracellular matrix. ADPKD is caused by mutations in PKD1 and PKD2, which, respectively, code for polycystin-1 (PC1) and polycystin-2 (PC2). PC1 is a large 11-transmembrane protein that functions like a G protein–coupled receptor. PC2 is a calcium-permeable six-transmembrane protein that structurally belongs to the transient receptor potential (TRP) cation channel family. PC1 and PC2 are widely expressed in almost all tissues and organs. PC1 expression is high in development and low in the adult, whereas PC2 expression is relatively constant. PC1 and PC2 are found on the primary cilium, a hair-like structure present on the apical membrane of a cell, in addition to the cell membranes and cell-cell junctions of tubular epithelial cells. Defects in the primary cilia are linked to a wide spectrum of human diseases, collectively termed ciliopathies. The most common phenotype shared by many ciliopathies is kidney cysts. PC1 and PC2 bind to each other via their respective C-terminal tails to form a receptor-channel complex and regulate each other’s function. The PC1/2 protein complex serves as a mechanosensor or chemical sensor and regulates calcium and G-protein signaling. The PC1/2 protein complex may also directly regulate a number of cellular functions including the cell cycle, the actin cytoskeleton, planar cell polarity (PCP), and cell migration. This protein complex has also been implicated in regulating a number of signaling pathways, including Wnt, mammalian target of rapamycin (mTOR), STAT3, cMET, phosphoinositide 3-kinase (PI3K)/AKT, G protein–coupled receptor (GPCR), and epidermal growth factor receptor (EGFR), as well as in the localization and activity of cystic fibrosis transmembrane conductance (CFTR). One hypothesis is that loss of ciliary function of PC1 and PC2 leads to reduced calcium signaling and a subsequent increase of adenylyl cyclase activity and decrease of phosphodiesterase activity, which, in turn, causes increased cellular cyclic AMP (cAMP). Increased cAMP promotes protein kinase A activity, among other effectors, and, in turn, leads to cyst growth by promoting proliferation and fluid secretion of cyst-lining cells through chloride and aquaporin channels in ADPKD kidneys.
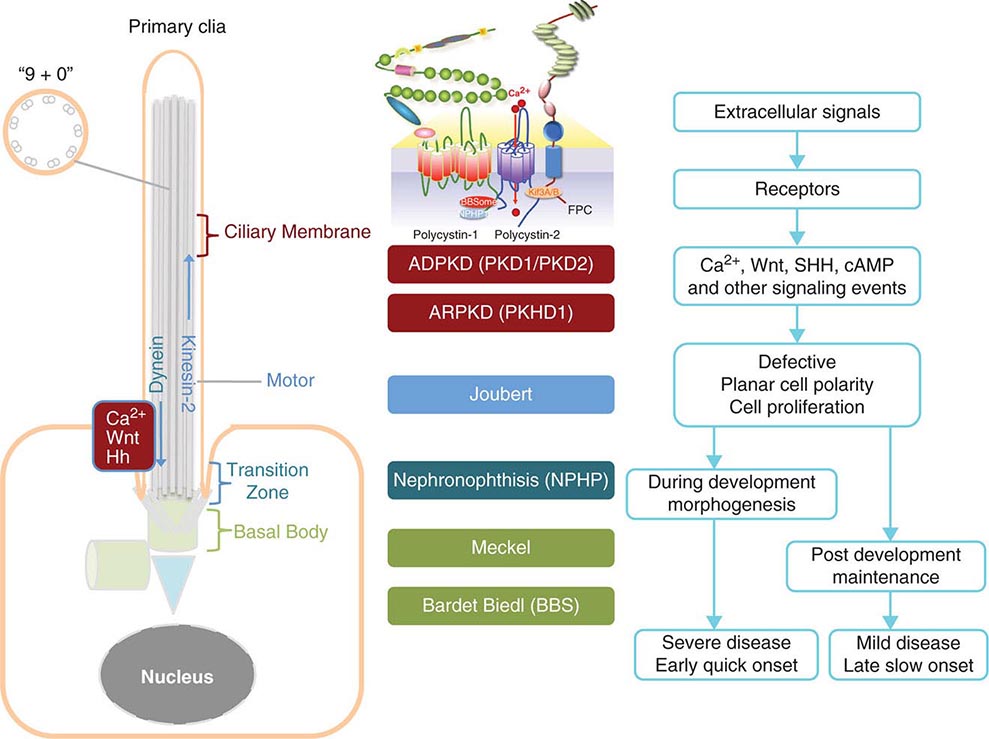
FIGURE 339-1 Scheme of the primary cilium and cystic kidney disease proteins. Left. A scheme of the primary cilium. Primary cilia share a “9+0” organization of microtubule doublets. Proteins are transported into the cilium by motor protein kinesin 2 and transported out of the cilium by dynein. The cilium is connected to the basal body through the transition zone. Middle. Topology of autosomal dominant polycystic kidney disease (ADPKD) and autosomal recessive polycystic kidney disease (ARPKD) proteins polycystin-1, polycystin-2, and fibrocystin/polyductin (FPC) are shown. PC1 also interacts with other proteins such as components of the BBSome and NPHP1. PC2 and FPC both interact with kinesin 2 (KIF 3A/B). Localization of disease proteins in the cilium, the transition zone, and the basal body is color coded. Right. Potential disease mechanisms due to cilium-mediated signaling events.
![]() Genetic Considerations ADPKD is inherited as an autosomal dominant trait with complete penetrance but variable expressivity. The disease affects all ethnic groups worldwide with an estimated prevalence of 1:1000 to 1:400. Only half of the patients with ADPKD are clinically diagnosed during their lifetime. ADPKD is genetically heterogeneous. The first disease gene (PKD1) was localized to the region of the α-globin gene on chromosome 16p13 in 1985, and a second disease gene (PKD2) locus was mapped to chromosome 4q21-q23 in 1993. Mutations of PKD1 and PKD2 are responsible for ~85% and ~15% of ADPKD cases, respectively. However, patients with PKD2 mutations may be higher than 15% because they tend to have milder clinical disease and, as a result, may be underdiagnosed. Embryonic lethality of Pkd1 and Pkd2 knockout mice suggests that human homozygotes may be lethal and thus not clinically recognized.
Genetic Considerations ADPKD is inherited as an autosomal dominant trait with complete penetrance but variable expressivity. The disease affects all ethnic groups worldwide with an estimated prevalence of 1:1000 to 1:400. Only half of the patients with ADPKD are clinically diagnosed during their lifetime. ADPKD is genetically heterogeneous. The first disease gene (PKD1) was localized to the region of the α-globin gene on chromosome 16p13 in 1985, and a second disease gene (PKD2) locus was mapped to chromosome 4q21-q23 in 1993. Mutations of PKD1 and PKD2 are responsible for ~85% and ~15% of ADPKD cases, respectively. However, patients with PKD2 mutations may be higher than 15% because they tend to have milder clinical disease and, as a result, may be underdiagnosed. Embryonic lethality of Pkd1 and Pkd2 knockout mice suggests that human homozygotes may be lethal and thus not clinically recognized.
