THE NORMAL PITUITARY
ANTERIOR LOBE
The anterior pituitary or adenohypophysis constitutes the largest portion of the pituitary gland, about 75% to 80%, and it is formed by the pars distalis, the pars intermedia, and the pars tuberalis. The adenohypophysis is epithelial in origin and arises from the Rathke pouch, an invagination in the oral ectoderm. The anterior pituitary has a great variety of cell types and functions (Table 12.2). A number of recognized transcription factors seem to be major participants in anterior pituitary organogenesis in a multistep, highly controlled process (1,2).
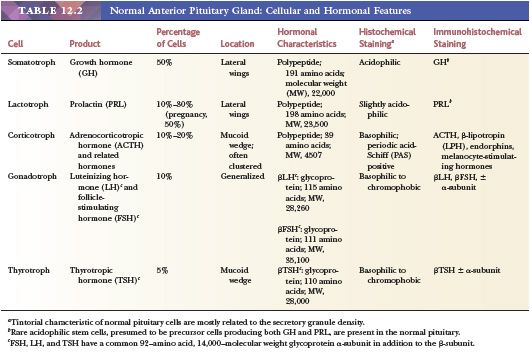
The pars distalis, corresponding to the largest portion of the adenophypophysis, is roughly divided into a central mucoid wedge and lateral components termed acidophilic wings (Fig. 12.1). The term mucoid refers to the abundance of basophilic, periodic acid-Schiff (PAS)–positive cells concentrated and often clustered in this mid portion of the gland. These cells are engaged in the production of adrenocorticotropic hormone (ACTH) and related molecules. The lateral wings contain the majority of growth hormone (GH) and prolactin (PRL) cells, both of which are variably granulated and eosinophilic. Follicle-stimulating hormone (FSH)– and luteinizing hormone (LH)–producing cells are widely and individually distributed throughout the gland. Thyrotrophic hormone (TSH)–producing cells are far smaller in number and more anteriorly situated. The morphologic features of anterior pituitary cells, as well as the biochemical characteristics of their hormone products, are summarized in Table 12.2. The region of the intermediate lobe (pars intermedia), a well-developed structure in lower animals, is vestigial in humans and consists primarily of a distinctive form of corticotropic cell and glandular spaces. The latter are remnants of the Rathke cleft and are lined by cuboidal to columnar, ciliated, or mucin-producing cells and only occasionally by granulated adenohypophyseal cells. A sleeve of anterior pituitary tissue extends upward along the anterior aspect of the pituitary stalk (pars tuberalis) and consists mainly of LH/FSH and ACTH cells, all of which are prone to squamous metaplasia with age (3). Salivary gland rests, serous in type, are encountered at the base of the pituitary stalk.
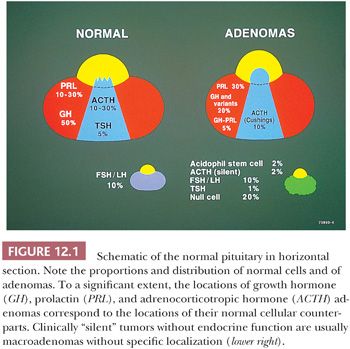
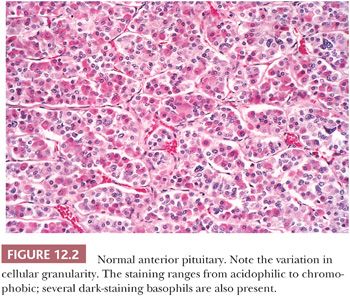
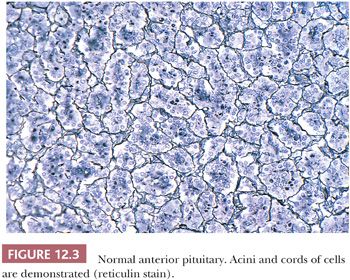
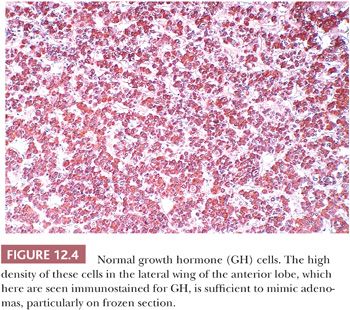
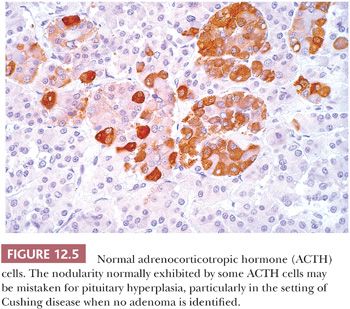
On hematoxylin and eosin (H&E) stain, anterior pituitary cells vary greatly in appearance (Fig. 12.2). Arranged in acini surrounded by a rich network of capillaries (Fig. 12.3), they were once grouped on the basis of tinctorial characteristics as acidophilic, basophilic, or chromophobic. The current cell type recognition is based on hormone content as visualized by immunohistochemistry associated with specific ultrastructural features (Figs. 12.4 and 12.5; Table 12.2). The same is true of pituitary adenomas (Tables 12.3 and 12.4) (4). Pituitary adenoma classification based solely on histochemical stains is no longer recommended because it provides little insight into functional differentiation, which can only be obtained by correlative immunohistochemical and, where needed, ultrastructural studies. These methods have shown GH and PRL cells to be histogenetically related, as are cells engaged in glycoprotein hormone (LH, FSH, and TSH) production. The cells of the anterior pituitary are not limited to the production of these hormones; they have been shown to contain various other peptides (5), as well as growth factors (6).
