http://evolve.elsevier.com/McCuistion/pharmacology/
Organ Transplantation
Organ transplantation is a life-saving procedure. In cadaveric transplantation, a healthy organ donated at the time of a person’s death is transplanted into the body of a patient with end-stage organ failure. In living-donor transplantation, a kidney or a portion of liver donated by a living person is transplanted into the body of a patient with end-stage kidney or liver disease. Organ transplant is an acceptable treatment option when organs fail (e.g., kidney, heart, liver, and lung). More than 122,000 people are currently waiting for an organ transplant. Every 10 minutes, another person is added to the wait list, and over 8000 people die each year while waiting for a donor organ.
Principles of Immunosuppression
The immune system remains the biggest barrier to transplantation as a routine medical treatment because it has effective mechanisms to fight off foreign organisms. These same mechanisms are involved in the rejection of transplanted organs, which are recognized as foreign by the recipient’s immune system. The underlying premise of immunosuppression is to use multiple drugs that alter different aspects of the immune system (Fig. 30.1), thereby reducing the chances of transplant rejection and enabling the use of lower doses of individual drugs, which reduces the likelihood of drug toxicity. Transplantation has revolutionized care for patients with end-stage organ failure, yet significant problems remain with treatments designed to promote transplant survival and prevent rejection. Immunosuppressant drugs are not always effective; in addition, they are expensive and must be taken daily, and they are associated with toxic effects.
Immunosuppressant Drugs
Induction Therapy
Induction therapy provides intense immunosuppression with drugs designed to diminish antigen presentation and T-cell response, thus reducing the risk for acute rejection during the initial transplant period.
Basiliximab is a monoclonal antibody that inhibits interleukin 2 (IL-2)–mediated activation of lymphocytes, a critical component of the cellular immune response involved in transplant rejection. By inhibiting activation of lymphocytes, it prevents the body from mounting an immune response against the transplanted organ. Basiliximab has been approved for induction therapy in kidney transplants. Complete pharmacokinetic data are not available, but drug half-life is known to be 7.2 days in adults and 9.5 days in children.
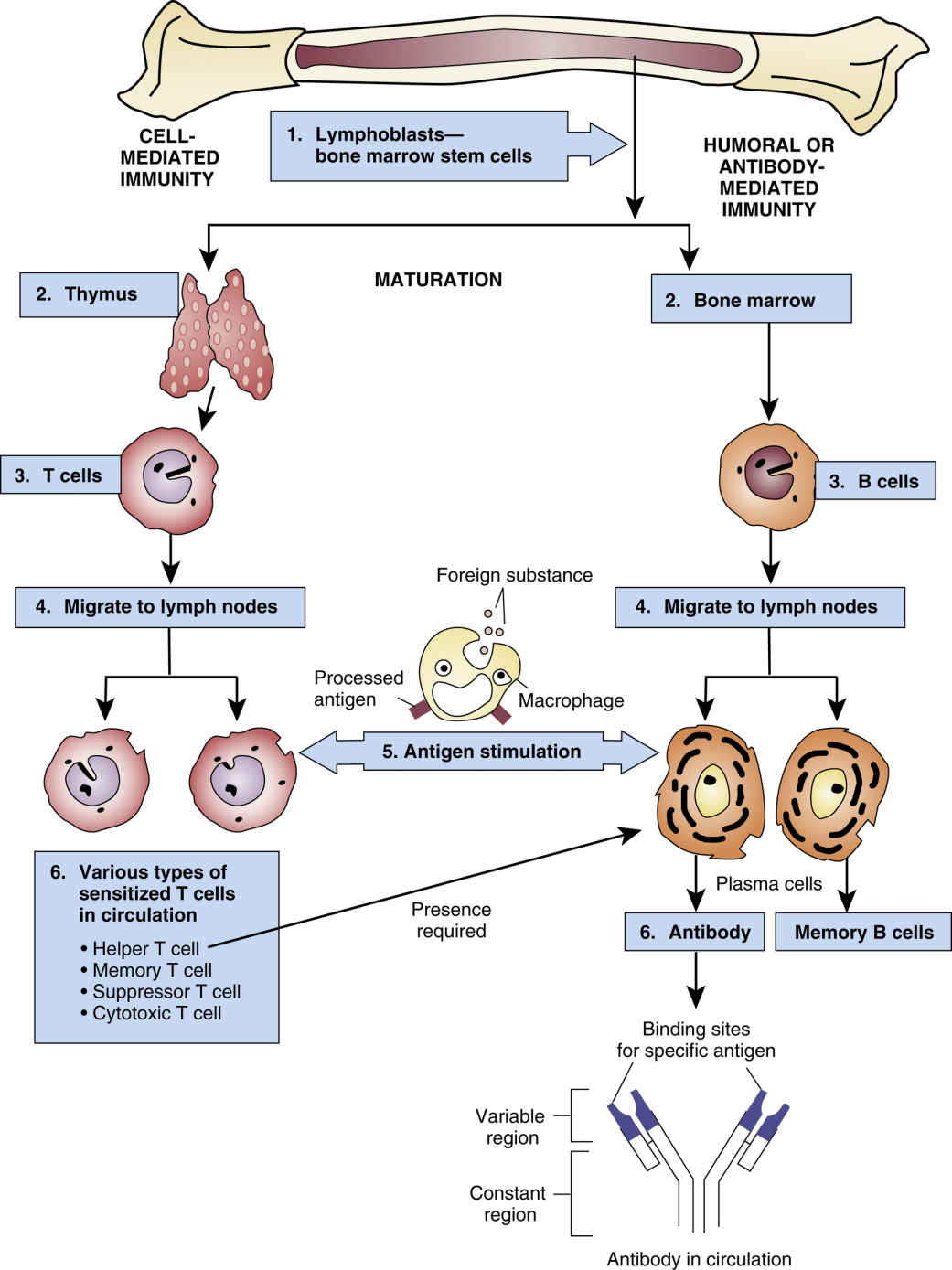
FIG. 30.1 The Immune Response.
From Gould, B., & Dyer, R. (2011). Pathophysiology for the health professions (4th ed.). St Louis: Saunders.
