DEVELOPMENTAL ANOMALIES
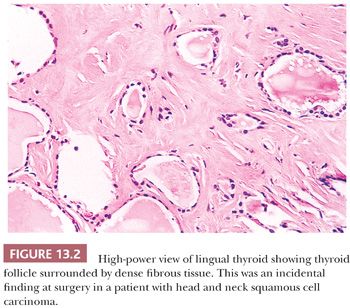
The most common anomalies encountered with the thyroid gland include abnormalities in gland development. Thyroid maldescent can result in the presence of lingual thyroid located in the back of the tongue; as this tissue grows, it could become a surgical emergency in young children (Fig. 13.2) (5). It is critical to be aware that in patients with lingual thyroid, this may be the only thyroid that is present. Its removal, therefore, could result in profound acute hypothyroidism. Thyroid tissue can be found in any location along the thyroglossal duct. In fact, more than 60% of thyroglossal duct cysts will contain thyroid tissue in the wall. Because of its close association with the development of other tissues, thyroid tissue may be seen in association with the esophagus, larynx, trachea, jugular carotid lymph nodes, soft tissues of the neck, and even in association with the heart and great vessels (6,7).
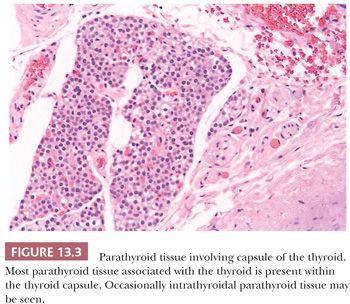
Because of the intimate developmental relationships, derivatives of the branchial or pharyngeal pouches (parathyroid tissue, salivary gland remnants, and thymus) can sometimes be found in the thyroid (Fig. 13.3). Developmental considerations also explain the finding of normal thyroid tissue in the cervical fat or muscle (8). Fat, cartilage, or muscle may occasionally be found within the thyroid capsule (9,10). These minor abnormalities of development must be remembered lest they be confused with infiltrative neoplastic growth.
Benign-appearing thyroid tissue in lymph node tissue should be evaluated with extreme caution. It is believed by some, but not all, that lymph nodes medial to the jugular vein may contain benign thyroid tissue within the capsule (11). These thyroid inclusions must be contained within the capsule and must be composed of normal-appearing thyroid without atypical nuclei, psammoma bodies, or papillary group formation (12,13). Although normal thyroid tissue may be present in soft tissues of the lateral neck, thyroid tissue in a lymph node lateral to the jugular vein, no matter how benign appearing, represents metastatic papillary carcinoma (14).
Occasionally, the thyroid gland may not develop or only partially develop. Thyroid hemiaplasia is much more common than complete aplasia, although hemiaplasia is quite rare. Thyroid hemiagenesis has been associated with Graves disease, although the exact nature of this association is not known (15).
ANATOMY OF THE THYROID
The normal adult thyroid consists of two lobes connected by an isthmus. The normal gland weight ranges from 14 to 18 g and depends on sex, size, and nutritional status of the individual, as well as appropriate iodine intake (4,16). The parathyroids and recurrent laryngeal nerves lie behind the gland (17). The superior and inferior thyroidal arteries supply the gland. The intraglandular and subcapsular lymphatics drain into the internal jugular lymph nodes (4,18).
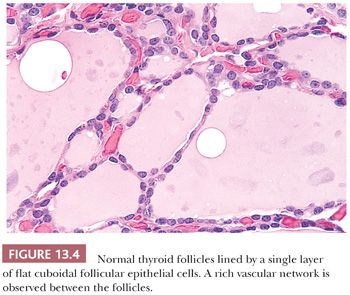
Thyroid tissue is light brown in color and firm in consistency. Nodules are not uncommon in the euthyroid adult population (19). Microscopically, the thyroid is composed of follicles lined by epithelial cells that surround the central colloid; 20 to 40 follicles make up a lobule (Fig. 13.4) (19). Birefringent crystalline material (shown by chemical analysis and x-ray diffraction analysis to be calcium oxalate) is commonly found in the colloid of normal or diseased thyroids (20). Small collections of C cells are situated within the confines of the basement membrane of the thyroid follicles. Difficult to identify by ordinary histologic stains, the normal C cells can be identified by immunostaining for calcitonin (21).
Ultrastructural studies of normal thyroid show that the follicular cells are arranged in a single layer around the central colloid (22). The cells contain liposomes and a complement of endoplasmic reticulum and small mitochondria. The nuclei are round with homogenous chromatin. Well-developed desmosomes and terminal bars separate cells. In the interstitium, numerous fenestrated capillaries are noted (22). The C cells are found within the confines of the basement membranes of the follicles. In the cytoplasm of these C cells, numerous double membrane-bound neurosecretory granules containing calcitonin are found (21,23).
DIFFUSE THYROID ENLARGEMENTS
THE THYROIDITIDES (TABLE 13.1)

Although occasionally presenting as nodules or asymmetric enlargement of the gland, thyroiditis commonly involves the thyroid diffusely.
Acute Thyroiditis
Acute thyroiditis is rare and is almost always a result of infection, although acute thyroiditis may be encountered in the thyroid shortly following radiation exposure (24–26). The disease is most commonly encountered in malnourished children, elderly debilitated adults, immunocompromised individuals, or in otherwise healthy patients following trauma to the neck. Most patients present with painful enlargement of the gland. Grossly, the gland often appears normal but may be focally or diffusely softened with areas of purulence. Microscopically, acute inflammation with microabscess formation is present. Microorganisms may be seen. A variety of organisms cause thyroiditis including bacteria, fungi, and viruses (26,27). In individuals with human immunodeficiency virus infection, the occurrence of Pneumocystis jiroveci infection has been reported.
Granulomatous Thyroiditis
Granulomatous subacute thyroiditis, also referred to as nonsuppurative thyroiditis or de Quervain disease, is a rare entity that usually presents in women and has been associated with HLA-Bw35 (26,28). The changes seen in the gland are most likely a result of the response of the thyroid to systemic viral infection; some authors suggest that it represents actual viral infection of the gland. A viral cause for this disorder can be supported by both clinical and epidemiologic studies. “Epidemics” of subacute thyroiditis have been reported (29). Most patients with subacute thyroiditis recover without any permanent damage to the thyroid. However, some studies have reported end-stage hypothyroidism in 5% to 9% of patients (28).
Grossly, the thyroid is asymmetrically enlarged and firm. Irregular white-tan lesions or several small poorly demarcated nodules may simulate carcinoma (26,30). Microscopically, early in the disease, there is loss of the follicular epithelium and colloid depletion. Leakage of stored hormone occurs, and clinical hyperthyroidism may result. The inflammatory response, composed initially of polymorphonuclear leukocytes and even microabscesses, progresses until lymphocytes, plasma cells, and histiocytes become the major inflammatory cells. A rim of histiocytes and giant cells replaces the follicular epithelium, giving rise to a granulomatous appearance. A central fibrotic reaction occurs. Recovery is associated with regeneration of follicles from the viable edges of the involved areas (26,31).
Palpation Thyroiditis
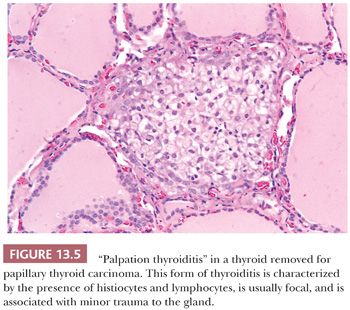
Palpation thyroiditis (multifocal granulomatous folliculitis) is found in 85% to 95% of surgically resected thyroids and probably represents the thyroid’s response to minor trauma. The histologic features of this lesion include multiple isolated follicles or small groups of follicles that show partial or circumferential loss of epithelium and replacement of the lost epithelium by inflammatory cells, predominantly macrophages (Fig. 13.5). The lesions most likely regress and sometimes need to be differentiated from C-cell hyperplasia and ultimobranchial body remnants (32).
AUTOIMMUNE THYROID DISEASE
Autoimmune thyroid disease (AITD) encompasses a spectrum of clinical and morphologic entities in which interrelationships are speculative but which share certain features suggesting their autoimmune etiology. The spectrum includes diffuse toxic goiter (Graves disease) associated with hyperthyroidism on the one hand and lymphocytic thyroiditis (Hashimoto disease) associated with hypothyroidism on the other. However, in between, various lesions associated with hyperthyroidism, hypothyroidism, or euthyroidism can be found. This section briefly summarizes the current thinking on autoimmunity as related to thyroid disease and discusses the pathologic aspects of the autoimmune entities most commonly seen in surgical practice.
The early work of Volpe and colleagues suggested that AITD involves an inherited defect in immune surveillance (33). The current understanding is that AITD is a polygenetic disease in which susceptibility genes and environmental factors act together to initiate both cellular and humoral immune responses against the thyroid gland (34,35). Genome-wide screening and linkage analyses have identified several chromosomal foci that are linked to AITD. These are HT-1 (chromosome 13q33) and HT-2 (chromosome 12q22) for Hashimoto thyroiditis and GD-1 (chromosome 14q31), GD-2 (chromosome 20q11.2), and GD-3 (chromosome Xq21) for Graves disease. In addition, several other genes have been proposed as susceptibility or immunoregulatory genes. These include major histocompatibility complex (chromosome 6), cytotoxic T-lymphocyte–associated antigen-4 (CTLA-4) gene (chromosome 2), the CD40 (chromosome 20), thyroglobulin gene (chromosome 8), and the autoimmune regulator gene (chromosome 21) (35–38). The exact mechanism of involvement of environmental factors in AITD is still not clearly understood; however, it is theorized to include dietary iodide, medication, and infection (38). Recently, Dittmar et al. (39) demonstrated downregulation of deoxyribonuclease I (DNase I) mRNA in patients with AITD, which may lead to decreased degradation of DNA from dying thyroid follicular cells causing development of thyroid autoimmunity.
PATHOLOGY OF AUTOIMMUNE THYROID DISEASE
The presence of lymphoid cells in the substance of the thyroid parenchyma probably reflects an abnormal immunologic state. However, the interrelationships among classic chronic thyroiditis, its variants, and “nonspecific” thyroiditis are problematic.
The morphologic and immunopathologic overlap between nonspecific lymphocytic thyroiditis and Hashimoto disease suggest that they represent a spectrum of autoimmune injury (37,54–56).
In Hashimoto thyroiditis (40,41), the gland is firm and symmetrically enlarged, weighing from 25 to 250 g (41). Normal thyroid lobulation is accentuated by interlobular fibrosis. The thyroid has a tan-yellow appearance attributed to the abundant lymphoid tissue. The thyroid follicles are small and atrophic. Colloid appears dense or may be absent. Follicular cells are metaplastic and include oncocytic (Hürthle cell), clear cell, and squamous types (Fig. 13.6) (25,26,41). In the stroma and in atrophic follicles, a lymphoplasmacytic infiltration with well-developed germinal centers is found. Variable degrees of interlobular fibrosis are seen. One may also encounter hyperplasia of the ultimobranchial body rests. The nuclei of the follicular epithelial cells often show nuclear clearing, enlargement, and overlapping; this reactive nuclear change can be mistaken for foci of PTC (Fig. 13.6) (26,42). Apoptosis is believed to be the mechanism for thyroid follicular cell destruction in Hashimoto disease (43).
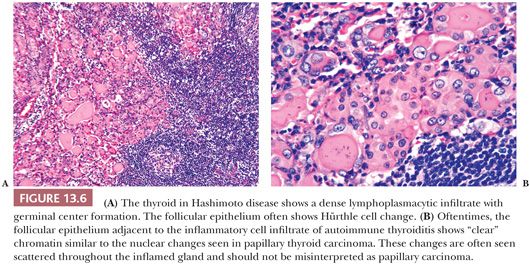
The lymphocytic infiltrate is composed of both T and B cells in an almost 1:1 ratio, differing from the peripheral blood, which shows T-cell predominance (44). T lymphocytes within the thyroid are predominantly suppressor type, whereas the peripheral blood of these patients contains mostly helper T cells (45). The B cells are usually of the immunoglobulin (Ig) G-κ subclass.
Patients with Hashimoto thyroiditis are at increased risk of neoplasia, with the most common malignancy being malignant lymphoma, B-cell type (46,47). In addition, patients with Hashimoto disease may be prone to the development of plasmacytomas within the gland (48). A peculiar variant of mucoepidermoid carcinoma, known as sclerosing mucoepidermoid carcinoma with eosinophilia, has been recognized in patients with Hashimoto disease (49). Whether there is an increase in the incidence of papillary carcinoma—especially microcarcinoma—is still debated.
CHRONIC LYMPHOCYTIC THYROIDITIS CLASSIFICATION
Mizukami and colleagues (50) established a new classification of chronic lymphocytic thyroiditis. This classification is useful because it allows one to see that the mere presence of lymphocytes in the thyroid does not indicate autoimmune disease. They divided their patients into four groups:
1. Chronic thyroiditis, oxyphilic: This group contains patients with classic Hashimoto disease histology.
2. Chronic thyroiditis, mixed: This group shows less of an infiltrate than group 1 with minimal fibrosis. Patients demonstrate normal thyroid, hyperthyroidism, or hypothyroidism.
3. Chronic thyroiditis, hyperplastic: This group shows glandular hyperplasia associated with only a small lymphocytic reaction. Most patients are hyperthyroid.
4. Chronic thyroiditis, focal: This group shows only a focal lymphocytic reaction, and most patients are euthyroid.
FIBROSING VARIANT OF HASHIMOTO THYROIDITIS
The fibrous or fibrosing variant of Hashimoto thyroiditis accounts for approximately 10% to 13% of all cases of Hashimoto disease. Pathologically, the thyroid architecture is destroyed with severe follicular atrophy, dense keloid-like fibrosis, and prominent squamous or epidermoid metaplasia of the follicular epithelium.
