10
Antimicrobial Drugs: Mechanism of Action
CHAPTER CONTENTS
PRINCIPLES OF ANTIMICROBIAL THERAPY
The most important concept underlying antimicrobial therapy is selective toxicity (i.e., selective inhibition of the growth of the microorganism without damage to the host). Selective toxicity is achieved by exploiting the differences between the metabolism and structure of the microorganism and the corresponding features of human cells. For example, penicillins and cephalosporins are effective antibacterial agents because they prevent the synthesis of peptidoglycan, thereby inhibiting the growth of bacterial but not human cells.
There are four major sites in the bacterial cell that are sufficiently different from the human cell that they serve as the basis for the action of clinically effective drugs: cell wall, ribosomes, nucleic acids, and cell membrane (Table 10–1).
TABLE 10–1 Mechanism of Action of Important Antibacterial and Antifungal Drugs
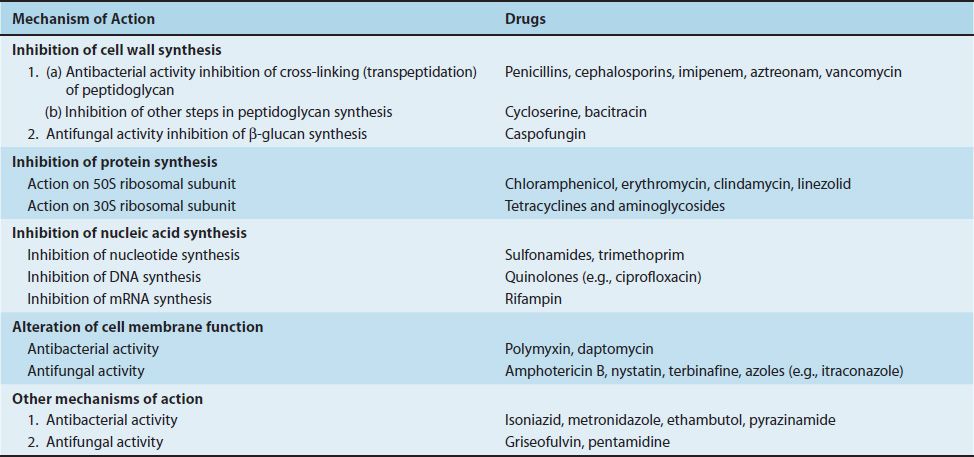
There are far more antibacterial drugs than antiviral drugs. This is a consequence of the difficulty of designing a drug that will selectively inhibit viral replication. Because viruses use many of the normal cellular functions of the host in their growth, it is not easy to develop a drug that specifically inhibits viral functions and does not damage the host cell.
Broad-spectrum antibiotics are active against several types of microorganisms (e.g., tetracyclines are active against many gram-negative rods, chlamydiae, mycoplasmas, and rickettsiae). Narrow-spectrum antibiotics are active against one or very few types (e.g., vancomycin is primarily used against certain gram-positive cocci, namely, staphylococci and enterococci).
Antifungal drugs are included in this chapter because they have similar unique sites of action such as cell walls, cell membranes, and nucleic acid synthesis. Additional information on antifungal drugs is given in Chapter 47.
BACTERICIDAL & BACTERIOSTATIC ACTIVITY
In some clinical situations, it is essential to use a bactericidal drug rather than a bacteriostatic one. A bactericidal drug kills bacteria, whereas a bacteriostatic drug inhibits their growth but does not kill them (Figure 10–1). The salient features of the behavior of bacteriostatic drugs are that (1) the bacteria can grow again when the drug is withdrawn, and (2) host defense mechanisms, such as phagocytosis, are required to kill the bacteria. Bactericidal drugs are particularly useful in certain infections (e.g., those that are immediately life-threatening; those in patients whose polymorphonuclear leukocyte count is below 500/μL; and endocarditis, in which phagocytosis is limited by the fibrinous network of the vegetations and bacteriostatic drugs do not effect a cure).