PKD1 is comprised of 46 exons occupying ~52 kb of genomic DNA. It produces an ~14-kb transcript that encodes PC1, a protein of ~4300 amino acids. A feature of the PKD1 gene is that the 5´ three-quarters of PKD1 have been duplicated at six other sites on chromosome 16p, and many of them produce mRNA transcripts, which provides a major challenge for genetic analysis of the duplicated region. PKD2 is a single-copy gene with 15 exons producing an ~5.3-kb mRNA transcript that encodes PC2, a protein of 968 amino acids. The presence of additional genes for ADPKD was suggested based on several families linked to neither PKD1 nor PKD2 genes. However, careful analyses have excluded the existence of a third ADPKD gene.
In ADPKD patients, every cell carries a germline mutant allele of either PKD1 or PKD2. However, cysts develop in only a small fraction of the nephrons. Cysts are thought to originate from clonal growth of single cells that have received a somatic “second hit” mutation in the “normal” allele of the PKD1 or PKD2 gene. Accumulating evidence in mouse models now shows that partial loss of function of the second allele of Pkd1 in a proliferative environment is sufficient for cystogenesis, suggesting that a critical amount of PKD1 is needed in a cell. Somatic inactivation of the second allele of Pkd1 in adult mice results in very slow onset of cyst development in the kidney, but a “third hit,” such as an additional genetic or epigenetic event, the inactivation of a growth-suppressor gene, the activation of a growth-promoting gene(s), or an event like renal injury that activates the developmental program, may promote rapid cyst formation.
Clinical Manifestations ADPKD is characterized by the progressive bilateral formation of renal cysts. Focal renal cysts are typically detected in affected subjects before 30 years of age. Hundreds to thousands of cysts are usually present in the kidneys of most patients in the fifth decade (Fig. 339-2). Enlarged kidneys can each reach a fourfold increase in length and weigh up to 20 times the normal weight. The clinical presentations of ADPKD are highly variable. Although many patients are asymptomatic until the fourth to fifth decade of life and are diagnosed by incidental discoveries of hypertension or abdominal masses, back or flank pain is a frequent symptom in ~60% of patients with ADPKD. The pain may result from renal cyst infection, hemorrhage, or nephrolithiasis. Gross hematuria resulting from cyst rupture occurs in ~40% of patients during the course of their disease, and many of them will have recurrent episodes. Flank pain and hematuria may coexist if the cyst that ruptures is connected with the collecting system. Proteinuria is usually a minor feature of ADPKD. Infection is the second most common cause of death for patients with ADPKD. Up to half of patients with ADPKD will have one or more episodes of renal infection during their lifetime. An infected cyst and acute pyelonephritis are the most common renal infections often due to gram-negative bacteria, which are associated with fever and flank pain, with or without bacteremia. These complications and renal insufficiency often correlate with structural abnormality of the renal parenchyma. Kidney stones occur in ~20% of patients with ADPKD. Different from the general population, more than half of the stones in patients with ADPKD are composed of uric acid, with the remainder due to calcium oxalate. Distal acidification defects, abnormal ammonium transport, low urine pH, and hypocitraturia may be important in the pathogenesis of renal stones in ADPKD. Renal cell carcinoma is a rare complication of ADPKD with no apparent increased frequency compared to the general population. However, in ADPKD, these tumors are more often bilateral at presentation, multicentric, and sarcomatoid in type. Radiologic imaging is often not helpful in distinguishing cyst infection and cyst hemorrhage because of their complexity. Computed tomography (CT) scan and magnetic resonance imaging (MRI) are often useful in distinguishing a malignancy from a complex cyst. Cardiovascular complications are the major cause of mortality in patients with ADPKD. Hypertension is common and typically occurs before any reduction in glomerular filtration rate (GFR). Hypertension is a risk factor for both cardiovascular and kidney disease progression in ADPKD. Notably, some normotensive patients with ADPKD may also have left ventricular hypertrophy. Hypertension in ADPKD may result from the increased activation of the renin-angiotensin-aldosterone system, increased sympathetic nerve activity, and impaired endothelial cilium function-dependent relaxation of small resistant blood vessels.
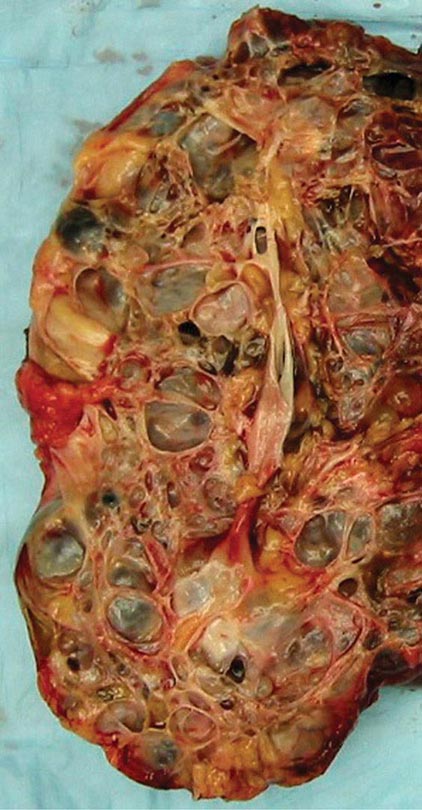
FIGURE 339-2 Photograph showing a kidney from a patient with autosomal dominant polycystic kidney disease. The kidney has been cut open to expose the parenchyma and internal aspects of cysts.
The progression of ADPKD has striking inter- and intrafamilial variability. The disease can present as early as in utero, but end-stage renal disease typically occurs in late middle age. Risk factors include early diagnosis of ADPKD, hypertension, gross hematuria, multiple pregnancies, and large kidney size. Liver cysts derived from the biliary epithelia are the most common extrarenal complication. Polycystic liver disease associated with ADPKD is different from autosomal dominant polycystic liver disease (ADPLD), which is caused by mutations in at least two distinct genes (PRKCSH and SEC63) and does not progress to renal failure. Massive polycystic liver disease occurs almost exclusively in women with ADPKD, particularly those with multiple pregnancies.
Intracranial aneurysm (ICA) occurs four to five times more frequently in ADPKD patients than in the general population and causes high mortality. The disease gene products PC1 and PC2 may be directly responsible for defects in arterial smooth muscle cells and myofibroblasts. The focal nature and the natural history of ICA in ADPKD remain unclear. A family history of ICA is a risk factor of aneurysm rupture in ADPKD, but whether hypertension and cigarette smoking are independent risk factors is not clear. About 20–50% of patients may experience “warning headaches” preceding the index episode of subarachnoid hemorrhage due to ruptured ICA. A CT scan is generally used as the first diagnostic test. A lumbar puncture may be used to confirm the diagnosis. The role of radiologic screening for ICA in asymptomatic patients with ADPKD remains unclear. ADPKD patients with a positive family history of ICAs may undergo presymptomatic screening of ICAs by magnetic resonance angiography. Other vascular abnormalities in ADPKD patients include diffuse arterial dolichoectasias of the anterior and posterior cerebral circulation, which can predispose to arterial dissection and stroke. Mitral valve prolapse occurs in up to 30% of patients with ADPKD, and tricuspid valve prolapse is less common. Other valvular abnormalities occurring with increased frequency in ADPKD patients include insufficiency of the mitral, aortic, and tricuspid valves. Most patients are asymptomatic, but some may progress and require valve replacement. The prevalence of colonic diverticulae and abdominal wall hernias is also increased in ADPKD patients.