Several other fibrosing entities, such as Riedel disease, tumefactive fibrosis of the head and neck, and fibrous atrophy (idiopathic myxedema), need to be differentiated from the fibrosing variant of Hashimoto disease. In fibrosing thyroiditis, the sclerosis is confined to the thyroid, and the lobular architecture of the gland is maintained (50,51).
PAINLESS/SILENT THYROIDITIS
Painless thyroiditis is an autoimmune disease that causes painless enlargement of thyroid gland along with brief hyperthyroidism followed by hypothyroidism. It can occur in the postpartum period and is called postpartum thyroiditis. Mizukami and colleagues (52) analyzed 26 biopsies from patients with painless thyroiditis. All showed follicular disruption and lymphocytic infiltration, but stromal fibrous and oxyphilic changes were rare. Subset analysis of the intrathyroidal lymphocytes showed similarities to Hashimoto thyroiditis (52,53).
FOCAL NONSPECIFIC THYROIDITIS
Lymphocytic infiltration of the thyroid is found more frequently at autopsy and in surgical specimens since the addition of iodide to the water supplies of the United States approximately 60 years ago (54). It has been suggested that iodide (iodine) may combine with a protein, act as an antigen, and evoke an immune response localized to the thyroid gland. Postmortem studies indicate an incidence of focal lymphocytic thyroiditis of approximately 15% to 20% in women and rarely in men. Kurashima and Hirokawa (55) believe that focal lymphocytic infiltration of the thyroid represents an immunologic disorder associated with aging.
Focal aggregates of lymphocytes are noted, and occasionally germinal-center formation, but oncocytes are rarely present. Follicular atrophy is also rare (55).
OTHER ENTITIES WITH LYMPHOCYTES IN THE THYROID
Diffuse Toxic Goiter/Graves Disease
Autoimmune hyperthyroidism occurs predominantly in women (female-to-male ratio of approximately 9:1) (56). Patients with Graves disease show a genetic predisposition to the disease, and their relatives have an unusually high incidence of other autoimmune disorders; the gland is infiltrated by lymphocytes (57,58). Similarities to Hashimoto thyroiditis include an apparently genetically determined defect in suppressor T cells with consequent proliferation of thyroid-directed B cells, which produce antithyroid autoantibodies. These antibodies (thyroid receptor antibodies [TRAbs]) are directed against the thyrotropin receptor complex of the follicular epithelial cell, which they stimulate to grow and function (34,59). Hence, the gland enlarges and secretes increased amounts of thyroid hormones, resulting in clinical hyperthyroidism.
Grossly, the thyroid in Graves disease is diffusely and usually symmetrically enlarged, with weights of 50 to 150 g. Vascularity is marked. Histologic examination shows retention of lobular architecture, prominent vascular congestion, and follicular hyperplasia. The follicular cells are columnar with enlarged nuclei. Colloid is virtually absent. Papillary infoldings of the follicular epithelium are also seen. The follicular cells in Graves disease can demonstrate marked nuclear chromatin clearing, which in combination with papillary hyperplasia can be mistaken for papillary carcinoma (26). However, the diffuse nature of the process can be a clue at low-power magnification that one is dealing with a nonneoplastic process. Lymphocytes are often found in the stroma. Within the follicular centers, B cells predominate; T lymphocytes, especially helper subsets, are seen in a perifollicular location (60). In a diffuse goiter removed from a hyperthyroid patient, the absence of stromal lymphocytes should raise the possibility of a secondary form of hyperthyroidism, such as a pituitary lesion (measurements of thyroid-stimulating hormone will resolve this issue).
Therapy-induced changes in microscopic appearance include decreased vascularity and involution of the follicular epithelium with repletion of colloid after iodide therapy. Beta-blocking drugs and thiouracil do not alter the morphology (61,62); however, radioactive iodine treatment can show follicular atrophy and fibrosis (63). Lymphoid infiltration, sometimes associated with follicular center formation, is often seen in patients with classic hyperthyroid Graves disease. The lymphoid cells are usually found only in the interfollicular stroma and do not encroach upon the follicles themselves. The follicles show marked epithelial hyperplasia, and unless presurgical iodide treatment has been administered, fibrosis is unusual (62).
Toxic Nodular Goiter
In glands with hyperfunctioning follicular nodules, lymphocytic collections can be seen at the periphery or outside the capsule of the nodule (64).
Drug-Associated Thyroiditis
Some medications may result in pathologic changes in the thyroid. It is often not possible to determine if the drug has induced thyroiditis or has uncovered preexisting subclinical thyroid disease. Certain drugs are associated with morphologic changes. Goiter commonly develops in patients treated with lithium for prolonged periods. Hypothyroidism occurs in 3% to 12% of such cases (65,66).
Lymphocytic thyroiditis is also believed to be associated with the addition of iodine to diets. In a study of 10,000 Japanese children between the ages of 6 and 18 years, the incidence of biopsy-proven lymphocytic thyroiditis was 5.3 per 1000 in a high-iodine area compared to a 1.4 per 1000 in a low-iodine zone (67).
Thyroid functional abnormalities have been recognized in patients taking amiodarone, an iodine-containing cardiotropic drug (68). Alves and colleagues (68) reported that in 104 patients on chronic amiodarone treatment, 32% developed hypothyroidism and 23% hyperthyroidism. The histologic changes described by Smyrk and colleagues (69) included follicular damage and disruption, epithelial cell vacuolization, and macrophagic and lymphocytic reaction to degenerating follicles. By electron microscopy, a lamellar configuration was seen in a few lysosomes (69).
Atkins and associates (70) described hypothyroidism occurring in several patients receiving interleukin-2 (IL-2) and lymphokine-activated killer cell therapy for advanced cancers and postulated that the therapy unmasked a subclinical autoimmune thyroiditis; the hypothyroidism appeared associated with a favorable tumor response in these patients.
Papillary Carcinoma
Scattered lymphocytic infiltrates are often seen at the periphery of thyroid neoplasms, particularly in the vicinity of the infiltrative edges of papillary carcinoma (71,72). Volpe (73) suggests that the lymphoid infiltrate results from a “local antigenic perturbation” to altered antigen on the tumor cells.
FIBROSING THYROID LESIONS
Several entities can result in significant fibrosis in the thyroid. Fibrosing thyroid lesions include the fibrosing variant of Hashimoto thyroiditis and fibrous atrophy, which were discussed earlier.
RIEDEL THYROIDITIS
Riedel thyroiditis (Riedel disease, invasive fibrous thyroiditis, Riedel struma) has been incorrectly included among the thyroiditides (74). It is not a disorder of the thyroid but one that involves the thyroid as well as other structures in the neck or even systemically. Riedel disease is an extremely rare entity with an incidence of 0.05% of surgical thyroid diseases and a female predominance. Most patients are euthyroid, although hypothyroidism and hyperthyroidism have been reported (75).
Descriptions of the thyroid range from stony hard or woody fixed; the term ligneous thyroiditis has been used (75). When the lesion is confined to the neck, no systemic abnormalities are found; however, in some patients, the neck lesion represents part of a systemic disease; retroperitoneal, mediastinal, or retroorbital fibrosis as well as sclerosing cholangitis may occur (76–78). In the review by Schwaegerle and colleagues (75), 34% of patients with Riedel disease reported after 1960 had other fibrosing lesions. It has been shown that Riedel disease belongs to the spectrum of IgG4-related systemic disease as they share the histopathologic features and both have similar pattern of multiple organ involvement (79–81). Recognition of this link between Riedel thyroiditis and IgG4-related systemic disease has implication for diagnosis, treatment, and prognosis (79).
Familial cases with multifocal involvement have been described (82). No etiologic relationship to drug intake has been suggested for isolated Riedel disease.
Grossly, the fibrosis involves all or part of the thyroid and is described as woody and very hard. Extension of the fibrosis beyond the thyroid is characteristic. Clinically and grossly, the lesion may be confused with carcinoma. Histologically, the involved portions of the gland are destroyed and replaced by keloid-like fibrous tissue associated with lymphocytes and plasma cells. The fibrous tissue extends into muscle, nerves, and fat and entraps blood vessels. In approximately 25% of cases, the parathyroid glands are also encased (75,78). There is an associated vasculitis (predominantly a phlebitis) with frequent thrombosis (74,78). In 25% of cases of Riedel disease, an adenoma is identified centrally in the fibrotic mass (74). Although the relationship between the adenoma and the fibrous reaction is unknown, the fibrous tissue proliferation may be a reaction to the adenoma or its products.
COMBINED RIEDEL DISEASE AND HASHIMOTO THYROIDITIS
In rare instances, the thyroid gland can show features of both Riedel disease and Hashimoto thyroiditis. The histologic picture resembles Riedel disease, whereas the serology shows thyroglobulin and microsomal antibodies seen in Hashimoto thyroiditis (83,84).
RADIATION FIBROSIS
Reaction of the thyroid to radiation can result in a variety of complications that are related to dose in cases of external radiation to the gland. When radioiodine is administered, hypothyroidism is common, and the incidence increases with time (85).
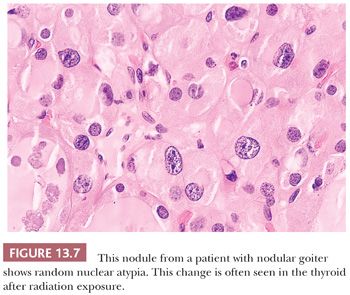
Pathologic changes occur in the thyroid following external radiation in approximately 75% of cases and include foci of follicular hyperplasia (88%), lymphocytic infiltration (67%), oncocytic metaplasia (42%), fibrosis (25%), and adenomatous nodule formation (51%) (Fig. 13.7) (86).
Months or years after radioiodine, a grossly shrunken and fibrotic gland results that histologically shows fibrosis, follicular atrophy, oncocytic and squamous metaplasia, lymphocytic infiltration, and nuclear abnormalities. Vascular change (intimal thickening and sclerosis of arterial walls often with inflammatory cell cuffing) is characteristic of radiation damage (86).
AMYLOIDOSIS
Amyloid is found in the thyroid in three different settings. The most common of these is the amyloid in the stroma of medullary thyroid carcinoma (87). Amyloid goiter is a tumefactive mass of amyloid associated with a foreign body giant cell response and with adipose tissue. It is associated with systemic amyloidosis. Another complication of systemic amyloidosis is amyloid deposition in the thyroid stroma and in glandular and periglandular blood vessels. Hypothyroidism may result (88,89).
FIBROSIS CAUSED BY COLLAGEN VASCULAR DISEASE
Approximately 14% to 24% of patients with scleroderma experience thyroid dysfunction owing to interfollicular fibrosis, and 5% show evidence of chronic lymphocytic thyroiditis. The fibrosis may be caused by vascular sclerosis, which is common in scleroderma (90,91).
PIGMENTS IN THE THYROID
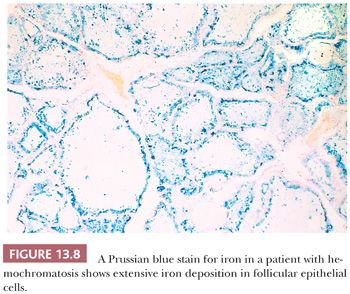
Pigmentation of the thyroid may be caused by iron (hemosiderin) deposition in sites of bleeding or may be found in disorders of iron metabolism such as hemochromatosis. In the former, the pigment is found in macrophages, whereas in the latter, it is present in the follicular epithelium (Fig. 13.8) (92). In patients on chronic minocycline therapy (or occasionally other tetracycline antibiotics), deposition of minocycline-associated pigment produces coal-black coloration to the thyroid. These effects appear to be related to interactions of minocycline with thyroid peroxidase, the key enzyme in thyroid hormone synthesis (93). Histologically, a granular, dustlike precipitate of black-brown pigment is noted in the apical portions of the follicular epithelial cells (92,94). Only rarely are abnormalities of thyroid function noted in these cases.
NODULAR THYROID ENLARGEMENTS
This category consists of thyroid lesions that present as solitary or multiple nodules: benign nodular goiter, toxic nodules, and benign and malignant neoplasms. The nodular thyroid lesions are of most interest to surgeons and patients because the major differential diagnostic possibility is carcinoma. This section describes the gross and microscopic morphology of the various thyroid nodular lesions.
LESIONS CHARACTERIZED BY A PAPILLARY PATTERN
Papillary thyroid lesions include papillary carcinoma, papillary hyperplasia (often associated with hyperfunction), and papillary change in an adenoma or adenomatous follicular nodule.
PAPILLARY CARCINOMA OF THE THYROID
In the United States, thyroid carcinoma comprises approximately 1% of all cancers and accounts for 0.2% of cancer deaths. Most of these carcinomas are of the papillary type. This is the most common malignant tumor of the gland in countries having iodine-sufficient or iodine-excess diets and comprises approximately 80% of thyroid malignancies in the United States (95). These common tumors tend to be biologically indolent and have an excellent prognosis (>90% survival at 20 years) (95,96). The tumors invade lymphatics, leading to multifocal lesions and to regional node metastases. Venous invasion rarely occurs, and metastases outside the neck are unusual (5% to 7% of cases) (97–99).
Papillary carcinoma can occur at any age and rarely has been diagnosed as a congenital tumor (100). Most tumors are diagnosed in patients in the third and fifth decades (101,102). Women are affected more than men in ratios of 2:1 to 4:1 (102).
Etiologic Factors
Etiologic factors for papillary carcinoma are not well established; various cellular and genetic mechanisms/targets have been studied in the development of papillary carcinoma.
Iodine
The addition of iodine to the diet in endemic goiter areas in Europe and South America has been associated with a decreased incidence of follicular cancer and an increase in papillary carcinoma (103,104).
External Radiation
External radiation probably plays a role in the development of papillary carcinoma (105). The average time from radiation exposure to tumor development has classically been reported as 20 years; however, development time is variable, particularly following major nuclear facility accidents such as that seen in Chernobyl. A great increase in the incidence of papillary carcinoma in Belarus and Ukraine has been apparent since the Chernobyl nuclear accident (106). The increased incidence was seen predominantly in young children from that area who had been exposed to the radiation. Most reported tumors following this nuclear disaster have been papillary carcinomas, many of which show aggressive histologic features including extracapsular extension and vascular invasion (107,108).