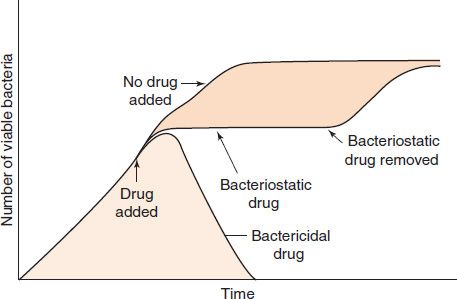
FIGURE 10–1 Bactericidal and bacteriostatic activity of antimicrobial drugs. Either a bactericidal or a bacteriostatic drug is added to the growing bacterial culture at the time indicated by the arrow. After a brief lag time during which the drug enters the bacteria, the bactericidal drug kills the bacteria, and a decrease in the number of viable bacteria occurs. The bacteriostatic drug causes the bacteria to stop growing, as indicated by the dotted line, but if the bacteriostatic drug is removed from the culture, the bacteria resume growing.
MECHANISMS OF ACTION
INHIBITION OF CELL WALL SYNTHESIS
1. Inhibition of Bacterial Cell Wall Synthesis
Penicillins
Penicillins (and cephalosporins) act by inhibiting transpeptidases, the enzymes that catalyze the final cross-linking step in the synthesis of peptidoglycan (see Figure 2–5). For example, in Staphylococcus aureus, transpeptidation occurs between the amino group on the end of the pentaglycine cross-link and the terminal carboxyl group of the D-alanine on the tetrapeptide side chain. Because the stereochemistry of penicillin is similar to that of a dipeptide, D-alanyl-D-alanine, penicillin can bind to the active site of the transpeptidase and inhibit its activity.
Two additional factors are involved in the action of penicillin:
(1) The first is that penicillin binds to a variety of receptors in the bacterial cell membrane and cell wall, called penicillin-binding proteins (PBPs). Some PBPs are transpeptidases; the others function in the synthesis of peptidoglycan. Their specific functions are beyond the scope of this book. Changes in PBPs are in part responsible for an organism’s becoming resistant to penicillin.
(2) The second factor is that autolytic enzymes called murein hydrolases (murein is a synonym for peptidoglycan) are activated in penicillin-treated cells and degrade the peptidoglycan. Some bacteria (e.g., strains of S. aureus) are tolerant to the action of penicillin, because these autolytic enzymes are not activated. A tolerant organism is one that is inhibited but not killed by a drug that is usually bactericidal, such as penicillin (see page 89).
Penicillin-treated cells die by rupture as a result of the influx of water into the high-osmotic-pressure interior of the bacterial cell. If the osmotic pressure of the medium is raised about threefold (e.g., by the addition of sufficient KCl), rupture will not occur and the organism can survive as a protoplast. Exposure of the bacterial cell to lysozyme, which is present in human tears, results in degradation of the peptidoglycan and osmotic rupture similar to that caused by penicillin.
Penicillin is bactericidal, but it kills cells only when they are growing. When cells are growing, new peptidoglycan is being synthesized, and transpeptidation occurs. However, in nongrowing cells, no new cross-linkages are required, and penicillin is inactive. Penicillins are therefore more active during the log phase of bacterial cell growth than during the stationary phase (see Chapter 3 for the bacterial cell growth cycle).