Diagnosis Diagnosis is typically made from a positive family history consistent with autosomal dominant inheritance and multiple kidney cysts bilaterally. Renal ultrasonography is often used for presymptomatic screening of at-risk subjects and for evaluation of potential living-related kidney donors from ADPKD families. The presence of at least two renal cysts (unilateral or bilateral) is sufficient for diagnosis among at-risk subjects between 15 and 29 years of age with a sensitivity of 96% and specificity of 100%. The presence of at least two cysts in each kidney and the presence at least four cysts in each kidney are required for the diagnosis of at-risk subjects age 30 to 59 years and age 60 years or older, respectively, with a sensitivity of 100% and specificity of 100%. This is because there is an increased frequency of developing simple renal cysts with age. Conversely, in subjects between age 30 and 59 years, the absence of at least two cysts in each kidney, which is associated with a false-negative rate of 0%, can be used for disease exclusion. These criteria have a lower sensitivity for patients with a PKD2 mutation because of a late onset of ADPKD2. CT scan and T2-weighted MRI, with and without contrast enhancement, are more sensitive than ultrasonography and can detect cysts of smaller size. However, a CT scan exposes the patient to radiation and radiocontrast, which may cause serious allergic reactions and nephrotoxicity in patients with renal insufficiency. T2-weighted MRI, with gadolinium as a contrast agent, has minimal renal toxicity and can detect cysts of only 2–3 mm in diameter. However, a large majority of cysts may still be below the detection level. Genetic testing by linkage analyses and mutational analyses is available for ambiguous cases. Because of the large size of the PKD1 gene and the presence of multiple highly homologous pseudogenes, mutational analysis of the PKD1 gene is difficult and costly. Application of new technologies, such as paired-end next-generation sequencing with multiplexing individually bar-coded long-range polymerase chain reaction libraries, may reduce the costs and improve the sensitivity for clinical genetic testing.
AUTOSOMAL RECESSIVE POLYCYSTIC KIDNEY DISEASE
![]() Genetic Considerations ARPKD is a significant hereditary renal disease in childhood, with an estimated prevalence of 1 in 20,000 live births. A carrier frequency of up to 1:70 has been reported. Mutations in a single gene, PKHD1, are responsible for all the clinical presentations of ARPKD. PKHD1, localized on human chromosome region 6p21.1-6p12.2, is one of the largest genes in the genome, occupies ~450 kb of DNA, and contains at least 86 exons. It produces multiple alternatively spliced transcripts. The largest transcript encodes fibrocystin/polyductin (FPC), which is a large receptor-like integral membrane protein of 4074 amino acids. FPC has a single transmembrane, a large N-terminal extracellular region, and a short intracellular cytoplasmic domain. FPC is localized on the primary cilia of epithelia cells of cortical and medullary collecting ducts and cholangiocytes of bile ducts, similar to polycystins and several other ciliopathy proteins. FPC is also expressed on the basal body and plasma membrane. The large extracellular domain of FPC is presumed to bind to an as yet unknown ligand(s) and is involved in cell-cell and cell-matrix interactions. FPC interacts with ADPKD protein PC2 and may also participate in regulation of the mechanosensory function of the primary cilia, calcium signaling, and PCP, suggesting a common mechanism underlying cystogenesis between ADPKD and ARPKD. FPC is also found on the centrosomes and mitotic spindle and may regulate centrosome duplication and mitotic spindle assembly during cell division. A large number of various mutations have been found throughout PKHD1 and are unique to individual families. Most patients are compound heterozygotes for PKHD1 mutations. Patients with two truncation mutations appear to have an earlier onset of the disease.
Genetic Considerations ARPKD is a significant hereditary renal disease in childhood, with an estimated prevalence of 1 in 20,000 live births. A carrier frequency of up to 1:70 has been reported. Mutations in a single gene, PKHD1, are responsible for all the clinical presentations of ARPKD. PKHD1, localized on human chromosome region 6p21.1-6p12.2, is one of the largest genes in the genome, occupies ~450 kb of DNA, and contains at least 86 exons. It produces multiple alternatively spliced transcripts. The largest transcript encodes fibrocystin/polyductin (FPC), which is a large receptor-like integral membrane protein of 4074 amino acids. FPC has a single transmembrane, a large N-terminal extracellular region, and a short intracellular cytoplasmic domain. FPC is localized on the primary cilia of epithelia cells of cortical and medullary collecting ducts and cholangiocytes of bile ducts, similar to polycystins and several other ciliopathy proteins. FPC is also expressed on the basal body and plasma membrane. The large extracellular domain of FPC is presumed to bind to an as yet unknown ligand(s) and is involved in cell-cell and cell-matrix interactions. FPC interacts with ADPKD protein PC2 and may also participate in regulation of the mechanosensory function of the primary cilia, calcium signaling, and PCP, suggesting a common mechanism underlying cystogenesis between ADPKD and ARPKD. FPC is also found on the centrosomes and mitotic spindle and may regulate centrosome duplication and mitotic spindle assembly during cell division. A large number of various mutations have been found throughout PKHD1 and are unique to individual families. Most patients are compound heterozygotes for PKHD1 mutations. Patients with two truncation mutations appear to have an earlier onset of the disease.
Clinical Features Classic ARPKD is generally diagnosed in utero or within the neonatal period and characterized by greatly enlarged echogenic kidneys in diseased fetuses. Reduced fetal urine production may contribute to oligohydramnios and pulmonary hypoplasia. About 30% of affected neonates die shortly after birth due to respiratory insufficiency. Close to 60% of mortality occurs within the first month of life. In the classic group, most patients are born with renal insufficiency and ESRD. However, infants often have a transient improvement in their GFR; death from renal insufficiency at this stage is rare. Some patients are diagnosed after the neonatal stage and form the older group. Morbidity and mortality in this group often involve systemic hypertension, progressive renal insufficiency, and liver manifestations. The hallmarks of ARPKD liver disease are biliary dysgenesis due to a primary ductal plate malformation with associated periportal fibrosis, namely congenital hepatic fibrosis (CHF) and dilatation of intrahepatic bile ducts (Caroli’s disease). CHF and Caroli’s disease can then lead to portal hypertension exhibiting hepatosplenomegaly, variceal bleeding, and cholangitis. Some patients with the diagnosis of ARPKD at 1 year of age with nephromegaly exhibit slowly declining renal function over 20 years with only minimally enlarged kidneys at ESRD and markedly atrophic kidneys following renal transplantation. The slow progression of renal disease is likely due to increasing fibrosis rather than the development of cysts. Systemic hypertension is common in all ARPKD patients, even those with normal renal function.