Basiliximab is administered intravenously (IV), 20 mg within 2 hours before transplant surgery, followed by a second 20-mg dose 4 days after transplantation. The second dose should be withheld if complications occur (including severe hypersensitivity reactions or loss of the transplanted organ). Children under 35 kg should receive 10 mg IV within 2 hours before transplant surgery, followed by a second 10-mg dose 4 days after transplantation; the second dose should be withheld if complications occur (including severe hypersensitivity reactions or loss of the transplanted organ); children over 35 kg should receive the adult dose.
Side effects of basiliximab include abdominal and back pain, coughing, dizziness, fever or chills, fatigue, weakness, dysuria, dyspnea, sore throat, edema, tremor, nausea and vomiting, and anemia. Serious reactions include sepsis, opportunistic infections, malignancy, lymphoproliferative disorders, thrombocytopenia, leukopenia, diabetes mellitus, anaphylaxis, capillary leak syndrome, and cytokine release syndrome (Box 30.1).
Transplant recipients who receive basiliximab should not receive live vaccines because they may produce an inadequate immune response and are at risk for disseminated infection resulting from the live virus. Caution is advised when basiliximab is administered with other drugs that lower the immune response because of the increased risk of serious infection.
Basiliximab was designated pregnancy category B; no adequate and well-controlled studies have been done on the drug’s use in pregnant women, therefore women of childbearing potential should use effective contraceptive measures before beginning treatment, during treatment, and for 4 months after completion of therapy. It is not known whether basiliximab is excreted in breast milk. Because of the potential for adverse drug reactions, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Maintenance Therapy
Calcineurin Inhibitors
The calcineurin inhibitors (CNIs) suppress the immune system by binding to cytoplasmic proteins that inhibit calcineurin phosphatase, resulting in a reduction in cytokine synthesis and inhibition of T-lymphocyte proliferation. There are two CNIs, cyclosporine and tacrolimus; the prototype CNI is cyclosporine. The drug became available in 1983 and was modified to improve bioavailability in 1994.  Cyclosporine oral solution USP modified and cyclosporine oral solution USP are not bioequivalent and cannot be used interchangeably.
Cyclosporine oral solution USP modified and cyclosporine oral solution USP are not bioequivalent and cannot be used interchangeably.
Cyclosporine oral solution USP modified and cyclosporine oral solution USP are not bioequivalent and cannot be used interchangeably.After oral administration, absorption of cyclosporine is incomplete and varies widely from patient to patient. Drug distribution is concentration dependent; cyclosporine is 90% protein bound. The drug is extensively metabolized by the cytochrome P450 (CYP450) 3A enzyme system in the liver. Elimination is primarily biliary with only 6% excreted in urine. The average drug half-life is 8.4 hours.