Penicillins (and cephalosporins) are called β-lactam drugs because of the importance of the β-lactam ring (Figure 10–2). An intact ring structure is essential for antibacterial activity; cleavage of the ring by penicillinases (β-lactamases) inactivates the drug. The most important naturally occurring compound is benzylpenicillin (penicillin G), which is composed of the 6-aminopenicillanic acid nucleus that all penicillins have, plus a benzyl side chain (see Figure 10–2). Penicillin G is available in three main forms:
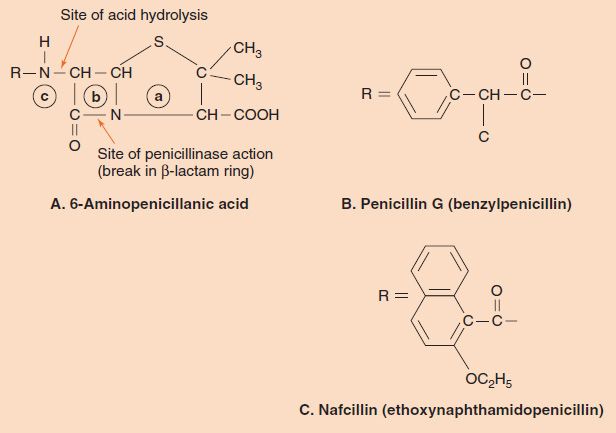
FIGURE 10–2 Penicillins. A: The 6-aminopenicillanic acid nucleus is composed of a thiazolidine ring (a), a β-lactam ring (b), and an amino group (c). The sites of inactivation by stomach acid and by penicillinase are indicated. B: The benzyl group, which forms benzylpenicillin (penicillin G) when attached at R. C: The large aromatic ring substituent that forms nafcillin, a β-lactamase–resistant penicillin, when attached at R. The large ring blocks the access of β-lactamase to the β-lactam ring.
(1) Aqueous penicillin G, which is metabolized most rapidly.
(2) Procaine penicillin G, in which penicillin G is conjugated to procaine. This form is metabolized more slowly and is less painful when injected intramuscularly because the procaine acts as an anesthetic.
(3) Benzathine penicillin G, in which penicillin G is conjugated to benzathine. This form is metabolized very slowly and is often called a “depot” preparation.
Benzylpenicillin is one of the most widely used and effective antibiotics. However, it has four disadvantages, three of which have been successfully overcome by chemical modification of the side chain. The three disadvantages are (1) limited effectiveness against many gram-negative rods, (2) hydrolysis by gastric acids, so that it cannot be taken orally, and (3) inactivation by β-lactamases. The limited effectiveness of penicillin G against gram-negative rods is due to the inability of the drug to penetrate the outer membrane of the organism. The fourth disadvantage common to all penicillins that has not been overcome is hypersensitivity, especially anaphylaxis, in some recipients of the drug.
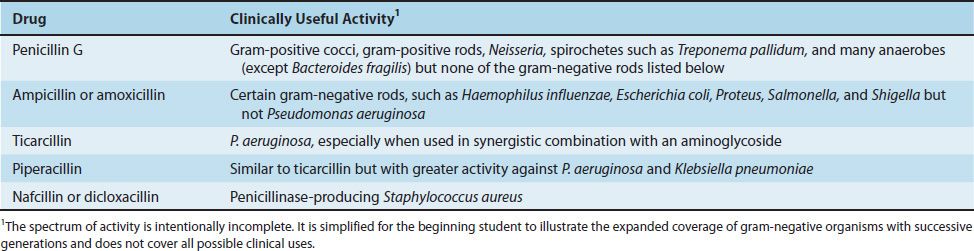
The effectiveness of penicillins against gram-negative rods has been increased by a series of chemical changes in the side chain (Table 10–2). It can be seen that ampicillin and amoxicillin have activity against several gram-negative rods that the earlier penicillins do not have. However, these drugs are not useful against Pseudomonas aeruginosa and Klebsiella pneumoniae. Hence other penicillins were introduced. Generally speaking, as the activity against gram-negative bacteria increases, the activity against gram-positive bacteria decreases.
TABLE 10–2 Activity of Selected Penicillins
The second important disadvantage—acid hydrolysis in the stomach—also has been addressed by modification of the side chain. The site of acid hydrolysis is the amide bond between the side chain and penicillanic acid nucleus (see Figure 10–1). Minor modifications of the side chain in that region, such as addition of an oxygen (to produce penicillin V) or an amino group (to produce ampicillin), prevent hydrolysis and allow the drug to be taken orally.