Diagnosis Ultrasonography, CT, and MRI all can be used for diagnosis. Ultrasonography reveals large, echogenic kidneys with poor corticomedullary differentiation. The diagnosis can be made in utero after 24 weeks of gestation in severe cases. Macrocysts generally are not common at birth in ARPKD patients. The absence of renal cysts in either parent, particularly if they are more than 40 years of age on ultrasonography, helps distinguish ARPKD from ADPKD in older patients. Clinical, laboratory, or radiographic evidence of hepatic fibrosis, hepatic pathology demonstrating characteristic ductal plate abnormalities, family history of affected siblings, or parental consanguinity suggestive of autosomal recessive inheritance is helpful. The lack of mutational hotspots and the large and complex genomic structure of PKHD1 make molecular diagnosis difficult; however, presymptomatic screening of other at-risk members in a family with already identified ARPKD mutations is straightforward and inexpensive.
OTHER DISEASES CHARACTERIZED BY LARGE KIDNEY CYSTS
TUBEROUS SCLEROSIS
Tuberous sclerosis (TS) is a rare autosomal dominant syndrome caused by mutations in one of two genes, TSC1, encoding hamartin, or, TSC2, encoding tuberin. Published estimates of prevalence vary widely, but it certainly occurs in less than 1:5000 births. Kidney cysts are a frequent feature of this condition, as are two other abnormalities of kidney growth, renal cell carcinoma and renal angiomyolipomas. TS is a syndrome affecting multiple organ systems. Other features of TS include benign growths in the nervous system, eyes, heart, lung, liver, and skin. Essentially all TS patients have associated skin lesions, and a large proportion of patients have neurologic and cognitive manifestations. The TSC2 gene is adjacent to PKD1 in the human genome. Some patients have deletions in their genomic DNA that inactivate these two genes. Such individuals may have manifestations of both ADPKD and TS.
The most common kidney finding in TS is the presence of angiomyolipomas. These growths tend to be multiple and bilateral. Although they are usually benign, they may bleed. Surgical removal is often recommended as a prophylactic measure in people with angiomyolipomas larger than 4 cm in diameter. The cysts in TS are radiographically similar to those seen in ADPKD. In contrast to ADPKD, there is a clearly increased risk of renal cell carcinoma in TS patients. Regular periodic imaging is recommended in TS patients with kidney involvement to screen for the development of renal cell carcinoma.
Although not common, TS may lead to significant chronic kidney disease (CKD) and progress to end-stage kidney failure. Patients with TS and CKD typically have an unremarkable urine sediment and only minimal to mild amounts of proteinuria.
Mechanistically, the TSC1 and TSC2 gene products tuberin and hamartin interact physically. This protein complex is localized to the base of the cilia and inhibits intracellular signaling processes mediated by mTOR, leading to abnormal growth in a number of tissues. Investigation of mTOR inhibitors as therapy for TS is ongoing.
VON HIPPEL-LINDAU DISEASE
Von Hippel-Lindau disease (VHL) is an inherited cancer syndrome with renal manifestations. VHL is an autosomal dominant condition caused by mutations in the VHL tumor-suppressor gene. VHL is localized to the primary cilia and is necessary for the formation of primary cilia. Like many autosomal dominant cancer syndromes, VHL is recessive at the cellular level: a somatic mutation in the second VHL allele leads to loss of VHL in the cell and abnormal growth. Kidney manifestations of VHL include multiple bilateral kidney cysts and renal cell carcinomas. Kidney cysts and carcinoma affect the majority of VHL patients. Nonrenal features of VHL include pheochromocytomas, cerebellar hemangioblastomas, and retinal hemangiomas.
Annual screening of the kidneys by imaging with CT or MRI is recommended for early detection of renal cell carcinomas. Increasingly, nephron-sparing surgical approaches are being used for removal of cancerous lesions in order to preserve kidney function.
OTHER INHERITED DISEASES OF TUBULE GROWTH AND DEVELOPMENT
ADPKD is by far the most common adult-onset, single-gene form of kidney disease. The large cysts that are sometimes seen in VHL and TS are similar in appearance to the cysts seen in ADPKD. A variety of other inherited disorders affecting primarily tubule and renal interstitial function can lead to CKD and eventual end-stage kidney disease in the absence of large tubule-derived cysts.
Inherited diseases affecting the tubulointerstitial compartment of the kidney can lead to secondary glomerular stress and glomerulosclerosis with some degree of concomitant proteinuria. Similarly, disorders of glomerular function will typically lead to secondary interstitial fibrosis and tubule atrophy. From a clinical perspective, therefore, distinguishing between a genetic disease of the renal tubules and a disease of the glomerulus may not be easy, particularly in the absence of a gross phenotype such as large kidney cysts.
MEDULLARY CYSTIC KIDNEY DISEASE (AUTOSOMAL DOMINANT INTERSTITIAL KIDNEY DISEASE)
The medullary cystic kidney diseases (MCKD) are autosomal dominant disorders. Despite the nosology, kidney cysts are not invariably present. Older literature often grouped MCKD together with the childhood-onset disorders known as the nephronophthises, but these are distinct clinical and genetic entities.
Medullary Cystic Kidney Disease Type I Patients with MCKD type I (MCKD I) have mutations in the mucin 1 gene MUC1. In contrast to MCKD type II (MCKD II) patients, individuals with MCKD I do not have elevated uric acid levels. The disease-causing MUC1 mutations that have been reported all alter a repeat region within the MUC1 gene, leading to a large “neoprotein” fragment that may lead to toxic effects on the kidney tubule.
Clinically, patients with MCKD I exhibit slowly progressive CKD in adulthood, with only minimal amounts of increased urine protein and occasional renal cysts seen on ultrasound examination. Kidney histology shows tubulointerstitial fibrosis and tubular atrophy. The mechanisms by which MUC1 mutations cause human kidney disease are not known.
Medullary Cystic Kidney Disease Type II MCKD II is caused by mutations in the UMOD gene, which encodes the protein uromodulin, also known as Tamm-Horsfall protein. Uromodulin is also found on the centrosome, the mitotic spindle, and the primary cilia; it colocalizes with nephrocystin-1 and KIF3A on the cilia. UMOD mutations also cause the conditions that have been referred to as familial juvenile hyperuricemic nephropathy (HNFJ1) and glomerulocystic kidney disease (GCKD), although it is not clear that these different names represent clearly distinct disorders. The term uromodulin-associated kidney disease (UAKD) has been suggested as a better name for MCKD II and the various other related UMOD-associated diseases. Despite the name, kidney cysts are not a common feature of MCKD II. MCKD II should be suspected clinically in patients with a family history of late-onset kidney disease, benign urine sediments, absence of significant proteinuria, and hyperuricemia. Large genome-wide association studies have suggested that certain common noncoding sequence variants in UMOD are associated with a moderately increased risk of CKD in the general population.
Other Forms of Familial Tubulointerstitial Kidney Disease A small number of families have been identified with autosomal dominant tubulointerstitial kidney disease and hyperuricemia who lack UMOD mutations. Some of these families carry disease-segregating mutations in the renin gene REN. There are other families who lack mutations in UMOD, MUC1, or REN. Thus, mutations in other yet-to-be identified genes are able to produce similar interstitial kidney disease, both with and without hyperuricemia.
Kidney biopsies in patients with any of the various forms of MCKD typically show interstitial fibrosis. These histologic features are not diagnostic of any particular genetic entity, and the specific diagnosis must be made by other means. Genetic tests for alterations in specific genes are increasingly available in the clinical setting.