Autoimmune Disease
Some authors believe that patients with Graves disease have a higher than expected incidence of papillary carcinoma (109); other studies disagree (110). Many studies indicate that up to one-third of papillary carcinomas arise in the setting of chronic thyroiditis. However, these studies tend to lack serologic proof of preexisting thyroiditis (111). Follow-up studies of patients with documented thyroiditis indicate that the tumor that arises much more frequently in these glands is malignant lymphoma, not papillary carcinoma (discussed subsequently) (112). Because papillary carcinoma and thyroiditis are both common conditions, the possibility of coincidental coexistence is more likely than a pathogenetic relationship (113). However, recent molecular data have shown that foci of atypical follicular epithelium in chronic thyroiditis do have loss of heterozygosity for various tumor suppressor genes and RET/PTC rearrangements (114).
Hormonal and Reproductive Factors
Papillary carcinoma is more common in women than men. Some studies have suggested the role of various hormonal factors in the development of papillary carcinoma; these include increased parity, late age at the onset of first pregnancy, fertility problems, and oral contraceptives (115). Lee et al. (116) have shown that estradiol promotes cell proliferation via enhancement of antiapoptotic signaling pathway in a human PTC cell line; interestingly, this growth-promoting effect of estradiol was attenuated by tamoxifen.
Genetic Syndromes
Papillary carcinomas have been described in patients with familial adenomatous polyposis coli (FAP), Cowden syndrome, hereditary nonpolyposis colon cancer syndrome (HNPCC), Peutz-Jeghers syndrome, and ataxia telangiectasia (117,118).
FAP is caused by germline mutations of the adenomatous polyposis coli (APC) gene. Thyroid carcinoma, mostly papillary carcinoma (>95% of cases), occurs in 1% to 2% of patients with FAP; all these patients show germline mutations of the APC gene; however, somatic mutations of or loss of heterozygosity (LOH) for APC gene are not found in thyroid tumors. Interestingly, a majority of these tumors do show activation of RET/PTC1 in thyroid tumors, suggesting a possible association between APC and RET/PTC in the development of this particular subset of familial papillary carcinoma (119).
Cowden syndrome is characterized by formation of hamartomas in several organs and a high risk of developing breast and thyroid cancer. The genetic locus for Cowden syndrome has been mapped to chromosome 10q23.3 and is also known as PTEN, which is a protein tyrosine phosphatase and exerts its tumor suppressor effects by antagonizing protein tyrosine kinase activity. Interestingly, PTEN mutation or gene deletion is noted in 26% of benign tumors but only in 6.1% of malignant tumors of the thyroid (120,121).
Thyroid and Parathyroid Adenomas
Occasionally, papillary carcinomas arise in benign nodules or adenomas (122,123). This may result merely from a malignant tumor arising in a nodular area of the gland instead of in a nonnodular zone (i.e., it is likely to be a random event of location and not indicate a casual relationship). Several authors have described the association of papillary carcinoma and parathyroid adenoma or hyperplasia. Both types of lesions are associated with a history of low-dose external radiation to the neck (122).
Pathology
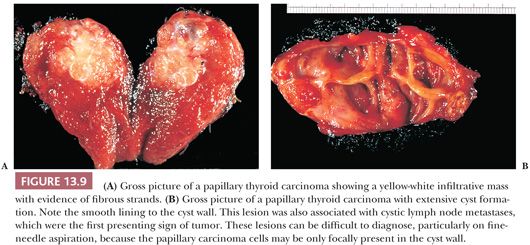
The gross appearance of PTC is quite variable (71,124). The lesions may appear anywhere within the gland. By definition, typical papillary carcinomas are greater than 1.0 to 1.5 cm, often averaging 2 to 3 cm, although lesions may be quite large (Fig. 13.9). The lesions are firm and usually white in color with an invasive appearance. Lesional calcification is a common feature. Because of extensive sclerosis, the lesion may grossly resemble a scar. In addition, cyst formation may be observed. In fact, some lesions may rarely be completely cystic, making diagnosis difficult (97). Necrosis (in the absence of a prior needle biopsy) is not a feature of typical papillary carcinoma and suggests a higher grade lesion (125).
Microscopically, papillary carcinomas share certain features (Table 13.2). The neoplastic papillae contain a central core of fibrovascular (occasionally just fibrous) tissue lined by one or occasionally several layers of cells with crowded oval nuclei (Fig. 13.10). In contrast, hyperplasia of thyroid follicles may sometimes exaggerate into papillary change; there is infolding of the lining epithelium composed of columnar cells with basal round and uniform nuclei. There is either no central core or a core of edematous or myxomatous paucicellular stroma often including small follicles (subfollicle formation) (71,110,124).
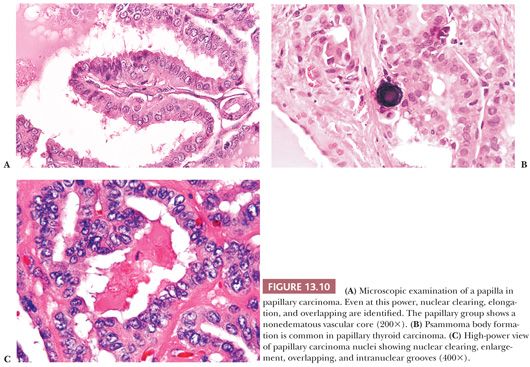


Psammoma bodies that represent the “ghosts” of dead papillae are differentiated from dystrophic calcifications by lamellations (Fig. 13.10) (126). True psammoma bodies are formed by focal areas of infarction of the tips of papillae attracting calcium that is deposited on the dying cells. Progressive infarction of the papillae and ensuing calcium deposition lead to lamellation (127). Psammoma bodies are usually present within the cores of papillae or in the tumor stroma, but not within the neoplastic follicles. Only rarely are psammoma bodies found in benign conditions in the thyroid (128).
The nuclei of papillary cancer have been described as clear, ground glass, empty, or Orphan Annie–eyed (Fig. 13.10). These nuclei are larger and more oval than normal follicular nuclei and contain hypodense chromatin (71,129). In papillary cancer, these nuclei often overlap one another. Although cleared nuclei are characteristic of papillary carcinoma, autoimmune thyroiditis, particularly Hashimoto disease, often shows similar nuclear changes (42). Intranuclear inclusions of cytoplasm are often found. Another characteristic of the papillary cancer nucleus is the nuclear groove (97,129). Nuclear grooves may be seen in other thyroid lesions including Hashimoto disease, adenomatous hyperplasia, and diffuse hyperplasia as well as in follicular adenomas (particularly hyalinizing trabecular neoplasm) (130,131). For the most part, nuclear grooves are more commonly seen in papillary carcinoma than in other thyroid lesions; however, the mere presence of nuclear grooves is not diagnostic for papillary carcinoma.
Most of these tumors will be composed predominantly or focally of papillary areas. A large number will contain follicular areas as well (132). The tumor cells are usually columnar. Clear nuclei are found in more than 80% of such lesions, intranuclear inclusions in approximately 80% to 85%, and nuclear grooves are seen in almost all the cases. Mitoses are exceptional in usual papillary carcinoma. Psammoma bodies are found in approximately 40% to 50% of cases (71), but their presence in thyroid tissue indicates that a papillary carcinoma is most likely present somewhere in the gland. The finding of psammoma bodies in a cervical lymph node is strong evidence of a papillary cancer in the thyroid (14).
Many papillary carcinomas (approximately 15% to 45%) contain foci of squamous differentiation (133). Almost all papillary carcinomas show areas of desmoplasia either in the central portions of the tumor or at the peripheral zones of the lesions. Even in encapsulated lesions, sclerosis is seen in areas of capsular penetration (124).
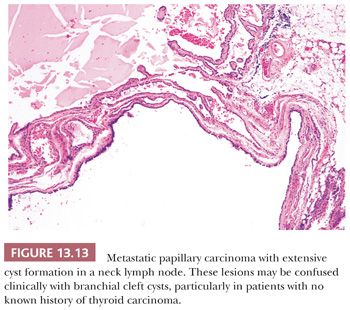
Scattered lymphocytes are common at the invasive edges of the tumor (124,134). Rarely, an intense lymphocytic infiltrate is seen, but it is localized to the invasive tumor foci and does not indicate a preexisting underlying chronic thyroiditis (124). Cyst formation may occur and in fact may be so striking that the diagnosis of papillary carcinoma is difficult to make, particularly if the lesion has metastasized to neck lymph nodes, making the distinction (particularly on a clinical basis) from a branchial cleft cyst difficult (135).
Papillary carcinoma invades the glandular lymphatics, which accounts for high incidence of regional node metastases (95,97). Papillary carcinoma can also present as multifocal tumors within the same gland. It has been shown that papillary carcinomas are clonal proliferations. The monoclonal nature of papillary carcinoma has been proven by molecular biology techniques. In view of these studies, it is believed that multifocality of papillary carcinoma must be owing to intrathyroidal lymphatic spread rather than multifocal primary tumors. Recent RET/PTC and LOH studies have shown that multifocal papillary microcarcinomas can be separate primaries instead of intraglandularly spreading from one tumor source (114,136). Hence, both possibilities can exist: true multiclonal lesions as well as intraglandular spread from one primary tumor.
What does this multifocality mean in regard to therapy and prognosis? Some argue that because of multifocal microscopic lesions, the entire gland should be excised; others indicate that more conservative surgery (lobectomy or lobectomy and isthmusectomy) followed by thyroid suppression is adequate because the tumors are often hormonally controllable (137,138). The low frequency of local recurrence in the opposite thyroid lobe and the long-term follow-up studies of conservatively treated patients showing excellent results suggest that the second viewpoint is the wiser one. Of course, treatment of extrathyroidal or massive papillary cancer requires more radical procedures (139).
Venous invasion can be identified in up to 7% of papillary cancers (71). It has been suggested that cases with histologic vascular invasion may be considered as a sign of an increased tendency toward hematogenic invasion and consequent increase in the relative percentage of metastases (140).
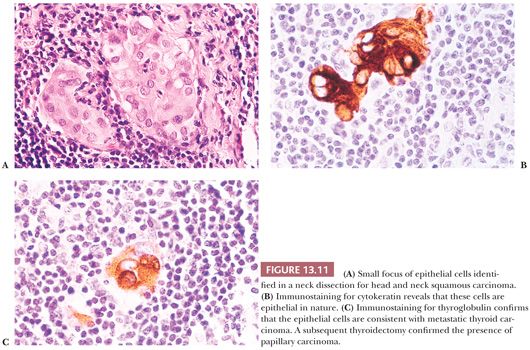
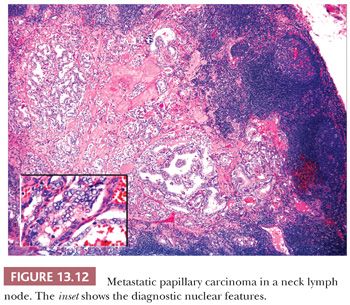
Regional lymph node metastases are extremely common (≥50%) at initial presentation of usual papillary cancer (Fig. 13.11). This feature does not adversely affect long-term prognosis (141,142). Hence, attempts at staging papillary carcinoma may have minimal clinical significance. Some patients will present with cervical node enlargement and will have no obvious thyroid tumor. Frequently, the nodal metastasis will involve one node that may be cystic (Fig. 13.12) (143). The histology of the nodal metastases in papillary cancer may appear papillary, mixed, or follicular (Fig. 13.13) (71).
Histologic grading is of no use in papillary carcinoma because more than 95% of these lesions are grade 1. In some tumors, either in the primary site or in recurrences, areas of poorly differentiated cancer characterized by solid growth of tumor, necrosis, mitotic activity, and cytologic atypia can be found. Such lesions have a much more guarded prognosis (125). Anaplastic change in a papillary cancer can occur, although it is uncommon (144).
Distant metastases of papillary carcinoma to lungs and bones occur in 5% to 7% of cases (95,98). Despite the presence of multiple metastases, however, survival may still be prolonged, especially if the metastases can be treated with radioiodine (145). In ordinary papillary carcinoma, death is uncommon (95).
Ultrastructure
The electron microscopic appearance of papillary carcinoma includes a nucleus with dispersed chromatin and highly infolded nuclear membrane, cytoplasmic intranuclear inclusions, and a cytoplasm that contain many mitochondria and numerous cytoplasmic filaments (22,146). Keratohyaline granules have been found in tumors with squamous foci (22).
Immunohistochemistry
Immunostaining shows that most papillary cancers contain thyroglobulin and thyroid transcription factor-1 (TTF-1). Several reports have been published regarding the use of various immunohistochemical markers that can differentiate papillary carcinoma from other follicular-derived lesions of the thyroid. From an extensive list of these markers, the ones that have shown some promise include cytokeratin-19, HBME-1, and galectin-3 (147–149). However, none of these have proven to be specific because all can be expressed in some benign lesions of thyroid. Therefore, some authors have proposed that diagnosis of papillary cancer should be carried out by employing an immunopanel consisting of markers previously mentioned (149).
The other markers that have been explored in the diagnosis of papillary carcinoma include S100 protein, blood group antigens, estrogen receptors, CD15, and CD57 (150–152).
Flow Cytometry
Although the great majority of PTCs are diploid, the literature suggests up to 20% may show aneuploid or at least nondiploid subpopulations (153). It has been shown that aneuploid tumors often are associated with a more aggressive clinical course (153,154); however, multivariate analysis has not shown that ploidy is an independent prognostic factor.
Molecular Pathology of Papillary Carcinoma
In the past decade, the literature on thyroid has been focused mainly on the role of various biologic events and genetic determinants in the pathogenesis of various thyroid tumors.