The inactivation of penicillin G by β-lactamases is another important disadvantage, especially in the treatment of S. aureus infections. Access of the enzyme to the β-lactam ring is blocked by modification of the side chain with the addition of large aromatic rings containing bulky methyl or ethyl groups (methicillin, oxacillin, nafcillin, etc.; Figure 10–2). Another defense against β-lactamases is inhibitors such as clavulanic acid and sulbactam. These are structural analogues of penicillin that have little antibacterial activity but bind strongly to β-lactamases and thus protect the penicillin. Combinations, such as amoxicillin and clavulanic acid (Augmentin), are in clinical use. Some bacteria resistant to these combinations have been isolated from patient specimens.
Penicillins are usually nontoxic at clinically effective levels. The major disadvantage of these compounds is hypersensitivity, which is estimated to occur in 1% to 10% of patients. The hypersensitivity reactions include anaphylaxis, skin rashes, hemolytic anemia, nephritis, and drug fever. A maculopapular drug-induced rash is quite common. Anaphylaxis, the most serious complication, occurs in 0.5% of patients. Death as a result of anaphylaxis occurs in 0.002% (1:50,000) of patients.
Cephalosporins
Cephalosporins are β-lactam drugs that act in the same manner as penicillins (i.e., they are bactericidal agents that inhibit the cross-linking of peptidoglycan). The structures, however, are different: Cephalosporins have a six-membered ring adjacent to the β-lactam ring and are substituted in two places on the 7-aminocephalosporanic acid nucleus (Figure 10–3), whereas penicillins have a five-membered ring and are substituted in only one place.
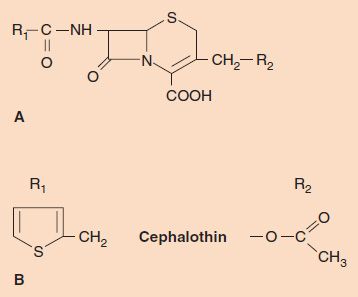
FIGURE 10–3 Cephalosporins. A: The 7-aminocephalosporanic acid nucleus. B: The two R groups in the drug cephalothin.
The first-generation cephalosporins are active primarily against gram-positive cocci (Table 10–3). Similar to the penicillins, new cephalosporins were synthesized with expansion of activity against gram-negative rods as the goal. These new cephalosporins have been categorized into second, third, and fourth generations, with each generation having expanded coverage against certain gram-negative rods. The fourth- and fifth-generation cephalosporins have activity against many gram-positive cocci as well.
TABLE 10–3 Activity of Selected Cephalosporins1

Cephalosporins are effective against a broad range of organisms, are generally well tolerated, and produce fewer hypersensitivity reactions than do the penicillins. Despite the structural similarity, a patient allergic to penicillin has only about a 10% chance of being hypersensitive to cephalosporins also. Most cephalosporins are the products of molds of the genus Cephalosporium; a few, such as cefoxitin, are made by the actinomycete Streptomyces.
Carbapenems
Carbapenems are β-lactam drugs that are structurally different from penicillins and cephalosporins. For example, imipenem (N-formimidoylthienamycin), a commonly used carbapenem, has a methylene group in the ring in place of the sulfur (Figure 10–4). Imipenem has one of the widest spectrums of activity of the β-lactam drugs. It has excellent bactericidal activity against many gram-positive, gram-negative, and anaerobic bacteria. It is effective against most gram-positive cocci (e.g., streptococci and staphylococci), most gram-negative cocci (e.g., Neisseria), many gram-negative rods (e.g., Pseudomonas, Haemophilus, and members of the family Enterobacteriaceae such as Escherichia coli), and various anaerobes (e.g., Bacteroides and Clostridium). Imipenem is especially useful in treating infections caused by gram-negative rods that produce extended-spectrum β-lactamases that make them resistant to all penicillins and cephalosporins. Carbapenems are often the “drugs of last resort” against bacteria resistant to multiple antibiotics.
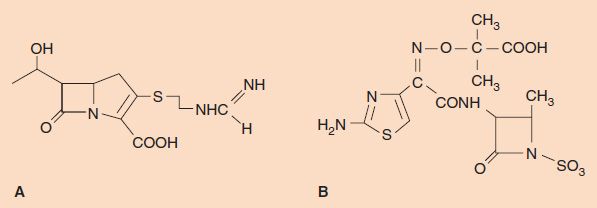
FIGURE 10–4 A: Imipenem. B: Aztreonam.