Patients with autosomal dominant interstitial kidney disease, UMOD or REN mutations, or hyperuricemia and gout should be treated similarly to others with these findings, with uric acid–lowering agents, such allopurinol or febuxostat.
NEPHRONOPHTHISIS
A large and growing number of genetically distinct but related autosomal recessive disorders are referred to as nephronophthises. These should not be confused with the adult-onset autosomal dominant medullary cystic kidney diseases discussed above, despite the often confusing nomenclature seen in older medical literature. Nephronophthisis is quite rare but is nevertheless the most common inherited childhood form of kidney failure requiring kidney replacement therapy.
Like ADPKD and ARPKD, the various genetically heterogeneous entities that fall under the category of nephronophthisis (NPHP) are disorders of ciliary function. Mutations in a very large number of genes have been identified that lead to NPHP under an autosomal recessive pattern of inheritance. The various forms of NPHP share common features, including tubulointerstitial fibrosis, corticomedullary cysts, and progressive CKD, leading to renal failure. Proteinuria is absent or mild, and the urine sediment is not active.
NPHP is often divided into infantile, juvenile, and adolescent forms. The juvenile form is the most frequent and usually caused by mutations in the NPHP2 gene. The infantile form, usually caused by NPHP2 mutations, is associated with end-stage kidney failure in early childhood. Patients with the adolescent form of NPHP typically develop end-stage kidney failure in early adulthood. The products of the NPHP genes are referred to as nephrocystins. NPHP1 through NPHP16 have been reported; some are referred to by other names as well.
NPHP can present as an isolated finding or be part of several multiorgan syndromes. Neurologic abnormalities are present in a significant number of patients. Bone and liver abnormities are seen in some NPHP patients. Senior-Løken syndrome is defined by the presence of NPHP with retinitis pigmentosa. Joubert’s syndrome is defined by multiple neurologic findings, including hypoplasia of the cerebellar vermis. Some forms of this genetically heterogeneous syndrome include NPHP as a component.
The multisystem disease Bardet-Biedl syndrome (BBS) is defined clinically by a spectrum of features, including truncal obesity, cognitive impairment, retinal dystrophy, polydactyly, developmental urogenital abnormalities, and kidney cysts. The kidney phenotype is NPHP-like, with small cysts deriving from the tubules, tubulointerstitial and often secondary glomerular disease, and urine concentrating defects. There are 18 BBS genes cloned. BBS follows autosomal recessive inheritance. Like ADPKD, ARPKD, and NPHP, BBS is a disease of abnormal ciliary function.
The multiple genes and gene products (nephrocystins) that are responsible for NPHP are expressed in cilia, basal bodies, and the centrosomes of kidney tubule cells. It has been hypothesized that all of the NPHP gene defects lead to a clinical phenotype by interfering with the regulation of PCP.
There are no specific clinical tests that define NPHP. Genetic diagnosis is possible but cumbersome because of the large number of genes that can be responsible. There are no specific therapies for NPHP. Rather, therapy is aimed at treating signs of these diseases as well as the systemic abnormalities seen with all CKDs. Chronic dialysis or kidney transplantation is eventually required for NPHP-affected individuals.
KARYOMEGALIC TUBULOINTERSTITIAL NEPHRITIS
Karyomegalic tubulointerstitial nephritis is an exceptionally rare form of kidney disease with adult-onset progressive kidney failure. Kidney biopsy shows chronic tubulointerstitial nephritis, as well as interstitial fibrosis. This is a recessive disorder caused by inheritance of two mutant copies of the FAN1 gene. FAN1 encodes a component of a DNA repair machinery complex. Individuals with two mutant FAN1 genes are genetically sensitized to the effect of DNA damage. Kidney histology shows karyomegaly in addition to the nonspecific findings of interstitial fibrosis and tubular atrophy.
MEDULLARY SPONGE KIDNEY
Medullary sponge kidney (MSK) is often grouped together with inherited disorders of the kidney affecting tubule growth and development, although it is usually a sporadic finding rather than an inherited phenotype. MSK is caused by developmental malformation and cystic dilatation of the renal collecting ducts. The medullary cysts seen in this entity can be quite variable in size.
MSK is usually a benign entity. The diagnosis of MSK is often made incidentally. In the past, the diagnosis of MSK was often made by intravenous pyelography (IVP). CT scans, which have replaced IVPs for much routine kidney imaging, are not as sensitive in detecting MSK.
MSK is associated with an increased frequency of calcium phosphate and calcium oxalate kidney stones. Altered flow characteristics in the kidney tubules may lead to the development of formation of a nidus for stone formation. Kidney stones in this group are treated the same as are kidney stones in the general population. MSK patients also often exhibit reduced kidney concentrating ability and an increased frequency of urinary tract infections.
CONGENITAL ABNORMALITIES OF THE KIDNEY AND URINARY TRACT
The structural abnormalities known as the congenital abnormalities of the kidney and urinary tract (CAKUTs) are a group of etiologically and phenotypically heterogeneous disorders. Some form of CAKUT is estimated to occur in up to 1 in 500 live births. Specific abnormalities classified as part of the CAKUT spectrum include kidney hypoplasia, kidney agenesis, ureteropelvic junction obstruction, and vesicoureteral reflux.
CAKUT can be the cause of clinically significant problems in both adults and children. However, it is a major contributor to kidney failure in children, accounting for more than one-third of end-stage kidney disease in this group.
CAKUT is typically a sporadic finding but can also cluster in families. Familial forms can be observed as parts of multisystem developmental syndromes. A growing list of specific genes have been identified, which when mutated lead to syndromic forms of CAKUT. For example, the branchio-oto-renal syndrome, characterized by developmental abnormalities in the neck, ears, and kidney, can be caused by mutations in the EYA1 and SIX1 genes. Mutations in the PAX2 transcription factor gene can cause the autosomal dominant renal coloboma syndrome, characterized by optic nerve malformations and hypoplastic kidneys.
In many instances, CAKUT is caused by environmental influences rather than genetic alterations. For example, renal tubular dysgenesis, defined by altered tubule development, can be caused by prenatal exposure to angiotensin-converting enzyme inhibitors or angiotensin receptor blockers.
MITOCHONDRIAL DISEASE
Inherited disorders of the mitochondrial genome (discussed elsewhere in this text [Chap. 85e]) commonly affect kidney function. Thirteen of the genes involved in encoding components of the mitochondrial respiratory chain are located on the mitochondrial genome that is inherited maternally. The remainder of these components is encoded by the nuclear genome. These defects of oxidative phosphorylation may affect multiple organs and tissues.
Neuromuscular disease is the best recognized part of this complex phenotype. Kidney disease is now recognized as a common component as well. Tubulointerstitial disease may be seen on kidney biopsy, and progression to kidney failure may occur. Glomerular involvement, manifest as proteinuria and glomerulosclerosis, can also develop. Changes in proximal tubule activity are the most common renal phenotype. Patients may have several defects in proximal tubule transport, including the Fanconi syndrome. Some patients may also have acidosis, hypophosphatemic rickets, hypercalciuria, glycosuria, and tubular proteinuria. Decreased urine concentrating ability is common.