Rearrangements of RET gene, known as RET/PTC, have been identified in papillary carcinoma of thyroid (155–158). The RET proto-oncogene is normally expressed in cells of neural crest origin and plays a role in kidney and gastrointestinal neuronal development. It is located on chromosome 10q11.2 and cell membrane receptor tyrosine kinase. In normal thyroid, wild-type RET is only expressed in C cells and not follicular cells. RET/PTC seen in papillary carcinomas occurs as a result of fusion of tyrosine kinase domain of RET to the 5′ portion of the various genes (155). To date, 11 novel types of rearrangements have been described in papillary carcinoma. RET/PTC1 and RET/PTC3 are the most common forms that occur in sporadic papillary carcinoma. RET/PTC1 is formed by fusion of RET to H4, and RET/PTC3 occurs as a result of fusion of RET to the ELE1 gene (159).
RET/PTC expression in thyroid follicular cells of transgenic mice leads to development of thyroid tumors with histologic features of PTC (160). Similarly, transfection of follicular cells in tissue culture by RET/PTC causes the cells to demonstrate nuclear features of papillary carcinoma (161). The prevalence of RET/PTC in papillary carcinoma varies significantly among various geographic regions (162,163); in the United States it ranges from 11% to 43% (164). In sporadic tumors, RET/PTC1 is the most common form of rearrangement (60% to 70%), followed by RET/PTC3 (20% to 30%) (164). The other rare forms of RET/PTC rearrangements have been mainly found in radiation-induced papillary carcinomas (165). Several studies have shown a strong association between radiation-induced papillary carcinoma and expression of RET/PTC; in papillary carcinoma, among children affected by the Chernobyl nuclear accident, RET/PTC3 was found to be the most common form of rearrangement, followed by RET/PTC1 (165,166).
Recently, it has been shown that RET/PTC expression can also occur in some benign lesions. These include hyalinizing trabecular adenoma, Hashimoto thyroiditis, and hyperplastic nodules and follicular adenoma (167–169).
Several authors have suggested an association between Hashimoto thyroiditis and papillary carcinoma (167,168); however, others have suggested that this association is most likely incidental because both are common (113). Recently, two independent studies have shown high prevalence of RET/PTC in histologically benign thyroid tissue affected by Hashimoto thyroiditis; these studies concluded that thyroiditic glands harbor multiple foci of papillary carcinoma which are not identified by histologic examination only (167,170). However, a study by Nikiforova et al. (113) have disputed these findings and failed to reproduce these results.
RET/PTC has been identified in benign thyroid nodules, especially the ones that are seen in patients with history of external radiation. However, the significance of this still remains controversial and needs to be further elucidated by examination of a large cohort of cases (169).
Activation of the RAS oncogene is considered to be an important mechanism by which human cancer develops. RAS has been shown to regulate several pathways that contribute to cellular transformation including the Raf/MEK/ERK pathway. Numerous studies confirm that the Raf/MEK/ERK pathway is a significant contributor to the malignant phenotype associated with deregulated RAS signaling.
An activating mutation in BRAF has been described in 29% to 69% of human PTCs. BRAF activating mutations in thyroid cancer are almost exclusively the BRAF (V600E) mutation and have been found in 29% to 69% of PTCs, 13% of poorly differentiated cancers, and 10% of anaplastic cancers (171–173). The role of BRAF as an oncogenic-initiating event in thyroid follicular cells has been confirmed in mice models (174).
There is practically no concordance between papillary carcinoma with RET/PTC, BRAF, or RAS mutations. The lack of this overlap provides strong evidence of this signaling pathway for thyroid follicular cells to develop into papillary carcinoma (175). Molecular analysis of fine-needle aspiration (FNA) samples for BRAF and RAS mutations and RET/PTC translocations have been shown to be of value in the preoperative diagnosis of PTC in cases diagnosed as indeterminate or suspicious for malignancy (176,177).
Other Oncogenes. Somatic rearrangements of NTRK gene give rise to four chimeric genes with transforming activity; these are designated as TRK, TRK-T1, TRK-T2, and TRK-T3. NTRK-1 proto-oncogene has been detected in 5% to 25% of spontaneous and radiation-induced papillary carcinomas (178,179). The other genes implicated in the pathogenesis of PTC include erbB-2, PTEN, p53, p16Ink4A, and p15Ink4B (121,180–182).
Prognostic Factors
Poor prognostic factors in papillary carcinoma include older age at diagnosis, male sex, large tumor size, and extrathyroidal growth. Pathologic variables associated with a more guarded prognosis include less differentiated or solid areas, vascular invasion, and aneuploid cell population (183,184).
The most widely accepted system of staging solid organ malignancies is the postoperative tumor-node-metastasis (pTNM) system, endorsed by both the International Union Against Cancer (UICC) and the American Joint Commission on Cancer (AJCC). Generally, this system stages malignant lesions according to the tumor size and invasiveness, nodal spread, and distant metastases. On the basis of this AJCC staging system, all patients younger than 45 years of age with PTC or follicular thyroid cancer have stage I disease unless they have distant metastases, in which circumstance the disease is classified as stage II. Older patients (≥45 years of age) with node-negative papillary or follicular microcarcinoma (T1N0M0) have stage I disease. Intrathyroidal tumors 1.1 cm or larger are stage II, and either nodal involvement or extrathyroid invasion in older patients with papillary or follicular thyroid carcinoma leads to stage III of classification scheme (185,186).
Some studies have shown that RET/PTC expression in papillary carcinoma can be associated with aggressive biologic behavior (187), whereas others have reported just the opposite (188): that RET/PTC expression is more commonly seen in slow-growing and clinically indolent tumors. It is also suggested that different rearrangements of RET/PTC are associated with different biologic behavior (189). Nikiforov (164) found a significant difference in local recurrence and distant metastases between tumors with RET/PTC1 and RET/PTC3 expression. Cetta and colleagues (190) reported similar findings.
It has been shown that papillary carcinoma with BRAF mutations follows an aggressive clinical course (i.e., extrathyroidal extension and more advanced clinical stage). In addition, aggressive phenotype of papillary carcinoma such as tall cell variant is associated with high prevalence of BRAF mutations (191).
Several other biologic markers have been suggested as prognostic predictors in papillary carcinoma; these include p53, Ki-67, cell cycle proteins, proliferating cell nuclear antigen (PCNA), bcl-2, cathepsin D, and topoisomerase II (192–194).
SUBTYPES OF PAPILLARY CARCINOMA (TABLE 13.2)
Papillary Microcarcinoma (Occult Papillary Carcinoma)
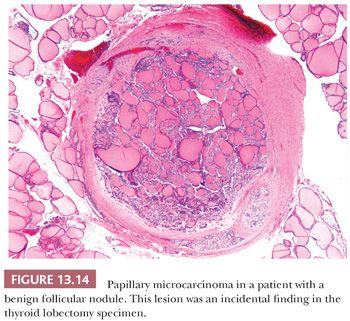
According to the World Health Organization (WHO), papillary microcarcinoma is defined as a tumor measuring 1 cm or less; however, some experts have also defined tumors measuring up to 1.5 cm as microcarcinomas (195,196). Other terms for these lesions have included occult papillary carcinoma, occult sclerosing carcinoma, small papillary carcinoma, and nonencapsulated sclerosing tumor. These lesions are quite common as incidental findings at autopsy or in thyroidectomy for benign disease or in completion thyroidectomies in patients with a history of carcinoma involving the opposite thyroid lobe. The incidence of these lesions has varied significantly with the study, but papillary microcarcinoma has been reported in up to 36% of carefully sectioned thyroid specimens (196–198). Harach and colleagues (199) suggested that tumors measuring less than 5 mm (minute papillary carcinomas) be considered a normal finding and be left untreated. However, lymph node metastases from lesions less than 0.5 cm are known (200,201). Distant metastases, although very rare, are also documented (201). The small papillary carcinoma is a nonencapsulated, sclerotic, white to tan nodule often located subcapsularly. Histologically, the tumors may be totally follicular or show papillary areas as well (Fig. 13.14). Sclerosis may be prominent; the lesions infiltrate the surrounding thyroid (202).
A familial form of papillary microcarcinoma has been recognized; these tumors are characterized by multifocality with increased tendency toward vascular and lymphatic invasion, distant metastasis, and even death (203).
It is important to recognize that the incidentally found microcarcinoma confined within the thyroid is probably of no clinical importance and should not be overtreated. Therefore, some authors have suggested the term papillary microtumor to prevent aggressive management (204).
Folllicular Variant of Papillary Cancer
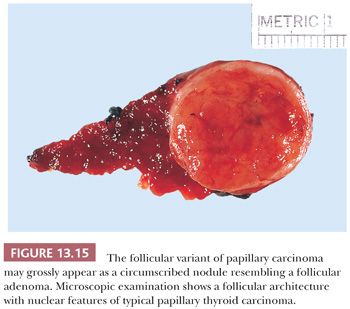
The follicular variant of papillary carcinoma is a distinctive papillary carcinoma variant. The incidence of this variant is difficult to determine because, in the past, some of these lesions have been classified as follicular carcinomas or adenomas. Grossly and histologically, the tumor may appear encapsulated (Fig. 13.15) (205,206). Despite the almost total follicular pattern in the primary site, features that suggest that a tumor is a follicular variant of papillary cancer include clear nuclei, psammoma bodies, and desmoplastic response at invasive areas (205). Immunohistochemical staining shows the presence of low– and high–molecular-weight cytokeratins and HBME-1, which may aid in differentiating this lesion from follicular adenomas and carcinomas (190,191,248). The prognosis of the follicular variant is apparently similar to usual papillary cancer, although there may be a greater risk for this variant to metastasize outside the neck and for vascular invasion; regional nodal metastases are less common than in classic papillary carcinoma (205,206).
Two distinct types of follicular variant include the diffuse follicular variant and the encapsulated follicular variant (205,207). In the diffuse follicular variant, the gland is diffusely replaced by tumor. Lymph node and distant metastases are common in these patients. The prognosis appears to be poor in these patients, although only a handful of cases have been described (207).
The encapsulated follicular variant, as the name implies, is characterized by the presence of a capsule around the lesion. These lesions are associated with an excellent prognosis. In some cases, the diagnosis of this particular variant of papillary carcinoma can be difficult because of the presence of multifocal rather than diffuse distribution of nuclear features of PTC (208,209). Because of this peculiar morphologic presentation, these tumors can be misdiagnosed as adenomatoid nodule or follicular adenoma (208). Some authors have suggested that these tumors be classified as “tumors of undetermined malignant potential” as a result of excellent prognosis (210); however, others have shown that some cases belonging in this category can lead to distant metastasis (211).
The work of Nikiforov (212,213) interestingly has shown that a significant number of follicular variant of papillary thyroid carcinoma (FVPTC) have mutations in NRAS rather than RET/PTC and less than 5% of these tumors demonstrate PAX8/peroxisome proliferator–activated receptor-γ (PPAR-γ) rearrangements, thus resembling more true follicular carcinoma than papillary subtype. It is unusual to find BRAF mutations in encapsulated follicular variant of papillary carcinoma (158,213,214). It has been shown that FVPTC demonstrates a distinct BRAF (K601E) mutation from the usual BRAF (V600E) mutation found in classic and tall cell variant of PTC (212,213,215).
Tall Cell Variant
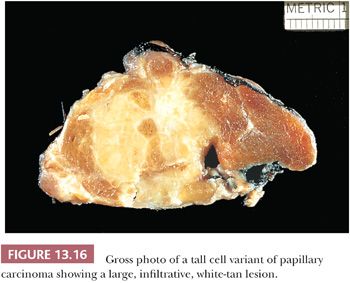
Hawk and Hazard (216) found that the tall cell variant (TCV) made up approximately 10% of the papillary cancers they studied. The tumor is large (>6 cm) (Fig. 13.16), extends extrathyroidally, and shows mitotic activity and vascular invasion more often than usual papillary cancer. The tumor tends to occur in older adult patients (216–218).
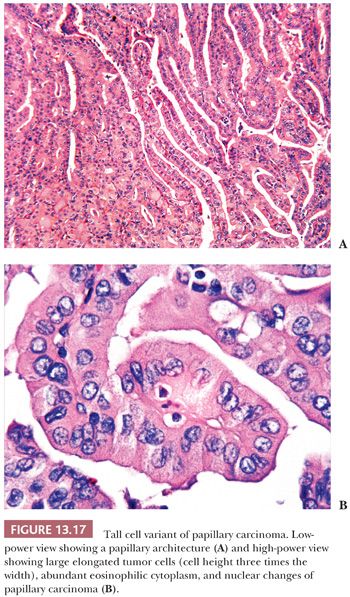
The tall cell is three times as tall as it is wide, and its cytoplasm is often eosinophilic (Fig. 13.17) (195). In fact, these tumors may be referred to as “pink-cell” variant of papillary carcinoma. Tall cells should represent 50% or more of the papillary carcinoma cells to make the diagnosis of TCV (219). The tumors show an extensive papillary pattern, often with a heavy lymphocytic infiltration present in or around the papillae. Some tall cell tumors arise in glands with extensive histologic evidence of chronic thyroiditis. Dedifferentiation to squamous cell carcinoma has been described. Local recurrences with invasion of the trachea can be seen, and this complication may be fatal (220). Flow cytometry studies, although limited in number, have failed to reveal differences between typical papillary carcinoma and the TCV (221). The prognosis for this variant is less favorable than for usual papillary cancer, although it is believed that the poor outcome in these tumors may be secondary to the fact that these tumors are often associated with poor prognostic variables such as older age, extrathyroidal spread, necrosis, high mitotic rate, and distant metastases (218,222). The importance of diagnosing TCV is accentuated by the fact that it is overrepresented in those thyroid carcinomas that are refractory to radioactive iodine (RAI) therapy (223,224).