Initial dosing of cyclosporine is 7 to 9 mg/kg orally per day in two divided doses; the first dose is given 4 to 12 hours before the transplant or postoperatively in both adult and pediatric patients. Adjustments to drug dosing are based on therapeutic drug monitoring (TDM), with the desired level between 100 and 500 ng/mL, depending on the organ transplanted and the length of time posttransplant.
The daily dose of cyclosporine oral solution USP modified should always be given in two divided doses and on a consistent schedule with regard to time of day and relation to meals. Grapefruit and grapefruit juice affect drug metabolism, increasing blood concentration of cyclosporine; for this reason, grapefruit should be avoided. To make cyclosporine oral solution USP modified more palatable, it should be diluted with room-temperature orange or apple juice; patients should avoid switching diluents frequently. When mixed with juice, the cyclosporine solution may appear cloudy. Cyclosporine is also available as immediate-release capsules (25 mg and 100 mg) and as an IV solution (250 mg/5 mL).
Common side effects of cyclosporine include elevated blood urea nitrogen (BUN) and creatinine, hypertension, hirsutism, infection, tremor, gingival hyperplasia, headache, hypertriglyceridemia, nausea and vomiting, diarrhea, hyperuricemia, hyperglycemia, arthralgia, edema, acne, hypomagnesemia, and hyperkalemia. Serious reactions include opportunistic infections, nephropathy and nephrotoxicity, diabetes mellitus, leukopenia, thrombocytopenia, hemolytic anemia, malignancy, and seizures.
Transplant recipients taking cyclosporine should not receive live vaccines because they may have inadequate immune response and are at risk for disseminated infection resulting from the live virus.
Multiple antibiotics, melphalan (an antineoplastic drug), antifungals, antiinflammatory drugs, cimetidine and ranitidine (histamine 2–receptor blockers), tacrolimus (an immunosuppressant), fibric acid derivatives, and methotrexate may potentiate kidney dysfunction when administered with cyclosporine. Calcium channel blockers, azole antifungals, macrolide antibiotics, and glucocorticoids can increase cyclosporine concentrations, as can allopurinol, amiodarone, bromocriptine, colchicine, danazol, imatinib, metoclopramide, nefazodone, and oral contraceptives. Other drugs—nafcillin, rifampin, anticonvulsants, bosentan, octreotide, orlistat, sulfinpyrazone, terbinafine, ticlopidine—and St. John’s wort can decrease cyclosporine concentrations.
Concomitant administration of cyclosporine with 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (e.g., lovastatin, simvastatin, pravastatin, fluvastatin, and atorvastatin) may increase the risk for rhabdomyolysis. The dosage of HMG-CoA reductase inhibitors should be reduced.
In addition to TDM (drug levels should be drawn just prior to the dose), patients taking cyclosporine should have frequent monitoring of BUN, creatinine, potassium, and magnesium levels in addition to liver function tests (LFTs) and lipid profiles.
Cyclosporine was designated pregnancy category C; it does not appear to be a major human teratogen; however, it may be associated with increased rates of prematurity. Cyclosporine is present in breast milk. Because of the potential for serious adverse drug reactions in nursing infants, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Tacrolimus
Approved by the FDA in 1994, tacrolimus is the second calcineurin inhibitor approved for prophylaxis of rejection in heart, liver and kidney transplants. It carries a boxed warning for malignancies and serious infections. Patients taking this drug are at increased risk for developing lymphoma and other malignancies. Additionally, patients taking tacrolimus are at risk for developing bacterial, viral, fungal, and protozoal infections.
Dosing for heart transplant recipients begins no sooner than 6 hours posttransplant with a continuous infusion at 0.01 mg/kg/day IV. When oral dosing begins, it should be at a dose of 0.075 mg/kg/day orally divided every 12 hours. For patients with a liver transplant, tacrolimus is administered with a continuous infusion at 0.03 to 0.05 mg/kg/day IV. When oral dosing begins, it should be dosed 0.1 to 0.15mg/kg/day orally divided every two hours. Kidney transplant recipients should begin tacrolimus within 24 hours of receiving the transplanted organ. Dosing ranges from 0.1 to 0.2 mg/kg/day orally divided every 12 hours, and may vary based on drugs used for induction.
For all transplant recipients, dosing is adjusted based on serum drug levels with trough levels ranging between 5 and 20 ng/mL, depending on organ transplanted and length of time since transplant.
The drug is highly nephrotoxic and should be administered at the lowest recommended dose for those with renal impairment. For those with postoperative oliguria, use of tacrolimus may be delayed until kidney function is adequate. All patients on tacrolimus should have their kidney function monitored periodically during therapy.