GLOBAL CONSIDERATIONS
![]() The disorders discussed above are all seen worldwide. In addition, a previously unrecognized epidemic of kidney disease is leading to very high rates of kidney failure in and near the western coast of Central America. This Mesoamerican nephropathy is particularly common in Nicaragua and El Salvador. Mesoamerican nephropathy patients do not have significant proteinuria, suggesting that this is a disease of the kidney tubules and interstitium. The cause is unknown, but some have suggested that a combination of toxic environmental factors and heat stress underlies the development of this kidney disease, which has a striking male predominance. However, the fact that, in many families, a large fraction of the men are affected with kidney disease has suggested that a strong genetic component may be involved as well.
The disorders discussed above are all seen worldwide. In addition, a previously unrecognized epidemic of kidney disease is leading to very high rates of kidney failure in and near the western coast of Central America. This Mesoamerican nephropathy is particularly common in Nicaragua and El Salvador. Mesoamerican nephropathy patients do not have significant proteinuria, suggesting that this is a disease of the kidney tubules and interstitium. The cause is unknown, but some have suggested that a combination of toxic environmental factors and heat stress underlies the development of this kidney disease, which has a striking male predominance. However, the fact that, in many families, a large fraction of the men are affected with kidney disease has suggested that a strong genetic component may be involved as well.
340 | Tubulointerstitial Diseases of the Kidney |
Inflammation or fibrosis of the renal interstitium and atrophy of the tubular compartment are common consequences of diseases that target the glomeruli or vasculature. Distinct from these secondary phenomena, however, are a group of disorders that primarily affect the tubules and interstitium, with relative sparing of the glomeruli and renal vessels. Such disorders are conveniently divided into acute and chronic tubulointerstitial nephritis (TIN) (Table 340-1).
CLASSIFICATION OF THE CAUSES OF TUBULOINTERSTITIAL DISEASES OF THE KIDNEY |
Abbreviations: CMV, cytomegalovirus; COX, cyclooxygenase; EBV, Epstein-Barr virus.
Acute TIN most often presents with acute renal failure (Chap. 334). The acute nature of this group of disorders may be caused by aggressive inflammatory infiltrates that lead to tissue edema, tubular cell injury, and compromised tubular flow, or by frank obstruction of the tubules with casts, cellular debris, or crystals. There is sometimes flank pain due to distention of the renal capsule. Urinary sediment is often active with leukocytes and cellular casts, but depends on the exact nature of the disorder in question.
The clinical features of chronic TIN are more indolent and may manifest with disorders of tubular function, including polyuria from impaired concentrating ability (nephrogenic diabetes insipidus), defective proximal tubular reabsorption leading to features of Fanconi’s syndrome (glycosuria, phosphaturia, aminoaciduria, hypokalemia, and type II renal tubular acidosis [RTA] from bicarbonaturia), or non-anion-gap metabolic acidosis and hyperkalemia (type IV RTA) due to impaired ammoniagenesis, as well as progressive azotemia (rising creatinine and blood urea nitrogen [BUN]). There is often modest proteinuria (rarely >2 g/d) attributable to decreased tubular reabsorption of filtered proteins; however, nephrotic-range albuminuria may occur in some conditions due to the development of secondary focal segmental glomerulosclerosis (FSGS). Renal ultrasonography may reveal changes of “medical renal disease,” such as increased echogenicity of the renal parenchyma with loss of corticomedullary differentiation, prominence of the renal pyramids, and cortical scarring in some conditions. The predominant pathology in chronic TIN is interstitial fibrosis with patchy mononuclear cell infiltration and widespread tubular atrophy, luminal dilation, and thickening of tubular basement membranes. Because of the nonspecific nature of the histopathology, biopsy specimens rarely provide a specific diagnosis. Thus, diagnosis relies on careful analysis of history, drug or toxin exposure, associated symptoms, and imaging studies.
ACUTE INTERSTITIAL NEPHRITIS
In 1897, Councilman reported on eight cases of acute interstitial nephritis (AIN) in the Medical and Surgical Reports of the Boston City Hospital; three as a postinfectious complication of scarlet fever and two from diphtheria. Later, he described the lesion as “an acute inflammation of the kidney characterized by cellular and fluid exudation in the interstitial tissue, accompanied by, but not dependant on, degeneration of the epithelium; the exudation is not purulent in character, and the lesions may be both diffuse and focal.” Today AIN is far more often encountered as an allergic reaction to a drug (Table 340-1). Immune-mediated AIN may also occur as part of a known autoimmune syndrome, but in some cases there is no identifiable cause despite features suggestive of an immunologic etiology (Table 340-1).
ALLERGIC INTERSTITIAL NEPHRITIS
Although biopsy-proven AIN accounts for no more than ~15% of cases of unexplained acute renal failure, this is likely a substantial underestimate of the true incidence. This is because potentially offending medications are more often identified and empirically discontinued in a patient noted to have a rising serum creatinine, without the benefit of a renal biopsy to establish the diagnosis of AIN.
Clinical Features The classic presentation of AIN, namely, fever, rash, peripheral eosinophilia, and oliguric renal failure occurring after 7–10 days of treatment with methicillin or another β-lactam antibiotic, is the exception rather than the rule. More often, patients are found incidentally to have a rising serum creatinine or present with symptoms attributable to acute renal failure (Chap. 334). Atypical reactions can occur, most notably nonsteroidal anti-inflammatory drug (NSAID)-induced AIN, in which fever, rash, and eosinophilia are rare, but acute renal failure with heavy proteinuria is common. A particularly severe and rapid-onset AIN may occur upon reintroduction of rifampin after a drug-free period. More insidious reactions to the agents listed in Table 340-1 may lead to progressive tubulointerstitial damage. Examples include proton pump inhibitors and, rarely, sulfonamide and 5-aminosalicylate (mesalazine and sulfasalazine) derivatives and antiretrovirals.
Diagnosis Finding otherwise unexplained renal failure with or without oliguria and exposure to a potentially offending agent usually points to the diagnosis. Peripheral blood eosinophilia adds supporting evidence but is present in only a minority of patients. Urinalysis reveals pyuria with white blood cell casts and hematuria. Urinary eosinophils are neither sensitive nor specific for AIN; therefore, testing is not recommended. Renal biopsy is generally not required for diagnosis but reveals extensive interstitial and tubular infiltration of leukocytes, including eosinophils.