The aggressive behavior of TCV-PTC can be correlated with the molecular profile (224) and certain factors elaborated by the tumor. The cases of TCV-PTC demonstrate higher prevalence of activating point mutations of the BRAF because these mutations in PTC have been associated with higher frequency of extraglandular extension and nodal metastases. LOH for chromosome 1 (D1S243) and the p53 gene (TP53) have been reported, although not consistently in TCV-PTC but not in classical PTC, and can be used as a potential tool for molecular discrimination between these two neoplasms (225). In another study, RET/PTC3 rearrangement, which leads to activation of downstream mitogenic signaling pathways, was found in 35.8% of TCV (157). The high expression of MUC1 and type IV collagenase (matrix metalloproteinase-2) in TCV may allow for degradation of stroma and greater invasive properties (226,227).
Columnar Cell Variant
The columnar cell variant (CCV) is a rare form of papillary carcinoma. (Some authors believe it is so unusual a tumor that it deserves its own category and should not be placed in the papillary group [228,229].) The tumor needs to be distinguished from other papillary carcinomas because this lesion is associated with an extremely poor outcome, with most deaths occurring within 5 years of diagnosis (228,230). Grossly, the tumors often measure more than 6 cm. The tumor is characterized microscopically by papillary growth. Tall columnar cells line the papillae. The nuclear features are usually not those of typical papillary carcinomas. The nuclei are hyperchromatic with a punctate chromatin; nuclear stratification is a prominent feature. The cells usually have scant cytoplasm which can be clear. Mitoses are frequently seen. Psammoma bodies are rare. Extrathyroidal extension is common as are distant metastases (230). Encapsulated variants, which may have a better prognosis, have been described (231). Because of its aggressive behavior, the actual classification of this lesion as a variant of papillary carcinoma has been controversial, although most authors include this lesion in the papillary carcinoma classification (230). CDX2, a nuclear transcription factor important in intestinal development, has been shown to be selectively expressed in 50% of cases of CCV (232,233). An encapsulated variant of columnar cell carcinoma has been described, which behaves similarly to conventional PTC (229,234). BRAF mutations can be seen in approximately 33% of CCV-PTC similar to conventional PTC (235).
Warthin-Like Variant
By light microscopy, these tumors resemble a “Warthin tumor” of the salivary gland. These tumors usually arise in a background of chronic lymphocytic thyroiditis and show papillary architecture. Cystic degeneration of variable degree can be commonly seen in the tumor nodule. Tumor cells with abundant eosinophilic cytoplasm line the papillae, and the papillary cores contain a brisk lymphoplasmacytic infiltrate. Some tumors may show transition to TCV, which usually occurs at the invasive edge of tumor. Limited follow-up have shown that these tumors in their pure form follow a clinical course similar to conventional papillary carcinoma (236,237). These do demonstrate BRAF (V600E) mutations (215).
Papillary Thyroid Carcinoma with Nodular Fasciitis-Like Stroma
In this rare variant of PTC, the tumor cells usually form cords, tubules, and papillae; show nuclear features of PTC with varying degrees of squamous metaplasia; and are embedded in a unique fasciitis-like stromal component. The low-power microscopy of these tumors appears similar to fibroadenoma, or phyllodes tumor of the breast. By immunohistochemistry, the stromal component shows myofibroblastic differentiation (negative for cytokeratin and positive for vimentin, desmin, and smooth muscle actin). This immunoprofile also helps to distinguish PTC with nodular fasciitis-like stroma from anaplastic transformation within a PTC, in which the spindle cells will stain positive for cytokeratins (238,239).
Cribriform-Morular Variant of Papillary Carcinoma
This rare variant of PTC can occur in 1% to 2% of patients with FAP. These tumors are exclusively seen in women, present as multifocal tumors with a cribriform solid and/or spindle cell growth pattern, and show nuclear features of PTC (240–242). All cases show APC germline mutations, and some cases also have shown RET/PTC activation; however, none have shown BRAF mutations (243). Some cases of PTC with similar morphology can also occur in patients without FAP; rare cases from this sporadic group have also shown APC mutations only in the tumoral tissue (240–242).
Diffuse Sclerosis Variant
The diffuse sclerosis variant (DSV) of papillary carcinoma is rare, representing only approximately 3% of all papillary carcinomas (244). The tumor, which most often affects children and young adults, may present as bilateral goiter. The tumor permeates the gland outlining the intraglandular lymphatics. Tumor papillae have associated areas of squamous metaplasia. Numerous psammoma bodies are found (Fig 13.18). Lymphocytic infiltrates are found around the tumor foci. The lesions tend to recur in the neck and have a somewhat more serious prognosis than usual childhood papillary cancer. These lesions appear to represent 10% of the papillary carcinomas seen in children exposed to the RAI released following the Chernobyl accident (244–246). Although the tumors often show extracapsular extension, distant and nodal metastases, and a decreased disease-free survival when compared with the usual type of papillary carcinoma, mortality is low.
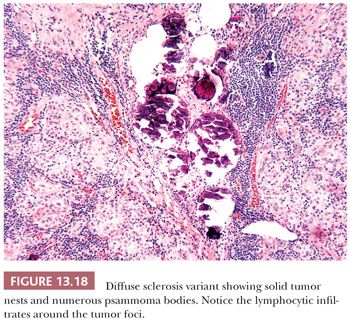
In a study of the genetic alterations in DSV, no BRAF (V600E) mutations were found, but all the cases showed RET proto-oncogene (RET/PTC) rearrangements (247). These rearrangements especially RET/PTC3 are more common in tumors seen in children and young adults, especially those associated with radiation exposure. The morphologic features of DSV have been described in some of thyroid tumors from children exposed to radiation after the Chernobyl accident (213,246). Therefore, it is not surprising that the presence of RET/PTC is a prominent genetic event—in DSV-PTC—and that these tumors are susceptible to RET therapy. Another study supporting the aforementioned findings identified that the profile of genomic mutations detected in DSV-PTC is different from that in classic PTC (248). None of genomic mutations of BRAF, 1p36, 3p12, 7p31, and 10q23 was detected in DSV, but a higher frequency of LOHs of 3p24, 9p21, 17q21, 21q22, and 22q13 was noted. It has been suggested that these mutations may play an important role in the different morphologic appearance and worse prognosis of DSV.
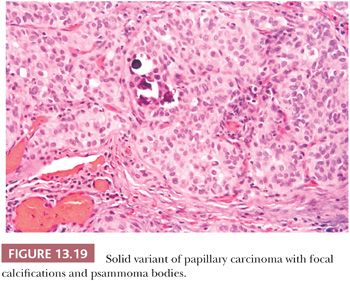
Solid Variant
A solid growth pattern is noted in many papillary carcinomas. When the solid growth represents greater than 50% of the tumor mass, a diagnosis of solid variant of papillary carcinoma may be made (Fig. 13.19) (249). The solid variant is most commonly seen in children and has been reported in more than 30% of patients with papillary carcinoma following the Chernobyl nuclear accident (108). Harach and Williams (250) designated papillary cancers with solid growth pattern in children as “childhood thyroid carcinoma.” These tumors are widely invasive throughout the thyroid. Almost half of these cases are associated with history of external radiation to the head and neck region (250).The nuclear features are those of papillary carcinoma. It is important to recognize these lesions as papillary carcinomas and not overdiagnose them as more aggressive tumors such as insular carcinoma (discussed later in the chapter). The prognosis is controversial, with some studies showing outcomes similar to typical papillary carcinoma and other studies showing more aggressive behavior (251). A study has shown that in adults, solid papillary carcinoma has a similar and only slightly less good prognosis than usual type. This differs from poorly differentiated thyroid cancer defined predominantly by the presence of necrosis (125). By molecular analysis, cases of solid variant of PTC arising in association with radiation exposure has shown RET/PTC3 rearrangements. Interestingly, a triplet deletion of the BRAF gene leading to the replacement of a valine and a lysine by a glutamate (BRAF [V600E + K601]) was first reported only in the solid variant of PTC by Trovisco et al. (252), which has been confirmed by other investigators (253).
Hobnail Variant
This rare variant of PTC was described by Asioli et al. (254). These tumors commonly occur in women and are associated with significant mortality—50% of the eight reported cases. The tumors in the reported series usually had more than 30% of the tumor with hobnail features. This peculiar feature is characterized by tumor cell nuclei located in the middle or in the apex of the cytoplasm, that is, bulging of the nuclei at the tip of the cell imparting the so-called hobnail appearance to the cells (255). Interestingly, this morphology is similar to that seen in papillary serous carcinoma; breast micropapillary carcinoma; and urinary bladder, kidney, and lung adenocarcinoma. Some authors have suggested that morphology also be encountered in oncocytic variant of PTC (256) as originally described by Rosai et al. (219). BRAF (V600E) mutations have been detected in more than 50% of these tumors, confirming the classification of these tumors as variants of PTC (254).
Encapsulated Variant
The encapsulated variant presents grossly as an adenoma and comprises from 8% to 13% of papillary cancers (231,508). Microscopically, such lesions usually show total encapsulation; however, there are cytologic features of papillary cancer including nuclear changes and psammoma bodies. Some of these lesions will show focal invasion into the capsule. The prognosis is excellent.
Other Variants
Rare variants of papillary cancer in which prognostic data are not well established include the spindle cell variant, the clear cell type, the oxyphilic (Hürthle cell) variant (257), papillary carcinoma with lipomatous stroma (258), and myxoid variant (259).
PAPILLARY HYPERPLASIA
Papillary hyperplasia, seen in untreated autoimmune hyperthyroidism (Graves disease), congenital errors of thyroid metabolism, and hyperfunctioning foci in goitrous glands, shows overgrowth of the follicular epithelium. The cell growth involves both hypertrophy and hyperplasia; infoldings (i.e., papillations) form. Diffuse papillary hyperplasia is distinguished from papillary carcinoma by the preservation of the gland architecture, the diffuse character of the lesion, and the normal nuclei. In toxic nodular goiter, the changes are similar but the lesion tends to be focal. Nuclear characteristics differentiate the lesion from cancer (132,260).
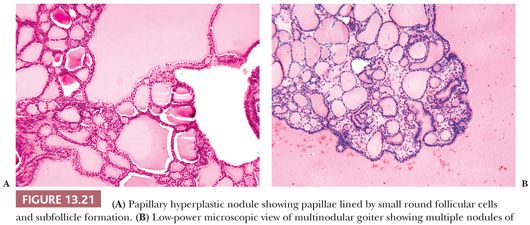
Papillary Hyperplasia in Follicular Nodule
Well-circumscribed solitary thyroid nodules may show a microscopic appearance of papillary growth. However, the papillae contain extremely edematous stalks with follicles in them (Fig. 13.20). The nuclei are round and not clear; there are no psammoma bodies. Some of these nodules produce a warm or hot appearance on scan (14,261). At times, areas in nodular goiters will show a focally similar papillary appearance; these areas are often associated with hemorrhage and edema. Although papillary adenoma has been used as a diagnostic term for these lesions, it should not be used because it has been applied to a number of lesions from hyperplastic ones to encapsulated papillary carcinomas. It has been shown by immunohistochemistry that papillary hyperplastic nodules are negative for HBME-1 and focally positive for CK-19. This can be helpful to differentiate these from papillary carcinoma in diagnostically challenging cases (262).
LESIONS WITH FOLLICULAR ARCHITECTURE
The majority of thyroid lesions (physiologic hyperplasia, benign nodular lesions, and neoplasms) display a macrofollicular or microfollicular pattern. The thyroid lesions in this histologic category include those found in patients with congenital disorders of thyroid metabolism (inborn errors of thyroid metabolism, dyshormonogenetic goiter) and disorders of the thyroid in diffuse toxic goiter (autoimmune hyperthyroidism [Graves disease] and Basedow disease). In some of these conditions, hypothyroidism is found; in others, hyperfunction; and in still others, euthyroidism is present.
When the thyroid cannot produce normal amounts of thyroid hormone, the pituitary secretes increased amounts of thyrotropin (thyroid-stimulating hormone [TSH]), which, in turn, directs the thyroid follicular cells to produce more thyroid hormones—thyroxine and triiodothyronine (T4 and T3). The cells undergo hyperplasia and hypertrophy. The thyroid gland enlarges and, in some cases, nodules form. Causes of this type of hyperplasia include dyshormonogenetic goiter, TSH-producing pituitary adenomas, trophoblastic disease, inadequate dietary iodine, and goitrogenic drugs.
In autoimmune hyperthyroidism (diffuse toxic goiter or Graves disease), hyperplasia and hypertrophy result from TSH-mimicking immunoglobulins (thyrotropin receptor antibodies) that attach to the receptor on the follicular cell membrane and trigger the intracellular sequence of events that enables increased thyroid hormone production.
INBORN ERRORS OF THYROID METABOLISM—GENETIC DISORDERS
Inborn errors of thyroid metabolism consist of a spectrum of inherited biochemical abnormalities that can affect any step in the pathway of thyroid hormone synthesis, secretion, or action. Differences in the clinical symptoms and presentations depend on whether the defect is complete or incomplete, whether the change is qualitative or quantitative, which enzyme or protein is involved, and the degree of compensation of the individual. If the defect is severe, neonatal hypothyroidism (cretinism) occurs (263–265).
The morphology of the thyroid in patients with dyshormonogenesis is similar despite the different biochemical abnormalities (266). The gland is usually enlarged and nodular. Secondary changes including hemorrhage and fibrosis can be found. Microscopically, the changes consist of extreme follicular hyperplasia. Most nodules show microfollicular or trabecular patterns (267). Clear cell change may be seen. Cytologic atypia is very common and may be so severe as to suggest a malignancy (268). However, true invasive growth is extremely rare, so care must be taken in order not to overdiagnose malignancy.
NODULAR THYROID DISEASE AND HYPERTHYROIDISM
Descriptions of clinical hyperthyroidism in patients with nodular glands have been attributed to Plummer and Goetsch. The gland may be multinodular, or only a solitary mass may be present (269).
Toxic Nodular Goiter
In toxic nodular goiter, the clinical symptoms complex of hyperthyroidism is associated with a nodular gland. This cause of hyperthyroidism is more commonly seen in older individuals. This histology is similar to diffuse toxic goiter with the changes occurring within nodules (270).
