66
Tolerance & Autoimmune Disease
CHAPTER CONTENTS
TOLERANCE
Tolerance is specific immunologic unresponsiveness (i.e., an immune response to a certain antigen [or epitope] does not occur, although the immune system is otherwise functioning normally). In general, antigens that are present during embryonic life are considered “self” and do not stimulate an immunologic response (i.e., we are tolerant to those antigens). The lack of an immune response in the fetus is caused by the deletion of self-reactive T-cell precursors in the thymus (Figure 66–1). On the other hand, antigens that are not present during the process of maturation (i.e., that are encountered first when the body is immunologically mature) are considered “nonself” and usually elicit an immunologic response. Although both B cells and T cells participate in tolerance, it is T-cell tolerance that plays the primary role.
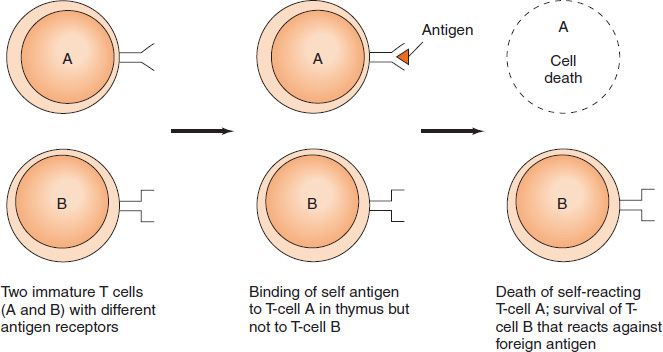
FIGURE 66–1 Production of T-cell tolerance in the thymus.
T-Cell Tolerance
The main process by which T lymphocytes acquire the ability to distinguish self from nonself occurs in the fetal thymus (see Chapter 58). This process, called clonal deletion, involves the killing of T cells (“negative selection”) that react against antigens (primarily self major histocompatibility complex [MHC] proteins) present in the fetus at that time. (Note that exogenous substances injected into the fetus early in development are treated as self.) The self-reactive cells die by a process of programmed cell death called apoptosis. Tolerance to self acquired within the thymus is called central tolerance, whereas tolerance acquired outside the thymus is called peripheral tolerance.
For negative selection and clonal deletion to be efficient, the thymic epithelial cells must display a vast repertoire of “self” proteins. A transcriptional regulator called the autoimmune regulator (AIRE) enhances the synthesis of this array of self proteins. Mutations in the gene encoding the AIRE protein result in the development of an autoimmune disease called autoimmune polyendocrinopathy. The AIRE transcription factor also functions in the peripheral lymphoid organs such as the spleen and lymph nodes, where it contributes to peripheral tolerance.
Peripheral tolerance is necessary because some antigens are not expressed in the thymus and therefore some self-reactive T cells are not killed in the thymus. There are several mechanisms involved in peripheral tolerance: Some self-reactive T cells are killed, some are not activated, and others are suppressed by regulatory T cells producing inhibitory cytokines. Clonal anergy is the term used to describe self-reactive T cells that are not activated because proper costimulation does not occur (Figure 66–2). Clonal ignorance refers to self-reactive T cells that ignore self antigens. These self-reactive T cells are either kept ignorant by physical separation from the target antigens (e.g., the blood–brain barrier) or ignore self antigens because the antigens are present in such small amounts.
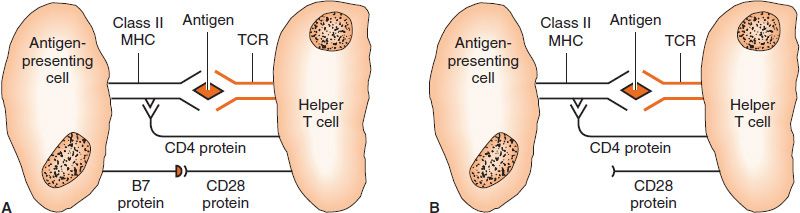
FIGURE 66–2 Clonal anergy outside the thymus. A: B7 protein on the antigen-presenting cell interacts with CD28 on the helper T cell, and full activation of the helper T cell occurs. B: B7 protein on the antigen-presenting cell is not produced; therefore, CD28 on the helper T cell does not get a costimulatory signal. Anergy of the helper T cell occurs despite interaction of the T-cell receptor (TCR) with the antigen.