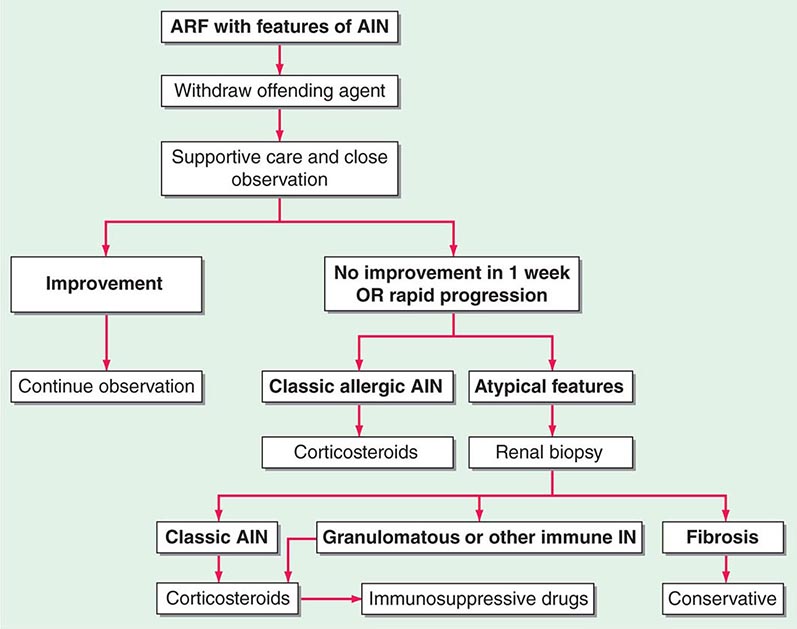
FIGURE 340-1 Algorithm for the treatment of allergic and other immune-mediated acute interstitial nephritis (AIN). ARF, acute renal failure; IN, interstitial nephritis. See text for immunosuppressive drugs used for refractory or relapsing AIN. (Modified from S Reddy, DJ Salant: Ren Fail 20:829, 1998.)
INDICATIONS FOR CORTICOSTEROIDS AND IMMUNOSUPPRESSIVES IN INTERSTITIAL NEPHRITIS |
Abbreviations: AIN, acute interstitial nephritis; SLE, systemic lupus erythematosus; TINU, tubulointerstitial nephritis with uveitis.
Source: Modified from S Reddy, DJ Salant: Ren Fail 20:829, 1998.
SJÖGREN’S SYNDROME
Sjögren’s syndrome is a systemic autoimmune disorder that primarily targets the exocrine glands, especially the lacrimal and salivary glands, and thus results in symptoms, such as dry eyes and mouth, that constitute the “sicca syndrome” (Chap. 383). Tubulointerstitial nephritis with a predominant lymphocytic infiltrate is the most common renal manifestation of Sjögren’s syndrome and can be associated with distal RTA, nephrogenic diabetes insipidus, and moderate renal failure. Diagnosis is strongly supported by positive serologic testing for anti-Ro (SS-A) and anti-La (SS-B) antibodies. A large proportion of patients with Sjögren’s syndrome also have polyclonal hypergammaglobulinemia. Treatment is initially with glucocorticoids, although patients may require maintenance therapy with azathioprine or mycophenolate mofetil to prevent relapse (Fig. 340-1 and Table 340-2).
TUBULOINTERSTITIAL NEPHRITIS WITH UVEITIS (TINU)
TINU is a systemic autoimmune disease of unknown etiology. It accounts for fewer than 5% of all cases of AIN, affects females three times more often than males, and has a median age of onset of 15 years. Its hallmark feature, in addition to a lymphocyte-predominant interstitial nephritis (Fig. 340-2), is a painful anterior uveitis, often bilateral and accompanied by blurred vision and photophobia. Diagnosis is often confounded by the fact that the ocular symptoms precede or accompany the renal disease in only one-third of cases. Additional extrarenal features include fever, anorexia, weight loss, abdominal pain, and arthralgia. The presence of such symptoms as well as elevated creatinine, sterile pyuria, mild proteinuria, features of Fanconi’s syndrome, and elevated erythrocyte sedimentation rate should raise suspicion for this disorder. Serologies suggestive of the more common autoimmune diseases are usually negative, and TINU is often a diagnosis of exclusion after other causes of uveitis and renal disease, such as Sjögren’s syndrome, Behçet’s disease, sarcoidosis, and systemic lupus erythematosus, have been considered. Clinical symptoms are typically self-limited in children, but are more apt to follow a relapsing course in adults. The renal and ocular manifestations generally respond well to oral glucocorticoids, although maintenance therapy with agents such as methotrexate, azathioprine, or mycophenolate may be necessary to prevent relapses (Fig. 340-1 and Table 340-2).
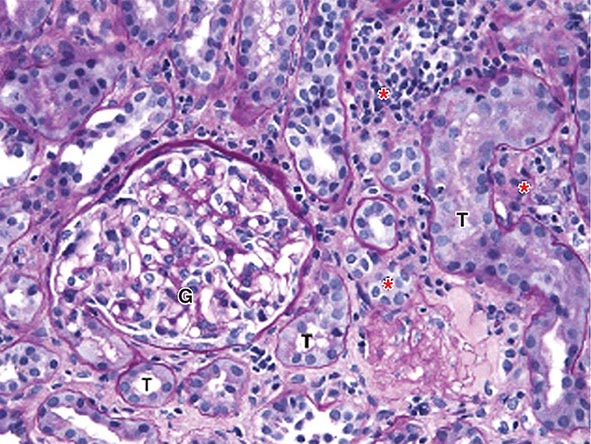
FIGURE 340-2 Acute interstitial nephritis (AIN) in a patient who presented with acute iritis, low-grade fever, erythrocyte sedimentation rate of 103, pyuria and cellular casts on urinalysis, and a newly elevated serum creatinine of 2.4 mg/dL. Both the iritis and AIN improved after intravenous methylprednisolone. This PAS-stained renal biopsy shows a mononuclear cell interstitial infiltrate (asterisks) and edema separating the tubules (T) and a normal glomerulus (G). Some of the tubules contain cellular debris and infiltrating inflammatory cells. The findings in this biopsy are indistinguishable from those that would be seen in a case of drug-induced AIN. PAS, Periodic acid–Schiff.
SYSTEMIC LUPUS ERYTHEMATOSUS
An interstitial mononuclear cell inflammatory reaction often accompanies the glomerular lesion in most cases of class III or IV lupus nephritis (Chap. 338), and deposits of immune complexes can be identified in tubule basement membranes in about 50% of cases. Occasionally, however, the tubulointerstitial inflammation predominates and may manifest with azotemia and type IV RTA rather than features of glomerulonephritis.
GRANULOMATOUS INTERSTITIAL NEPHRITIS
Some patients may present with features of AIN but follow a protracted and relapsing course. Renal biopsy in such patients reveals a more chronic inflammatory infiltrate with granulomas and multinucleated giant cells. Most often, no associated disease or cause is found; however, some of these cases may have or subsequently develop the pulmonary, cutaneous, or other systemic manifestations of sarcoidosis such as hypercalcemia. Most patients experience some improvement in renal function if treated early with glucocorticoids before the development of significant interstitial fibrosis and tubular atrophy (Table 340-2). Other immunosuppressive agents may be required for those who relapse frequently upon steroid withdrawal. Other immunosuppressive agents may be required for those who relapse frequently upon steroid withdrawal (Fig. 340-1). Tuberculosis should be ruled out before starting treatment because this too is a rare cause of granulomatous interstitial nephritis.