Toxic Nodule (Adenoma, Carcinoma)
Rarely, a solitary nodule will function autonomously and produce clinical hyperfunction. Most of these are benign nodules; only rare examples of documented follicular carcinomas producing hyperthyroidism are known (271).
Central Hyperthyroidism
The pituitary and, more rarely, hypothalamic causes of hyperthyroidism have been documented in some patients (272). The thyroid in these cases is enlarged but is often only slightly so. Histologically, the appearance mimics the hyperplasia seen in autoimmune hyperthyroidism, except that lymphocytic infiltration is absent or minimal.
Other Causes of Hyperthyroidism
Other causes of clinical hyperthyroidism include gestational trophoblastic disease (273), struma ovarii (274), subacute thyroiditis (discussed previously), painless thyroiditis (discussed previously), rapidly growing tumors in the gland with thyroid destruction, excess exogenous thyroid hormone ingestion (275), ectopic production of TSH and thyrotropin-releasing hormone (TRH), and peripheral and generalized resistance to thyroid hormone (276). In some of these conditions, the histologic changes are well described (i.e., subacute thyroiditis and rapidly growing tumor in the gland produce destruction of the thyroid with release of stored hormone into the circulation); in others, the histology is poorly documented.
FOLLICULAR THYROID NODULES
NODULAR GOITER
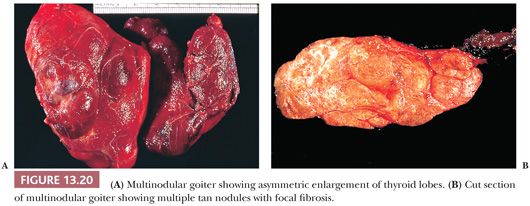
The incidence of nodular goiter depends on the criteria used to define it. Approximately 2% to 4% of the population has a clinical mass. Approximately 10% of thyroids removed at autopsy will contain nodules, usually multiple, but up to 40% to 50% of thyroids will harbor microscopic nodules (277,278). Although multinodular goiter can be associated with hyperfunction (279), usually the patient is euthyroid.
Grossly, nodular goiters range from slightly to massively enlarged glands (weights of 50 to >800 g can be found) with intact capsules and a bumpy external surface (Fig. 13.21). Sectioning will show multiple nodules of varying consistency separated by variable amounts of normal-appearing thyroid (Fig. 13.21). The nodules are composed chiefly of brown thyroid tissue; fibrous bands and calcification are often noted. Microscopically, colloid lakes alternating with normal to hyperplastic-appearing foci of thyroid, hemorrhage, siderosis, fibrosis, calcification, and even bone are found (Fig. 13.21). Variable amounts of lymphocytic infiltrate will also be found, possibly reflecting an immunologic abnormality (278).
Why do nodules occur? The work of Studer and colleagues (280,281) suggests that certain follicular cells or groups of cells are intrinsically more rapidly growing than their neighbors. The initial proliferation is polyclonal, involving one or more likely a group of follicles. Adjacent follicles remain quiescent. In the process, vascular compression in the stroma leads to focal ischemia, necrosis, and inflammatory changes. At later times, the same process may affect another group of follicles until large zones of the thyroid are affected. As the process continues, the secondary phenomena—hemorrhage, fibrosis, and calcification—take place. While these changes occur, the hormonal stimuli to the gland continue. Distortion of the vascular supply and the presence of dilated follicles filled with colloid interfere with the distribution of iodide and thyrotropin. Some parts of the gland will be exposed to excess thyrotropin, and focal hyperplasia may occur; other areas will have relative iodide or thyrotropin deficiency leading to zones of atrophy (280,281).
What can be said about solitary follicular nodules that histologically, at least, appear identical to the multiple nodules seen in nodular goiter? Are these then regenerative, proliferative nodules, but not neoplasms; or alternatively, do these represent true benign follicular adenomas? Many pathologists prefer the less definitive term “adenomatous or adenomatoid follicular nodule” for such lesions, avoiding the issue of histogenesis.
Hicks and coworkers (282) and Namba et al. (283), using genetic analysis technology that had previously proved clonal origin of colonic adenomas and parathyroid adenomas, found that many solitary thyroid follicular nodules were clonal; hence, they are follicular adenomas.
FOLLICULAR ADENOMA
A follicular adenoma or solitary adenomatous or adenomatoid nodule is defined as a benign encapsulated mass of follicles, usually showing a uniform pattern throughout the confined nodule (Fig. 13.22) (219,284). Adenomas are solitary; indeed, if there are multiple nodules in a lobe or a thyroid gland, it is probably more appropriate to diagnose multinodular goiter with adenomatous change (adenomatous hyperplasia). The features Meissner used to distinguish histologically between adenoma and adenomatous nodules included encapsulation, uniformity of pattern within the adenoma, and compression of the surrounding gland by the adenoma and its capsule.
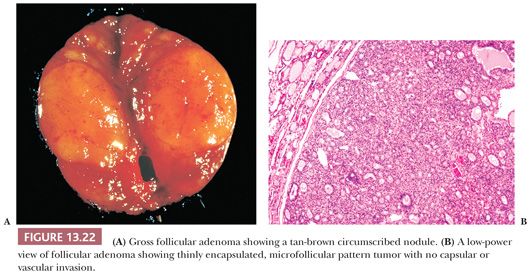
Descriptive terms that have been used to delineate the patterns seen in follicular adenomas include macrofollicular, simple, microfollicular, fetal, embryonal, and trabecular. However, because these patterns have no clinical import, it is not necessary to subdivide thyroid adenomas. Relatively common changes found in adenomas include hemorrhage, edema, and fibrosis, especially in the central portions of the tumor. Calcification may be seen. Lesions that have undergone fine-needle aspiration biopsy (FNAB) may show necrosis, increased mitotic activity, and cellular atypia in the area of the needle tract. Rare adenomas with fat, cartilage, or signet ring cells may be seen (14,219).
Whether or not some solitary follicular nodules have the biologic potential to become carcinoma is unknown; the findings of aneuploid cell populations in 27% of such lesions suggest that some of these may represent carcinoma in situ (153). The solitary follicular lesion removed by lobectomy, when it is adequately studied, shows no evidence of invasion and will neither recur nor metastasize. (Enucleation of follicular adenomas should be mentioned here only to be condemned as a surgical procedure. The pathologic evaluation of these lesions requires analysis of the tumor-capsule-thyroid interface.)
Occasionally, a follicular adenoma will contain bizarre cells (i.e., will show random, often marked cytologically atypical cells with huge hyperchromatic nuclei); sometimes multinucleated cells may be seen. These lesions are benign.
HYALINIZING TRABECULAR ADENOMA/NEOPLASM OF THE THYROID
The hyalinizing trabecular adenoma is a follicular-derived lesion that has a distinctive histology (285). Microscopically, these adenomas grow in nests that are surrounded by dense hyaline stroma. The histology is reminiscent of that seen in paragangliomas; however, the tumor is derived from the follicular epithelium (Fig. 13.23). The nuclear features of the follicular cells are similar to those seen in papillary carcinoma (285). By immunohistochemistry, the cells of hyalinizing trabecular adenoma stain positive for thyroglobulin and cytokeratin 19 and negative for calcitonin, although the presence of other neuroendocrine markers has been described (286,287).
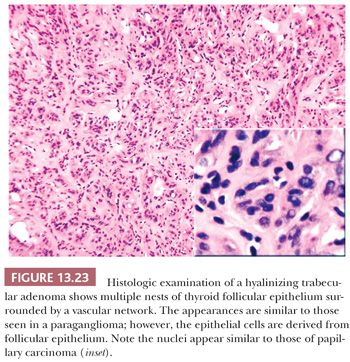
Some authors have proposed that these adenomas actually represent a variant of papillary carcinoma. This is because of similar nuclear cytology, immunoprofile, and RET oncogene rearrangements in both tumors (288,289). However, to date, no BRAF mutations have been seen in these tumors, and a benign behavior has thus far been described in all cases of hyalinizing trabecular adenoma (290). Therefore, until metastatic behavior is described in a case of hyalinizing trabecular adenoma, these tumors can be designated as hyalinizing trabecular neoplasm.
ATYPICAL FOLLICULAR ADENOMA
Atypical follicular adenoma, a term proposed by Hazard and Kenyon (291), includes those follicular tumors that show pathologically disturbing features (spontaneous necrosis, infarction, numerous mitoses, or unusual cellularity) but do not show invasive characteristics on careful examination. The overwhelming majority of the atypical adenomas behave clinically in a benign fashion.
FOLLICULAR CARCINOMA OF THE THYROID
Follicular carcinoma comprises approximately 5% of thyroid cancers; however, in iodide-deficient areas, this tumor is more prevalent, making up 25% to 40% of thyroid cancers (292,293). When iodide is added to the diet, papillary cancer increases and follicular tumors decrease (294). The true incidence of follicular carcinoma is difficult to determine because the follicular variant of papillary carcinoma may still be placed into this category (295). Risk factors include iodine deficiency, older age, female gender, and radiation exposure. Clinically, follicular carcinoma usually presents as a solitary mass in the thyroid (293).
Follicular carcinoma has a marked propensity for vascular invasion (not lymphatics). It is possible that follicular cancers produce certain factors that alter the venous endothelium, allowing the tumor easy access (208,292,293). In any event, follicular carcinoma avoids lymphatics; hence, true embolic lymph node metastases are exceedingly rare (296).
Follicular carcinoma disseminates hematogenously and metastasizes to bone, lungs, brain, and liver (Fig. 13.24A) (297). Follicular cancer metastases will usually be treatable by radioiodine if there is no normal thyroid tissue present in the neck. For this reason, many surgeons consider total thyroidectomy appropriate therapy for encapsulated follicular cancers (298). Because it is very difficult, if not impossible, to render a definitive cancer diagnosis on an FNA sample or frozen section of such a tumor, delayed completion thyroidectomy may be necessary (299).
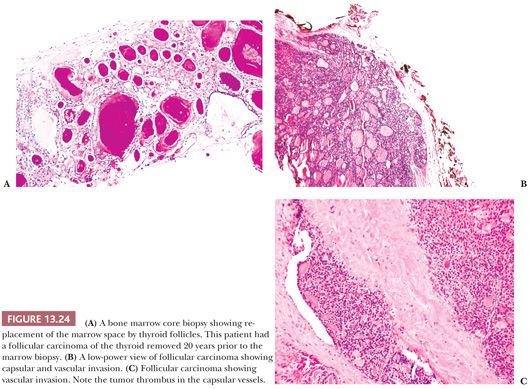
Alternatively, because the disease-free interval after lobectomy or cure of these encapsulated lesions is so great, completion thyroidectomy may represent “overkill” in a large percentage of these patients (300). Some therapists prefer radioablation of the residual thyroid either when the definite diagnosis of cancer is made or if and when metastases first appear (301). Total thyroidectomy appears warranted in those individuals who present with metastatic disease and in whom the primary is still untreated (302). Certainly, lymph node dissection is not warranted because these tumors do not spread to nodes (302).
Patients who have follicular carcinoma that is widely invasive fare poorly (292). However, those individuals with encapsulated follicular tumors confined to the thyroid enjoy a prolonged survival (>80% at 10 years) (293). Several studies have addressed those factors (clinical and pathologic) that are associated with a worse outcome (303,304). Poor prognostic factors include the presence of metastases, multiple sites of metastases, age over 50 years, large tumor size, extensive vascular invasion, extracapsular extension, and poorly differentiated areas of tumor (305). Studies using multivariate analysis have identified age over 45 years, extrathyroidal extension, distant metastases, and tumor size greater than 4 cm as independent prognostic factors in follicular carcinoma (306). An extremely significant complication that may occur in patients with follicular cancer is transformation into anaplastic cancer (307); this may occur de novo in an untreated follicular lesion or in metastatic foci (308).
Pathology
The widely invasive follicular carcinoma is a tumor that is clinically and surgically recognized as a cancer; the role of the pathologist in its diagnosis is to confirm that it is of thyroid origin and is a follicular neoplasm (14). Lang and coworkers (303) noted 80% of the patients with widely invasive cancers developed metastases and approximately 20% died of tumor. Woolner (309) found a 50% fatality rate for widely invasive tumors compared with only 3% of those with minimal invasion. Many of these tumors contain poorly differentiated areas and should probably be so designated rather than as follicular carcinoma.
Only the pathologist, upon examining well-fixed histologic sections, can diagnose the minimally invasive follicular carcinoma. These lesions are not diagnosable by FNA cytology because the diagnosis requires the demonstration of invasion at the edges of the lesion; therefore, sampling of the center, as in obtaining a cytologic sample, cannot be diagnostic (310).
Similar problems exist in evaluating such lesions by frozen section. Some authors recommend that intraoperative assessment of such lesions involve the examination of frozen sections from three or four separate areas of the nodule (311). This wastes resources and rarely gives useful diagnostic information. The surgeon should have removed the lobe involved by the nodule, and if it is a follicular carcinoma that is only minimally invasive, the appropriate therapy has probably already been accomplished. Because only a small number of these lesions will show evidence of invasion at the time of permanent section (i.e., the majority of them are benign) and because overdiagnosis is more dangerous for the patient than is the delay in making a definitive diagnosis, we discourage frozen section evaluation of these nodules (208).
Grossly, the minimally invasive follicular carcinoma resembles a follicular adenoma; the lesion is well encapsulated. The thickness of the capsule is prominent and is often thicker than that seen in follicular adenoma. Microscopically, the tumor demonstrates a microfollicular or trabecular pattern with regular, small round follicles (219,293,312). Hemorrhage, necrosis, or even tumor infarction may be noted, and significant mitotic activity is often found (310).