Imipenem is prescribed in combination with cilastatin, which is an inhibitor of dehydropeptidase, a kidney enzyme that inactivates imipenem. Imipenem is not inactivated by most β-lactamases; however, carbapenemases produced by K. pneumoniae that degrade imipenem and other carbapenemases have emerged. Other carbapenems, such as ertapenem and meropenem, are not inactivated by dehydropeptidase and are not prescribed in combination with cilastatin.
Monobactams
Monobactams are also β-lactam drugs that are structurally different from penicillins and cephalosporins. Monobactams are characterized by a β-lactam ring without an adjacent sulfur-containing ring structure (i.e., they are monocyclic) (Figure 10–4). Aztreonam, currently the most useful monobactam, has excellent activity against many gram-negative rods, such as Enterobacteriaceae and Pseudomonas, but is inactive against gram-positive and anaerobic bacteria. It is resistant to most β-lactamases. It is very useful in patients who are hypersensitive to penicillin, because there is no cross-reactivity.
Vancomycin
Vancomycin is a glycopeptide that inhibits cell wall peptidoglycan synthesis by blocking transpeptidation but by a mechanism different from that of the β-lactam drugs. Vancomycin binds directly to the D-alanyl-D-alanine portion of the pentapeptide, which blocks the transpeptidase from binding, whereas the β-lactam drugs bind to the transpeptidase itself. Vancomycin also inhibits a second enzyme, the bacterial transglycosylase, which also functions in synthesizing the peptidoglycan, but this appears to be less important than inhibition of the transpeptidase.
Vancomycin is a bactericidal agent effective against certain gram-positive bacteria. Its most important use is in the treatment of infections caused by S. aureus strains that are resistant to the penicillinase-resistant penicillins such as nafcillin and methicillin (e.g., methicillin-resistant S. aureus [MRSA]). Note that vancomycin is not a β-lactam drug and, therefore, is not degraded by β-lactamase. Vancomycin is also used in the treatment of infections caused by Staphylococcus epidermidis and enterococci. Strains of S. aureus, S. epidermidis, and enterococci with partial or complete resistance to vancomycin have been recovered from patients.
Telavancin is a synthetic derivative of vancomycin that both inhibits peptidoglycan synthesis and disrupts bacterial cell membranes. It is used for the treatment of skin and soft tissue infections, especially those caused by MRSA.
A well-known adverse effect of vancomycin is “red man” syndrome. “Red” refers to the flushing caused by vasodilation induced by histamine release from mast cells and basophils. This is a direct effect of vancomycin on these cells and is not an IgE–mediated response.
Cycloserine & Bacitracin
Cycloserine is a structural analogue of D-alanine that inhibits the synthesis of the cell wall dipeptide D-alanyl-D-alanine. It is used as a second-line drug in the treatment of tuberculosis. Bacitracin is a cyclic polypeptide antibiotic that prevents the dephosphorylation of the phospholipid that carries the peptidoglycan subunit across the cell membrane. This blocks the regeneration of the lipid carrier and inhibits cell wall synthesis. Bacitracin is a bactericidal drug useful in the treatment of superficial skin infections but too toxic for systemic use.
2. Inhibition of Fungal Cell Wall Synthesis
Echinocandins, such as caspofungin (Cancidas) and micafungin (Mycamine), are lipopeptides that block fungal cell wall synthesis by inhibiting the enzyme that synthesizes β-glucan. β-Glucan is a polysaccharide composed of long chains of D-glucose, which is an essential component of certain medically important fungal pathogens.
Caspofungin inhibits the growth of Aspergillus and Candida but not Cryptococcus or Mucor. Caspofungin is used for the treatment of disseminated candidiasis and for the treatment of invasive aspergillosis that does not respond to amphotericin B. Micafungin is approved for the treatment of esophageal candidiasis and the prophylaxis of invasive Candida infections in bone marrow transplant patients. Anidulafungin is approved for the treatment of esophageal candidiasis and other serious Candida infections.