Tacrolimus may prolong the QT/QTc interval and may cause Torsade de Pointes. Avoid Tacrolimus in patients with congenital long QT syndrome. In patients with congestive heart failure, bradyarrhythmias, those taking certain antiarrhythmic medications or other medicinal products that lead to QT prolongation, and those with electrolyte disturbances such as hypokalemia, hypocalcemia, or hypomagnesemia, consider obtaining electrocardiograms and monitoring electrolytes (magnesium, potassium, calcium) periodically during treatment.
Tacrolimus is slowly absorbed in the GI tract. Bioavailability averages 25% and peak serum concentration is reached in 3 hours. Food, especially food high in fat, slows absorption and reduces bioavailability. It is metabolized extensively in the liver and is over 90% excreted in the feces, with the remaining excreted in the urine. Genetic variations in activity of the CYP3A5 protein can affect serum concentrations of tacrolimus. Its half-life is variable depending on organ transplanted (3.5 to 40.6 hours).
Tacrolimus has multiple common side effects, including tremor, diarrhea, headache, hypertension, nephrotoxicity, infection, insomnia, electrolyte, metabolic and lipid abnormalities, constipation, edema, fever, anemia, hyperglycemia, hepatotoxicity, anorexia, dyspepsia, dyspnea, pruritus, dizziness, cough, leukopenia, and photosensitivity. Serious reactions include malignancy, posttransplant lymphoproliferative disorder, severe infections, Stevens-Johnson syndrome, toxic epidermal necrolysis, anaphylaxis, neurotoxicity, seizures, myocardial hypertrophy, QT prolongation, torsades de pointes, pericardial effusion, diabetes, myelosuppression, DIC, thrombocytopenic purpura, and hemolytic anemia.
Persons receiving tracrolimus have absolute contraindication for live vaccines, mifepristone, pimozide, quinidine, saquinavir, streptozocin, talimogene laherparepvec (a genetically modified oncolytic viral therapy used in patients with recurrent melanoma), and ziprasidone. Multiple other drugs should be used with caution. Protease inhibitors may increase serum drug levels, as can antifungal agents, calcium channel blockers, gastric acid suppressors/antacids, and antibacterials. Anticonvulsants can decrease serum drug levels, as can St. John’s wort. Patients receiving tacrolimus should avoid grapefruit juice, as it too, may increase serum drug levels.
Costimulation Blockers
Belatacept is a first-in-class selective T-cell costimulation blocking agent indicated for use in combination with basiliximab induction, mycophenolate mofetil, and corticosteroids to prevent kidney transplant rejection. It inhibits T-cell CD28 activation and proliferation by binding costimulatory ligands (CD80, CD86) of antigen-presenting cells, thereby inhibiting T-lymphocyte proliferation and the production of the cytokines IL-2 and IL-4, interferon-alfa, and tumor necrosis factor alpha (TNF-α), which are involved in systemic inflammation. Metabolism and excretion of belatacept are unknown, but half-life is 8.2 to 9.8 days.
The prescribed dose must be divisible by 12.5 to accurately prepare the dose from the reconstituted solution. Initial dosing is 10 mg/kg IV prior to surgery on the day of transplant; the dose is repeated on day 5 and at the end of weeks 2, 4, 8, and 12 after transplantation. Maintenance dosing is 5 mg/kg IV at the end of week 16 after transplantation and then every 4 weeks thereafter. Belatacept is not approved for pediatric use.
 This drug carries a boxed warning for increased risk of developing posttransplant lymphoproliferative disorder (PTLD), predominantly involving the central nervous system (CNS). Additionally, recipients without immunity to EBV are at a particularly increased risk, therefore belatacept is for use in EBV-seropositive patients only.
This drug carries a boxed warning for increased risk of developing posttransplant lymphoproliferative disorder (PTLD), predominantly involving the central nervous system (CNS). Additionally, recipients without immunity to EBV are at a particularly increased risk, therefore belatacept is for use in EBV-seropositive patients only.Common side effects of belatacept include infection, anemia, diarrhea, peripheral edema, hypertension, constipation, fever, cough, nausea and vomiting, altered potassium levels, headache, leukopenia, abdominal pain, dyslipidemia, hypophosphatemia, arthralgia, hyperglycemia, proteinuria, increased creatinine, insomnia, hypocalcemia, back pain, dysuria, and anxiety. Serious reactions include PTLD, malignancy, serious infections, progressive multifocal leukoencephalopathy (PML), neutropenia, acute renal failure, nephropathy, and diabetes mellitus.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