What are the minimum criteria for making this diagnosis? Invasion of the capsule, invasion through the capsule, and invasion into veins in or beyond the capsule represent the diagnostic criteria for carcinoma in a follicular thyroid neoplasm (Fig. 13.24B,C). The criterion for vascular invasion applies solely and strictly to veins in or beyond the capsule because tumor plugs within capillaries in the substance of the tumor have no apparent diagnostic and prognostic importance (293,310). The definition of capsular invasion is controversial (208). Lang and Georgii (313) and Franssila and coworkers (292) require penetration of the capsule to diagnose a follicular tumor as carcinoma. Iida (314) noted that distinguishing capsular invasion from trapping may prove difficult; he required that the invasive tongues of tumor sever and deflect the collagen fibers in the capsule.
Is capsular invasion insufficient for the diagnosis of follicular cancer? Kahn and Perzin (312) found that only one in seven patients (14%) with capsular invasion demonstrated metastases. Evans (297) noted that capsular invasion only was present in three of seven patients (43%) with metastases. However, in these three patients, metastases were already present at initial diagnosis. On the other hand, those authors who diagnose tumors with capsular invasion only as atypical adenomas indicate a benign clinical course after thyroid lobectomy. Periods of follow-up differ among various series.
The presence of vascular invasion is also indicative of malignancy in a follicular tumor. Invasion of vessels within or beyond the lesional capsule is necessary for a definitive diagnosis of vascular invasion because vascular invasion may be seen within the lesion itself and is not believed to be of prognostic significance (315). The authors believe that those lesions with vascular invasion should be separated from the minimally invasive follicular carcinomas, which show capsular invasion only. Perhaps use of the term angioinvasive follicular carcinoma is warranted because it is believed that the angioinvasive lesions have a greater probability of recurrence and metastasis (208).
Although most recurrences or metastases will appear within 5 years after thyroidectomy, encapsulated follicular carcinomas notoriously can present as metastases many years after initial resection (293,297).
In our practice, we use the terms minimally invasive and angioinvasive carcinoma. The former is applied to those cases which show only capsular or transcapsular invasion; the latter is used for tumors in which vascular invasion is found with or without capsular invasion. As previously mentioned, we propose this distinction based on the belief that angioinvasive tumors have greater propensity toward distant metastasis (208).
None of the ancillary techniques assist in defining benign from malignant follicular tumors. Ultrastructural, morphometric, and flow cytometric analyses have not helped in distinguishing these lesions (316,317). Approximately 60% of follicular carcinomas will show aneuploid cell populations. Backdahl et al. (318) analyzed 65 follicular thyroid tumors (26 benign and 39 carcinomas). He noted that of the 20 patients with cancer who survived, 19 had diploid tumors, whereas 17 of 19 patients who died of carcinoma had tumors with aneuploid DNA patterns (318).
All follicular carcinomas express thyroglobulin and show a similar cytokeratin profile to normal thyroid parenchyma. Some authors have shown that HBME-1 is exclusively expressed in 90% to 100% of follicular carcinomas and not adenomas. However, others have reported HBME-1 expression in adenomatoid nodules and follicular adenomas (151,286,319).
Molecular Biology of Follicular Carcinoma
A specific translocation t(2;3) leads to the expression of PAX8-PPAR-γ chimeric protein; initial studies by Kroll and colleagues (320,321) demonstrated this translocation is specific to follicular carcinoma. However, follow-up studies employing immunohistochemistry and molecular biology have shown that PPAR-γ expression can occur in some cases of follicular adenoma, FVPTC, and even benign thyroid parenchyma (322–324). RAS mutations are more frequent in follicular carcinoma as compared to follicular adenoma; some authors have found an association between RAS mutations and clinically aggressive follicular carcinomas (325). LOH on chromosomes 10q and 3p can be seen in follicular carcinoma, suggesting a role of tumor suppressor genes in its pathogenesis (326,327). RET oncogene rearrangements, which were initially thought to be specific to PTC, has been reported in 45% of adenomas in patients with history of radiation exposure (169).
WELL-DIFFERENTIATED FOLLICULAR “TUMORS OF UNDETERMINED MALIGNANT POTENTIAL”
This designation has been recently proposed in thyroid pathology for follicular-patterned encapsulated tumors that have been controversial and difficult to diagnose as a result of (a) questionable or minimal nuclear features of PTC or (b) questionable or one focus of capsular invasion that is confined to tumor capsule and does not traverse the entire thickness of capsule and lacks any nuclear features of PTC (210).
This terminology may be extremely helpful to pathologists in the diagnoses of certain follicular-patterned lesions; however, these terms are proposed on the basis of data which lack complete clinical follow-up. Therefore, clinicians may find it problematic to establish treatment strategies.
ONCOCYTIC FOLLICULAR (ALSO KNOWN AS HÜRTHLE CELL LESIONS) LESIONS
Oncocytic follicular cells are derived from follicular epithelium and are characterized morphologically by large size, distinct cell borders; voluminous granular cytoplasm; large nucleus; and prominent nucleolus (328). Ultrastructural studies have shown that the cytoplasmic granularity is produced by huge mitochondria filling the cell (329). Studies by Harcourt-Webster and Stott (330) and Tremblay and Pearse (331) have shown that these cells contain high levels of oxidative enzymes.
Oncocytic follicular cells can be found in a number of conditions in the thyroid (nodular goiter, nonspecific chronic thyroiditis, long-standing hyperthyroidism, chronic lymphocytic thyroiditis [Hashimoto disease], and nodules and neoplasms); they cannot be considered specific for any disease entity (332,333).
Nodules of oncocytic follicular cells are not all neoplastic. In fact, the majority represents oncocytic change/metaplasia of preexisting follicular adenomatous nodules in goiters or thyroiditis (332).
The clinical behavior in oncocytic follicular neoplasms has elicited debate in the literature. Some authors cite 80% or more of these lesions as benign, whereas others consider all such lesions malignant (334–336). Because most oncocytic neoplasms are follicular in pattern, the criterion for distinguishing benign from malignant is the same as for other follicular neoplasms (i.e., the identification of capsular or vascular invasion) (328,337). However, the pathologic criterion for malignancy is met more frequently for tumors composed of oncocytic follicular cells than for their nononcocytic counterparts (328,337). Thus, whereas 2% to 3% of solitary encapsulated follicular tumors of the thyroid show invasive characteristics, 30% to 40% of such lesions with Hürthle cell cytology will have such features. In addition, whereas true follicular carcinomas of the thyroid rarely, if ever, metastasize embolically to lymph nodes, approximately 30% of oncocytic follicular carcinomas do (328,335,337).
Most oncocytic follicular neoplasms of the thyroid are solitary mass lesions that show complete or partial encapsulation. They are distinguished from the surrounding thyroid by their distinctive brown to mahogany color (Figs. 13.25 and 13.26). Rarely, an oncocytic follicular neoplasm may undergo spontaneous infarction. Extensive infarction may also be seen following FNAB (Fig. 13.27) (333,337).
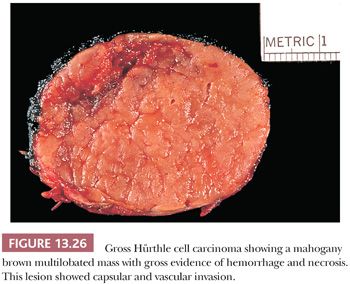
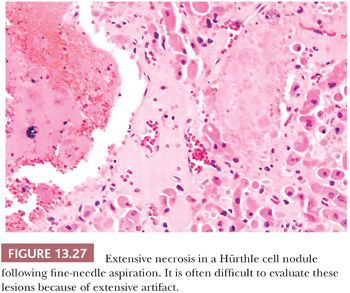
The claim that all oncocytic follicular neoplasms should be considered malignant or potentially malignant, especially if 2 cm or greater in size, is no longer considered valid. Many studies from the United States and Europe (338,339) indicate that benign Hürthle cell neoplasms exist. Size, nuclear atypia, multinucleation, cellular pleomorphism, mitotic figures, or histologic pattern of the lesion are not predictive of behavior (328,337). Both benign and malignant tumors can show calcifications, which may be confused with psammoma bodies, but these calcifications are present within the colloid (333).
Pathologic criteria for malignancy—vascular invasion, transcapsular penetration, and destructive capsular invasion—can predict the clinical behavior of these tumors. The distinction between benign and malignant oncocytic follicular neoplasm is made by the pathologic evaluation of the mass, applying strict criteria as previously noted for the non-Hürthle follicular tumors. In the absence of invasion, an oncocytic follicular thyroid tumor should be considered benign (333,337,340). Total thyroidectomy is not needed for all oncocytic follicular neoplasms (341,342).
By immunohistochemistry, oncocytic follicular lesions are positive for thyroglobulin and TTF-1. Carcinoembryonic antigen (CEA) expression has been described in some but not all series. Hürthle cell lesions are positive for S100 protein (343,344). In addition, galectin-3 may potentially serve as a marker in difficult differential diagnosis cases involving oncocytic follicular adenomas and carcinomas (345).
In the histologically defined carcinomas, flow cytometric data may provide prognostic aid. Carcinomas that show an aneuploid pattern may behave more aggressively than those that are diploid. Biologically and histologically benign oncocytic follicular tumors of the thyroid can show aneuploid DNA patterns. Such a finding, however, does not indicate malignant behavior. Approximately 20% to 50% of oncocytic follicular tumors that are histologically malignant and aneuploid are more aggressive biologically and clinically than diploid oncocytic follicular carcinomas (346,347).
Molecular Biology of Oncocytic Follicular Neoplasms
Oncocytic follicular tumors are biologically different from other follicular-derived tumors (348,349). H–ras mutations are more frequent in oncocytic carcinoma than follicular carcinoma (325,350), and there are significant differences in the expression of transforming growth factor-alpha (TGF-α), TGF-β, insulin-like growth factor-1 (IGF-1), and N-myc between oncocytic follicular and nononcocytic follicular carcinoma (350).
Maximo and colleagues (351) studied the relationship between mitochondrial DNA alterations and thyroid tumorigenesis. This study showed that oncocytic follicular tumors display a relatively higher percentage of common deletions of mitochondrial DNA as compared to other follicular-derived tumors. In addition, oncocytic tumors also showed germline polymorphisms of the ATPase 6 gene, which is required for the maintenance of mitochondrial DNA (351). Gasparre et al. (352) have shown that disruptive mutations in mitochondrial DNA were found in complex I subunit genes in oncocytic neoplasms and this finding appears to be a marker for thyroid oncocytic tumors. A somatic missense mutations in GRIM19 has been identified in approximately 11% of sporadic oncocytic follicular carcinomas. GRIM19 is believed to promote apoptosis as part of the interferon and retinoic acid–induced pathway of cell death, is a negative regulator of cell growth, is involved in mitochondrial metabolism, and is linked in part to mitochondrial complex I assembly (353–355).
It is postulated that oncocytic follicular tumors may develop in thyroid gland via two different mechanisms. The presence of abundant and abnormal mitochondria in the cytoplasm of the neoplastic oncocytic follicular cells may be seen either throughout the tumor, suggesting that the neoplastic transformation occurred in cells with abnormal mitochondrial DNA (i.e., de novo transformation), or limited to some portions of tumor, indicating that abnormalities in mitochondrial DNA occurred after the tumor originated from the follicular cells (351,356).
C-CELL LESIONS
The hormone calcitonin was discovered and characterized in the early 1960s; the thyroid C cell had been described in many animal species by the end of the nineteenth and early twentieth centuries. In the early 1960s, pathologists were defining the morphology of the tumor now known as medullary carcinoma (357,358). In 1966, Williams (359) proposed that medullary carcinoma might be derived from or at least differentiated toward the C cell and predicted that if the C cell was the source of calcitonin, the tumors might also produce this hormone.
Normal C cells are thought to be derived embryologically from the neural crest and migrate into the thyroid along with the ultimobranchial body. Hence, these two elements may be closely associated in the adult thyroid; however, the ultimobranchial body itself does not show calcitonin immunoreactivity and should not be considered as hyperplastic C cells (360,361). In humans, C cells are found along the lateral aspects of the thyroid lobes in the upper two-thirds of the gland. The C cells comprise less than 0.1% of the thyroid mass in humans (362,363). Wolfe and colleagues (362,363) showed C-cell distribution in the thyroid by immunohistochemistry. The number of C cells in the thyroid differs according to age. They identified larger numbers of these cells in infants and children under the age of 6 years than in adults. In children, groups of up to six cells can be seen, with as many as 100 cells noted in a low-power microscopic field. In adult glands, no more than 10 cells should be found in a low-power field (362,363).
Clusters of C cells in adults have been described in endocrinologically normal adults. O’Toole and colleagues (364) examined thyroid glands from forensic autopsies and recognized a trend toward increased numbers of these cells in older individuals (over age 60 years); however, they noted large standard deviations. This remains a problem area for pathologists.
MEDULLARY THYROID CARCINOMA
Medullary thyroid carcinoma (MTC) is rare and comprises fewer than 10% of all thyroid malignancies (365,366). This tumor is of great diagnostic importance because of its potential aggressiveness; its close association with multiple endocrine neoplasia (MEN) syndromes type 2A (MEN2A or Sipple syndrome), type 2B (MEN2B, MEN3, or mucosal neuroma syndrome), or familial non-MEN MTC (FMTC); and a relationship to a C-cell hyperplasia as probable precursor lesion (367). Although the majority of medullary carcinomas are sporadic, approximately 10% to 20% are familial. Because these familial cases have been identified, a gene associated with medullary carcinoma has been identified on chromosome 10 and involves mutations in the RET oncogene, which codes for a tyrosine kinase receptor (368,369). The clinical features are similar in both sporadic and familial cases that are symptomatic (365). Medullary carcinoma can affect patients of any age; however, most affected individuals are adults with an average age of approximately 50 years (365,366). In familial cases, though, children can be affected; also in these instances, the age of diagnosis tends to be younger (mean age: approximately 20 years). Although sporadic medullary carcinomas are seen more commonly in women, familial cases have an equal sex ratio because an autosomal dominant mode of inheritance is present (370,371).