INHIBITION OF PROTEIN SYNTHESIS
Several drugs inhibit protein synthesis in bacteria without significantly interfering with protein synthesis in human cells. This selectivity is due to the differences between bacterial and human ribosomal proteins, RNAs, and associated enzymes. Bacteria have 70S1 ribosomes with 50S and 30S subunits, whereas human cells have 80S ribosomes with 60S and 40S subunits.
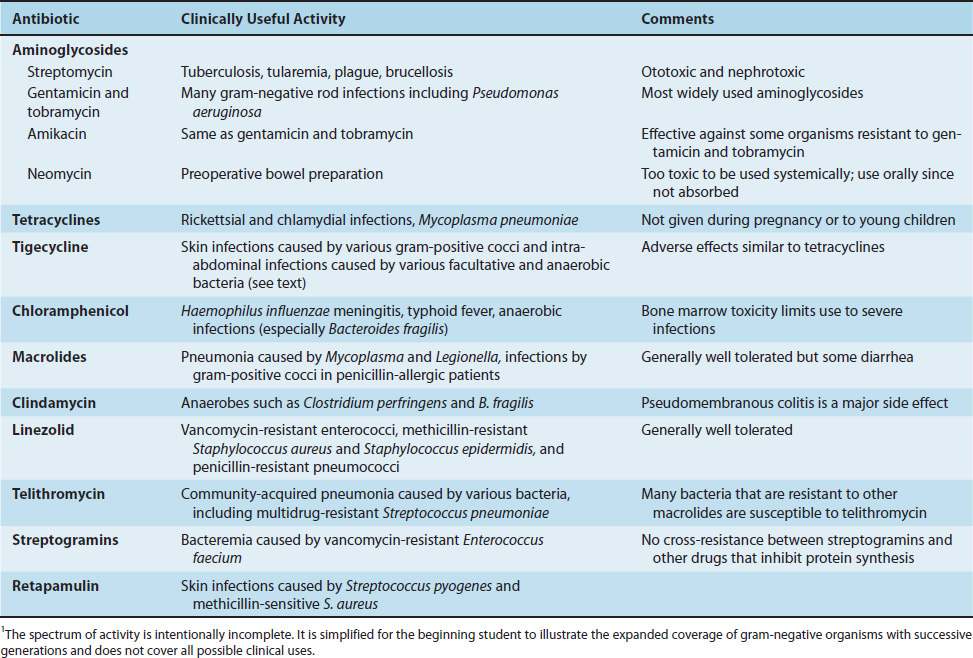
Chloramphenicol, erythromycin, clindamycin, and linezolid act on the 50S subunit, whereas tetracyclines and aminoglycosides act on the 30S subunit. A summary of the modes of action of these drugs is presented in Table 10–4, and a summary of their clinically useful activity is presented in Table 10–5.
TABLE 10–4 Mode of Action of Antibiotics That Inhibit Protein Synthesis
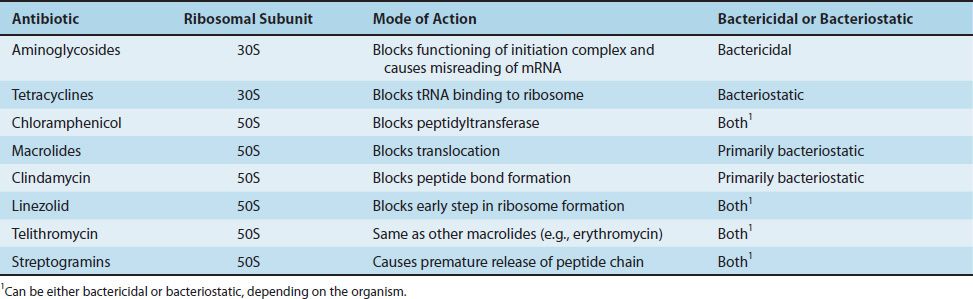
TABLE 10–5 Spectrum of Activity of Antibiotics That Inhibit Protein Synthesis1
1. Drugs That Act on the 30S Subunit
Aminoglycosides
Aminoglycosides are bactericidal drugs especially useful against many gram-negative rods. Certain aminoglycosides are used against other organisms (e.g., streptomycin is used in the multidrug therapy of tuberculosis, and gentamicin is used in combination with penicillin G against enterococci). Aminoglycosides are named for the amino sugar component of the molecule, which is connected by a glycosidic linkage to other sugar derivatives (Figure 10–5).