Although T cells that are clonally anergic are nonreactive, they can become reactive and initiate an autoimmune disease if conditions change later in life. The mechanism of clonal anergy involves the inappropriate presentation of antigen, leading to a failure of interleukin-2 (IL-2) production. Inappropriate presentation is due to a failure of “costimulatory signals” (e.g., sufficient amounts of IL-1 might not be made, or cell surface proteins, such as CD28 on the T cell and B7 on the B cell, might not interact properly, leading to a failure of signal transduction by ras proteins). For example, the inhibitory protein CTLA-4 on the surface of the T cells may displace CD28 and interact with B7, resulting in a failure of T-cell activation. Furthermore, B7 is an inducible protein, and failure to induce it in sufficient amounts can lead to anergy. In addition, the costimulatory proteins, CD40 on the B cell and CD40L on the helper T cell, may fail to interact properly.
The failure of costimulatory signals most often occurs when there is an insufficient inflammatory response at the site of infection. The presence of microbes typically stimulates the production of proinflammatory cytokines such as tumor necrosis factor (TNF) and IL-1. However, if the inflammatory response is insufficient (i.e., if the adjuvant effect of the cytokines is inadequate, the T cells will die instead of being activated).
B-Cell Tolerance
B cells also become tolerant to self by two mechanisms: (1) clonal deletion, probably while the B-cell precursors are in the bone marrow, and (2) clonal anergy of B cells in the periphery. However, tolerance in B cells is less complete than in T cells, an observation supported by the finding that most autoimmune diseases are mediated by antibodies.
B cells bearing an antigen receptor for a self protein can escape clonal deletion (apoptosis) by a process called receptor editing. In this process, a new, different light chain is produced that changes the specificity of the receptor so that it no longer recognizes a self protein. This reduces the risk of autoimmune diseases and increases the repertoire of B cells that can react against foreign proteins. It is estimated that as many as 50% of self-reactive B cells undergo receptor editing. T cells do not undergo receptor editing.
INDUCTION OF TOLERANCE
Whether an antigen will induce tolerance rather than an immunologic response is largely determined by the following:
(1) The immunologic maturity of the host (e.g., neonatal animals are immunologically immature and do not respond well to foreign antigens; for instance, neonates will accept allografts that would be rejected by mature animals).
(2) The structure and dose of the antigen (e.g., a very simple molecule induces tolerance more readily than a complex one, and very high or very low doses of antigen may result in tolerance instead of an immune response). Purified polysaccharides or amino acid copolymers injected in very large doses result in “immune paralysis”—a lack of response.
Other aspects of the induction or maintenance of tolerance are as follows:
(1) T cells become tolerant more readily and remain tolerant longer than B cells.
(2) Administration of a cross-reacting antigen tends to terminate tolerance.
(3) Administration of immunosuppressive drugs enhances tolerance (e.g., in patients who have received organ transplants).
(4) Tolerance is maintained best if the antigen to which the immune system is tolerant continues to be present.
AUTOIMMUNE DISEASES
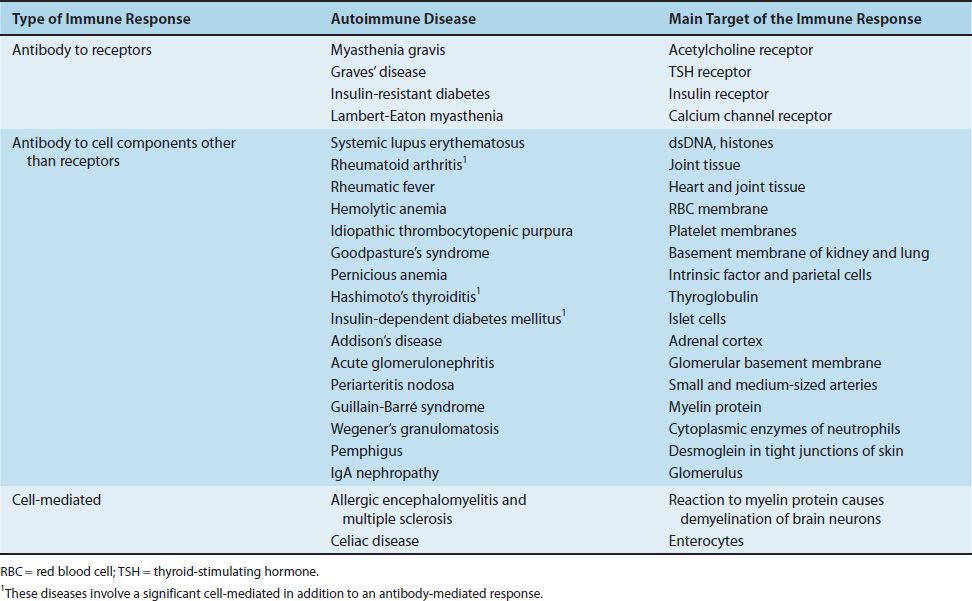
The adult host usually exhibits tolerance to tissue antigens present during fetal life that are recognized as “self.” However, in certain circumstances, tolerance may be lost and immune reactions to host antigens may develop, resulting in autoimmune diseases. The most important step in the production of autoimmune disease is the activation of self-reactive helper (CD4) T cells. These self-reactive Th-1 or Th-2 cells can induce either cell-mediated or antibody-mediated autoimmune reactions, respectively. As described in Table 66–1, most autoimmune diseases are antibody-mediated.