Most patients with medullary carcinoma will present with a thyroid nodule that is painless but firm. In up to 50% of cases, obvious nodal metastases will be present at the time of diagnosis. Distant metastases to lung, bone, or liver may also be noted initially in approximately 15% to 25% of cases (372). When the tumor produces excess hormone other than calcitonin, the presenting symptoms may be related to that hormone hypersecretion (adrenocorticotropic hormone [ACTH], prostaglandin) (373,374).
In the familial forms, there are associated endocrine or neuroendocrine lesions. RET mutations in families with MEN2A (95% of families) and FMTC (85% of families) have been seen in one of the five cysteine codons in exon 10 and exon 11 (375–378).
Sipple syndrome (MEN2 or MEN2A) consists of medullary thyroid cancer and C-cell hyperplasia, adrenal pheochromocytoma and adrenal medullary hyperplasia, and parathyroid hyperplasia (379,380). Although most affected patients will have the complete syndrome, not every patient will manifest each of these lesions. In MEN2A, RET mutations in codon 634 are identified frequently (>60%) and have been associated with the presence of pheochromocytoma and hyperparathyroidism and, rarely, cutaneous lichen amyloidosis (381–383). It has been shown that a variety of phenotypic expressions in MEN2A families can be seen with the same RET mutations (382,384). Mutations in specific codons have been correlated with clinical behavior and symptomatology in some families (385–387).
MEN2B consists of MTC and C-cell hyperplasia, pheochromocytoma and adrenal medullary hyperplasia, mucosal neuromas, gastrointestinal ganglioneuromas, and musculoskeletal abnormalities (388–392). These patients may have familial disease (>50% do); some cases arise apparently as spontaneous mutations. A point mutation at codon M918T (exon 16) has been noted in 95% of cases with MEN2B. The other rare mutations reported in MEN2B patients include genotype A883F (exon 15) and double mutation V804M/Y806C at codon 804 (exon 14) and 806 in the same allele (376,393–395). Mutations in codons 918 and 883 are associated with MTC presenting at younger age with high risk of mortality and disease-specific mortality (396–398). Because MTC is generally considered a potentially lethal tumor, especially in MEN2B patients, it is recommended that first-degree relatives of the affected family member should undergo genetic screen; a positive result should be followed by prophylactic total thyroidectomy (399). Although still controversial, most endocrinologists and pediatric endocrinologists recommend total thyroidectomy before the age of 6 years in children found to have certain “aggressive” RET mutations in their germline (399,400). MEN2B shows similarity to von Recklinghausen disease because in neurofibromatosis, similar lesions are found in the gastrointestinal tract and pheochromocytomas are common. Nerve growth factor has been identified in some medullary carcinomas of these patients; it has been postulated that this product of the tumor may be responsible for the neural lesions seen in patients with MEN2B (401). However, the neural lesions often precede the development of medullary cancer by many years.
RET mutations can also be seen in sporadic medullary cancer, usually in the tumor cells and rarely in germline (indeed if found in the latter, the possibility of familial disease needs to be considered) (402). In sporadic MTC, somatic mutations have been seen in exon 16 of the RET (M918T) in 50% to 80% of cases. The rare somatic mutation in sporadic MTC is seen in codon 618, 603, 634, 768, 804, 883, and partial deletion of RET gene (160,369,397,402–406).
The pathologist may contribute to the determination of familial rather than sporadic disease if, upon examining a medullary carcinoma of the thyroid, he or she notes multifocal or bilateral tumors and the presence of C-cell hyperplasia (365).
Pathology
Medullary carcinoma is usually located in the area of highest C-cell concentration (i.e., the lateral upper two-thirds of the gland). In familial cases, multiple small nodules may be detected grossly, and rarely, lesions may be found in the isthmus. The tumors range in size from barely visible to several centimeters. Many medullary carcinomas are grossly circumscribed, but some will show infiltrative borders (Fig. 13.28). Some tumors will show gross necrosis and hemorrhage (219,365).
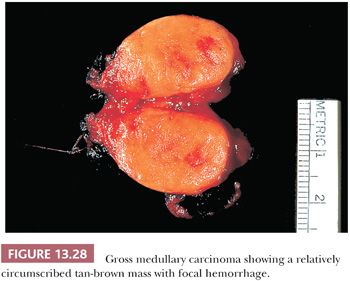
The typical medullary carcinoma may be microscopically circumscribed or more likely will be freely infiltrating into the surrounding thyroid. The pattern of growth is of tumor cells arranged in nests separated by varying amounts of stroma. The tumor nests are composed of round, oval, or spindle-shaped cells; there often is isolated cellular pleomorphism or even multinucleated cells (Fig. 13.29). The nuclei are uniform; the nuclear-to-cytoplasmic ratio is low. Intranuclear cytoplasmic inclusions are commonly noted. Mitotic figures can be seen. The tumor stroma characteristically contains amyloid, although this is not necessary for the diagnosis; approximately 25% of medullary carcinomas do not contain amyloid (Fig. 13.29). The amyloid is most likely derived from precalcitonin and, indeed, immunohistochemical stains for calcitonin often stain the amyloid. Calcifications in areas of amyloid deposition are characteristically present. The tumors commonly invade lymphatics and veins (219,365,407).
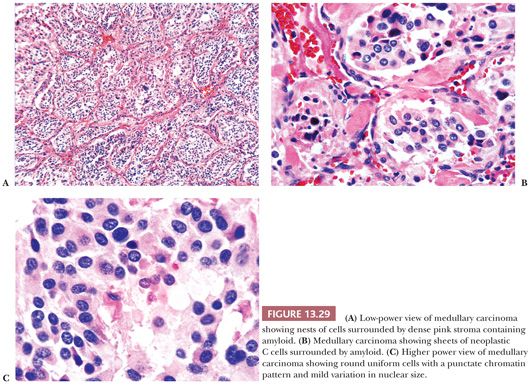
Several medullary carcinoma variants have been described. In the papillary variant, a papillary or pseudopapillary growth pattern is identified. The pseudopapillary variant is more common and probably results from fixation artifact (408). The true papillary variant is extremely rare and needs to be differentiated from typical PTC; nuclear morphology is the most important distinguishing feature. The follicular variant is characterized by the presence of follicles, glands, or tubules. Care must be rendered to determine that the follicular structures are not just entrapped normal thyroid within the lesion (408,409). Some medullary carcinomas are grossly and microscopically encapsulated (410,411). Mendelsohn and Oertel (412) reported a series of encapsulated thyroid lesions classified as atypical adenomas and showed that many of these were medullary cancers containing immunoreactive calcitonin. The follow-up in encapsulated medullary carcinomas indicates that they have a more benign prognosis than usual medullary tumors (412). The histologic differential diagnosis for the encapsulated variant includes hyalinizing trabecular adenoma. Immunohistochemistry for calcitonin will be positive in the medullary carcinoma but not within the hyalinizing trabecular adenoma. In the past, some authors have used the term C-cell adenoma to describe encapsulated variants of medullary carcinoma; however, this terminology is not favored (413). The small cell variant of medullary carcinoma has also been described (414). These tumors look like pulmonary small cell carcinoma from which they need to be distinguished, if possible. The prognosis is worse than for typical medullary carcinoma. Although calcitonin expression may not always be seen, the small cell variant of medullary carcinoma often expresses CEA and calcitonin gene–related peptide, just as other types of medullary carcinoma do (414). The giant cell variant is rare and is characterized by large atypical cells admixed with areas of typical medullary carcinoma (415). Because of the presence of large atypical cells, this variant needs to be differentiated from anaplastic thyroid carcinoma, a tumor with a worse prognosis when compared to medullary carcinoma. The clear cell variant is a rare form of medullary carcinoma and is characterized by cells with abundant clear cytoplasm (416). Immunohistochemical stains reveal the presence of calcitonin in the lesional cells. Differential diagnostic consideration for this variant includes follicular-derived neoplasms with clear cell cytoplasm as well as metastatic renal cell carcinoma. Other variants of medullary carcinoma include oncocytic and squamous variants (417). Immunohistochemical stains are often needed to establish the correct diagnosis.
Up to 40% of medullary carcinomas contain mucin, most of which is extracellular; intracytoplasmic mucin can be seen in approximately 15% of medullary carcinomas (418). Rare tumor may contain melanin pigment, the significance of which is not known (419). By immunohistochemistry, the majority of medullary carcinomas express low–molecular-weight cytokeratin, calcitonin, and calcitonin gene–related peptide (Fig. 13.30) (420). In addition, many tumors express CEA, which may also be elevated in the serum (Fig. 13.30) (421). A variety of other peptides may be found in tumor cells including somatostatin, vasoactive intestinal peptide, and synaptophysin (422,423). Some studies have also identified polysialic acid (neural cell adhesion molecule) in medullary carcinomas but not in other thyroid tumors (424).
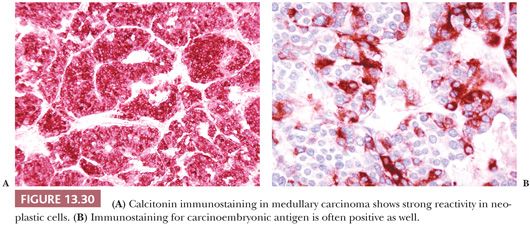
Occasional lesions (and often these are small cell type) do not contain immunoreactive calcitonin. In order to accept a calcitonin-free tumor of the thyroid as a medullary carcinoma, it should arise in a familial setting or occur in a thyroid with unequivocal C-cell hyperplasia. Immunoreactivity for calcitonin gene–related peptide would add proof to the histogenetic nature of such a lesion. The existence of a true small cell nonmedullary neuroendocrine tumor of the thyroid is accepted.
Prognostic Factors
From the clinical standpoint, stage is the most important variable for prognosis (425–427). A tumor confined to the thyroid without nodal or distant metastases is associated with prolonged survival (427). Several researchers have found that younger patients (under age 40 years), especially women, fare somewhat better than the whole group of medullary cancer patients (426,428). Patients who are discovered by screening because they are members of affected families often have very small tumors and can be cured by thyroidectomy. Patients with Sipple syndrome tend to have less aggressive tumors than the sporadic group, whereas the patients with MEN2B have aggressive lesions (427,429). Pathologic features that have been related to prognosis include tumor pattern, amyloid content, pleomorphism, necrosis, and mitotic activity (410,427). Small cell medullary carcinomas and those tumors with extensive areas of necrosis, marked cellular pleomorphism, and high mitotic activity are associated with poor prognosis (410). Encapsulated tumors and tumors with uniform cytology and abundant amyloid tend to be indolent tumors. Schroder and colleagues (410) found that aneuploidy was associated with a poor outcome.
MIXED FOLLICULAR AND MEDULLARY CARCINOMA
These controversial tumors show thyroglobulin and calcitonin immunoreactivity and ultrastructural evidence of differentiation along two cell lines (430,431). Some of the series of these tumors may have been confusing, with trapping of follicles at the invading edge of the medullary carcinoma and diffusion of thyroglobulin into the medullary carcinoma; this may result in diagnosis of mixed tumors showing immunostaining for both hormones (431). Caution should be taken when making the diagnosis of mixed medullary and follicular-derived carcinomas.
C-CELL HYPERPLASIA
The definition of C-cell hyperplasia is difficult. The lower limit of C-cell hyperplasia and the upper limit of normal C-cell mass are not clear. Various studies show C-cell clusters in adults that, taken alone, could fit into the category of C-cell hyperplasia. Yet O’Toole and colleagues (364) and Gibson and colleagues (432) noted these clusters of C cells at autopsy in apparently endocrinologically normal individuals. Conversely, the lower limit of medullary carcinoma and upper limit of C-cell hyperplasia is difficult to define. DeLellis and Wolfe (433) state that C-cell hyperplasia ranges from diffuse increase in the cells to nodules of C-cell–replacing follicles, and once the basement membrane of the follicle is breached, medullary carcinoma should be diagnosed. However, Carney and associates (434) point out it is not always obvious that the basement membrane has been crossed.
In the classic case of C-cell hyperplasia, the lesion appears as multifocal areas of increased numbers of amphophilic large cells replacing follicular epithelium and also replacing follicles completely forming nodules (Fig. 13.31). More specific definitions of C-cell hyperplasia include more than 50 C cells per low-power field and more than 40 C cells/cm2 to more than 50 C cells per three low-power fields (435,436). With these definitions, C-cell hyperplasia is not only seen in patients with MEN syndromes or familial medullary carcinoma (437) but also in patients with hyperparathyroidism, chronic hypercalcemia of other causes, Hashimoto disease, in residual thyroid tissue following removal of medullary cancer (sporadic type), and even in thyroid tissue adjacent to nonmedullary carcinomas (436,438). Because C-cell hyperplasia may be associated with so many lesions, the exact significance using these various definitions cannot be determined. One of the best ways to distinguish C-cell hyperplasia is by routine histologic examination. C-cell hyperplasia associated with familial medullary carcinoma and MEN syndromes is readily observed on routine hematoxylin and eosin (H&E) stains (439,440). The cells are often large and show significant nuclear atypia as well as occasional features of medullary carcinoma. On the other hand, secondary C-cell hyperplasia is often only observed by immunohistochemical staining for calcitonin and quantitative analysis. The actual diagnosis of C-cell hyperplasia may therefore be made by routine histologic examination for the presence of C cells by H&E stains. Micromedullary carcinoma can be found in glands removed prophylactically because of positive genetic testing for RET mutations (441). These are similar to micropapillary carcinomas; that is, they measure 1 cm or less. In the familial setting, there is usually associated C-cell hyperplasia (442).
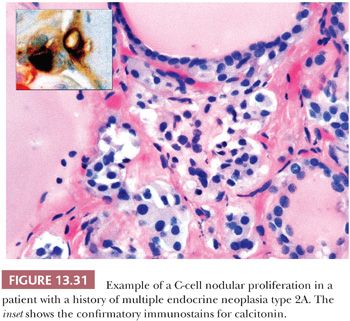
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