FIGURE 10–5 Aminoglycosides. Aminoglycosides consist of amino sugars joined by a glycosidic linkage. The structure of gentamicin is shown.
The two important modes of action of aminoglycosides have been documented best for streptomycin; other aminoglycosides probably act similarly. Both inhibition of the initiation complex and misreading of messenger RNA (mRNA) occur; the former is probably more important for the bactericidal activity of the drug. An initiation complex composed of a streptomycin-treated 30S subunit, a 50S subunit, and mRNA will not function—that is, no peptide bonds are formed, no polysomes are made, and a frozen “streptomycin monosome” results. Misreading of the triplet codon of mRNA so that the wrong amino acid is inserted into the protein also occurs in streptomycin-treated bacteria. The site of action on the 30S subunit includes both a ribosomal protein and the ribosomal RNA (rRNA). As a result of inhibition of initiation and misreading, membrane damage occurs and the bacterium dies. (In 1993, another possible mode of action was described, namely, that aminoglycosides inhibit ribozyme-mediated self-splicing of rRNA.)
Aminoglycosides have certain limitations in their use: (1) They have a toxic effect both on the kidneys and on the auditory and vestibular portions of the eighth cranial nerve. To avoid toxicity, serum levels of the drug, blood urea nitrogen, and creatinine should be measured. (2) They are poorly absorbed from the gastrointestinal tract and cannot be given orally. (3) They penetrate the spinal fluid poorly and must be given intrathecally in the treatment of meningitis. (4) They are ineffective against anaerobes, because their transport into the bacterial cell requires oxygen.
Tetracyclines
Tetracyclines are a family of antibiotics with bacteriostatic activity against a variety of gram-positive and gram-negative bacteria, mycoplasmas, chlamydiae, and rickettsiae. They inhibit protein synthesis by binding to the 30S ribosomal subunit and by blocking the aminoacyl transfer RNA (tRNA) from entering the acceptor site on the ribosome. However, the selective action of tetracycline on bacteria is not at the level of the ribosome, because tetracycline in vitro will inhibit protein synthesis equally well in purified ribosomes from both bacterial and human cells. Its selectivity is based on its greatly increased uptake into susceptible bacterial cells compared with human cells.
Tetracyclines, as the name indicates, have four cyclic rings with different substituents at the three R groups (Figure 10–6). The various tetracyclines (e.g., doxycycline, minocycline, oxytetracycline) have similar antimicrobial activity but different pharmacologic properties. In general, tetracyclines have low toxicity but are associated with some important side effects. One is suppression of the normal flora of the intestinal tract, which can lead to diarrhea and overgrowth by drug-resistant bacteria and fungi. Second is that suppression of Lactobacillus in the vaginal normal flora results in a rise in pH, which allows Candida albicans to grow and cause vaginitis. Third is brown staining of the teeth of fetuses and young children as a result of deposition of the drug in developing teeth; tetracyclines are avid calcium chelators. For this reason, tetracyclines are contraindicated for use in pregnant women and in children younger than 8 years of age. Tetracyclines also chelate iron, and so products containing iron, such as iron-containing vitamins, should not be taken during therapy with tetracyclines. Photosensitivity (rash upon exposure to sunlight) can also occur during tetracycline therapy.
FIGURE 10–6 Tetracycline structure. The four-ring structure is depicted with its three R sites. Chlortetracycline, for example, has R = Cl, R1 = CH3, and R2 = H.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