TABLE 66–1 Important Autoimmune Diseases
Genetic Factors
Many autoimmune diseases exhibit a marked familial incidence, which suggests a genetic predisposition to these disorders. There is a strong association of some diseases with certain human leukocyte antigen (HLA) specificities, especially the class II genes. For example, rheumatoid arthritis occurs predominantly in individuals carrying the HLA-DR4 gene. Ankylosing spondylitis is 100 times more likely to occur in people who carry HLA-B27, a class I gene, than in those who do not carry that gene.
There are two hypotheses offered to explain the relationship between certain HLA genes and autoimmune diseases. One is that those genes encode class I or class II MHC proteins that present autoantigens with greater efficiency than do the MHC proteins that are not associated with autoimmune diseases. The other hypothesis is that autoreactive T cells escape negative selection in the thymus because they bind poorly to those class I or class II MHC proteins on the surface of the thymic epithelium.
It should be noted, however, that whether a person develops an autoimmune disease or not is clearly multifactorial, because people with HLA genes known to predispose to certain autoimmune diseases nevertheless do not develop the disease (e.g., many people carrying the HLA-DR4 gene do not develop rheumatoid arthritis). That is to say, HLA genes appear to be necessary but not sufficient to cause autoimmune diseases. In general, class II MHC-related diseases (e.g., rheumatoid arthritis, Graves’ disease [hyperthyroidism], and systemic lupus erythematosus) occur more commonly in women, whereas class I MHC-related diseases (e.g., ankylosing spondylitis and Reiter’s syndrome) occur more commonly in men.
Hormonal Factors
Approximately 90% of all autoimmune diseases occur in women. Although the explanation for this markedly unequal gender ratio is unclear, there is some evidence from animal models that estrogen can alter the B-cell repertoire and enhance the formation of antibody to DNA. Clinically, the observation that systemic lupus erythematosus either appears or exacerbates during pregnancy (or immediately postpartum) supports the idea that hormones play an important role in predisposing women to autoimmune diseases.
Environmental Factors
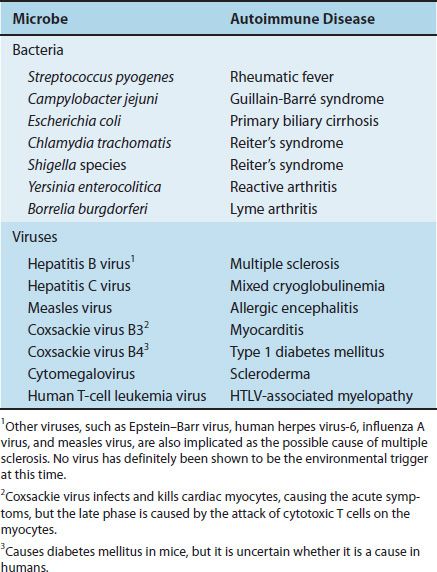
There are several environmental agents that trigger autoimmune diseases, most of which are either bacteria or viruses. For example, pharyngitis caused by Streptococcus pyogenes predisposes to rheumatic fever. Other examples are described in Table 66–2. It is speculative at this time, but members of the normal flora of the bowel are thought to play a role in the genesis of inflammatory bowel diseases, such as Crohn’s disease and ulcerative colitis.
TABLE 66–2 Microbial Infections Associated with Autoimmune Diseases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