IgG4-RELATED SYSTEMIC DISEASE
A form of AIN characterized by a dense inflammatory infiltrate containing IgG4-expressing plasma cells can occur as a part of a syndrome known as IgG4-related systemic disease. Autoimmune pancreatitis, sclerosing cholangitis, retroperitoneal fibrosis, and a chronic sclerosing sialadenitis (mimicking Sjögren’s syndrome) may variably be present as well. Fibrotic lesions that form pseudotumors in the affected organs soon replace the initial inflammatory infiltrates and often lead to biopsy or excision for fear of true malignancy. Although the involvement of IgG4 in the pathogenesis is not understood, glucocorticoids have been successfully used as first-line treatment in this group of disorders, once they are correctly diagnosed.
IDIOPATHIC AIN
Some patients present with typical clinical and histologic features of AIN but have no evidence of drug exposure or clinical or serologic features of an autoimmune disease. The presence in some cases of autoantibodies to a tubular antigen, similar to that identified in rats with an induced form of interstitial nephritis, suggests that an autoimmune response may be involved. Like TINU and granulomatous interstitial nephritis, idiopathic AIN is responsive to glucocorticoid therapy but may follow a relapsing course requiring maintenance treatment with another immunosuppressive agent (Fig. 340-1 and Table 340-2).
INFECTION-ASSOCIATED AIN
AIN may also occur as a local inflammatory reaction to microbial infection (Table 340-1) and should be distinguished from acute bacterial pyelonephritis (Chap. 162). Acute bacterial pyelonephritis does not generally cause acute renal failure unless it affects both kidneys or causes septic shock. Presently, infection-associated AIN is most often seen in immunocompromised patients, particularly renal transplant recipients with reactivation of polyomavirus BK (Chaps. 169 and 337).
CRYSTAL DEPOSITION DISORDERS AND OBSTRUCTIVE TUBULOPATHIES
Acute renal failure may occur when crystals of various types are deposited in tubular cells and interstitium or when they obstruct tubules. Oliguric acute renal failure, often accompanied by flank pain from tubular obstruction, may occur in patients treated with sulfadiazine for toxoplasmosis, indinavir and atazanavir for HIV, and intravenous acyclovir for severe herpesvirus infections. Urinalysis reveals “sheaf of wheat” sulfonamide crystals, individual or parallel clusters of needle-shaped indinavir crystals, or red-green birefringement needle-shaped crystals of acyclovir. This adverse effect is generally precipitated by hypovolemia and is reversible with saline volume repletion and drug withdrawal. Distinct from the obstructive disease, a frank AIN from indinavir crystal deposition has also been reported.
Acute tubular obstruction is also the cause of oliguric renal failure in patients with acute urate nephropathy. It typically results from severe hyperuricemia from tumor lysis syndrome in patients with lympho- or myeloproliferative disorders treated with cytotoxic agents, but also may occur spontaneously before the treatment has been initiated (Chap. 331). Uric acid crystallization in the tubules and collecting system leads to partial or complete obstruction of the collecting ducts, renal pelvis, or ureter. A dense precipitate of birefringent uric acid crystals is found in the urine, usually in association with microscopic or gross hematuria. Prophylactic allopurinol reduces the risk of uric acid nephropathy but is of no benefit once tumor lysis has occurred. Once oliguria has developed, attempts to increase tubular flow and solubility of uric acid with alkaline diuresis may be of some benefit; however, emergent treatment with hemodialysis or rasburicase, a recombinant urate oxidase, is usually required to rapidly lower uric acid levels and restore renal function.
Calcium oxalate crystal deposition in tubular cells and interstitium may lead to permanent renal dysfunction in patients who survive ethylene glycol intoxication, in patients with enteric hyperoxaluria from ileal resection or small-bowel bypass surgery, and in patients with hereditary hyperoxaluria (Chap. 342). Acute phosphate nephropathy is an uncommon but serious complication of oral Phospho-soda used as a laxative or for bowel preparation for colonoscopy. It results from calcium phosphate crystal deposition in tubules and interstitium and occurs especially in subjects with underlying renal impairment and hypovolemia. Consequently, Phospho-soda should be avoided in patients with chronic kidney disease.
LIGHT CHAIN CAST NEPHROPATHY
Patients with multiple myeloma may develop acute renal failure in the setting of hypovolemia, infection, or hypercalcemia or after exposure to NSAIDs or radiographic contrast media. The diagnosis of light chain cast nephropathy (LCCN)—commonly known as myeloma kidney—should be considered in patients who fail to recover when the precipitating factor is corrected or in any elderly patient with otherwise unexplained acute renal failure.
In this disorder, filtered monoclonal immunoglobulin light chains (Bence-Jones proteins) form intratubular aggregates with secreted Tamm-Horsfall protein in the distal tubule. Casts, in addition to obstructing the tubular flow in affected nephrons, incite a giant cell or foreign body reaction and can lead to tubular rupture, resulting in interstitial fibrosis (Fig. 340-3). Although LCCN generally occurs in patients with known multiple myeloma and a large plasma cell burden, the disorder should also be considered as a possible diagnosis in patients who have known monoclonal gammopathy even in the absence of frank myeloma. Filtered monoclonal light chains may also cause less pronounced renal manifestations in the absence of obstruction, due to direct toxicity to proximal tubular cells and intracellular crystal formation. This may result in isolated tubular disorders such as RTA or full Fanconi’s syndrome.
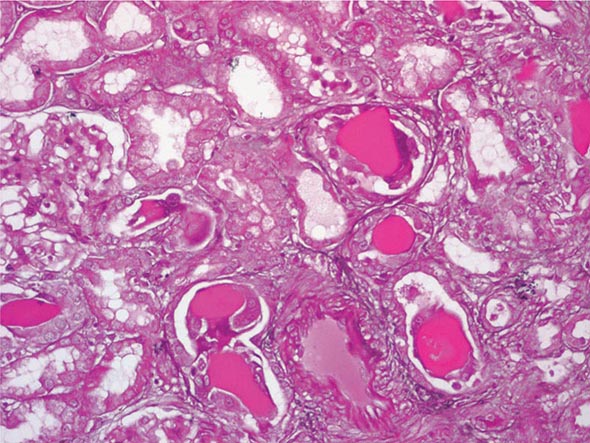
FIGURE 340-3 Histologic appearance of myeloma cast nephropathy. A hematoxylin-eosin–stained kidney biopsy shows many atrophic tubules filled with eosinophilic casts (consisting of Bence-Jones protein), which are surrounded by giant cell reactions. (Courtesy of Dr. Michael N. Koss, University of Southern California Keck School of Medicine; with permission.)
Diagnosis Clinical clues to the diagnosis include anemia, bone pain, hypercalcemia, and an abnormally narrow anion gap due to hypoalbuminemia and hypergammaglobulinemia. Urinary dipsticks detect albumin but not immunoglobulin light chains; however, laboratory detection of increased amounts of protein in a spot urine specimen and a negative dipstick result are highly suggestive that the urine contains Bence-Jones protein. Serum and urine should both be sent for protein electrophoresis and for immunofixation for the detection and identification of a potential monoclonal band. A sensitive method is available to detect urine and serum free light chains.
TREATMENT | LIGHT CHAIN CAST NEPHROPATHY |
The goals of treatment are to correct precipitating factors such as hypovolemia and hypercalcemia, discontinue potential nephrotoxic agents, and treat the underlying plasma cell dyscrasia (Chap. 136); plasmapheresis to remove light chains is of questionable value for LCCN.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