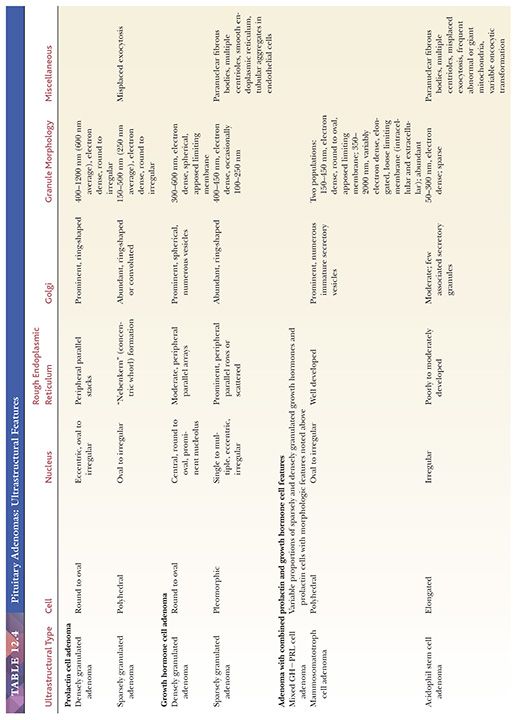
Electron microscopy has played a pivotal role in categorizing pituitary cells and their tumors. Ultrastructurally, anterior pituitary cells are epithelial in nature and possess the full complement of organelles required for hormone production and export. Identification of normal and neoplastic pituitary cell types requires attention to such features as cell shape; the content and disposition of organelles; the presence or absence and arrangement of filaments; and of course, the number, size, electron density, and shape of secretory granules (Table 12.4). The mechanisms of hormone secretion vary from transmembrane diffusion to the actual expulsion of secretory granules, a characteristic of PRL cells (7).
Folliculostellate cells akin to sustentacular cells in the paraganglia are also encountered in the anterior pituitary (5,8). Small in number, their immunoreactivity for S-100 protein, glial fibrillary acidic protein (GFAP), galectin 3, and annexin 1, and their lack of neuroendocrine marker staining distinguish them from the hormone-producing cells. The function of folliculostellate cells is still controversial, but it appears to be diverse and related to phagocytosis, secretion of growth factors, and intercellular communication (9). It has even been suggested that they represent stem cells (8).
POSTERIOR LOBE
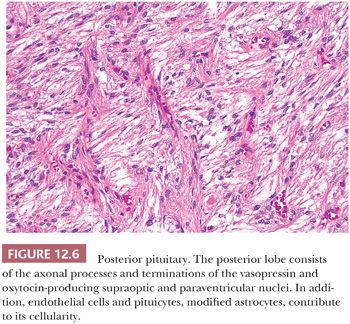
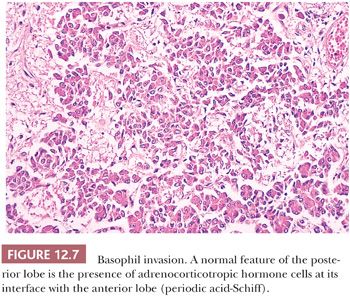
The pituitary stalk and the posterior lobe or neurohypophysis represent part of a neurosecretory unit that begins in the magnocellular neurons of the supraoptic and paraventricular nuclei. Coursing via the stalk to the posterior lobe, their unmyelinated axons and terminations carry and store the hormones vasopressin and oxytocin. In their course, the 1-nm diameter axons often show the formation of swellings (Herring bodies) in which neurosecretory materials accumulate. Pituicytes, modified glial cells of primarily astrocytic type, are found throughout the posterior lobe (Fig. 12.6). At the ultrastructural level, axons and their swellings (Herring bodies) and terminations contain secretory granules of varying electron density, as well as multilamellar bodies and electron-lucent vesicles (10). In intimate association with the pituicyte processes, axonal terminations are seen to abut or lie within perivascular spaces. Lastly, scattered corticotroph cells are a normal feature of the posterior lobe (Fig. 12.7). Derived from the intermediate lobe, they appear to be physiologically distinct from anterior lobe corticotrophs (ACTH-secreting cells). Their accumulation with age, a process termed basophil invasion of the posterior lobe, is of unknown clinical significance. Such cells are believed by some to give rise to so-called silent corticotroph cell adenomas (5,11).
BASIC CONCEPTS ON PITUITARY ADENOMAS
In neurosurgical series, pituitary adenomas represent 10% to 20% of intracranial neoplasms (12). A higher proportion of clinically functioning adenomas (65%) occurs in surgical series (13,14). Incidental or subclinical adenomas are encountered in nearly 25% of autopsies (15,16). The majority are either null cell adenomas (50%) or prolactinomas (45%). For an in-depth discussion of the endocrinologic aspects of pituitary tumors, the reader is referred to authoritative texts (17,18).
Pituitary adenomas show somewhat of a predilection for women in the third to sixth decades of life. However, no age group is exempt, including childhood, a period wherein 2% of all adenomas occur (19,20). ACTH-producing tumors are those most often encountered in pediatric patients (19).
The vast majority of pituitary adenomas are sporadic, but a small percentage of adenomas are associated with hereditary syndromes, including multiple endocrine neoplasia type 1 (MEN1), Carney complex, McCune-Albright syndrome, and a few other rare familial syndromes described as familial isolated pituitary adenoma group (21). The majority of adenomas arising in these hereditary syndromes are GH-secreting adenomas (22), but somatotroph adenomas linked to either MEN1 or Carney complex correspond to only 3% of all GH-secreting tumors (23).
Multiple adenomas are encountered in less than 1% of surgical specimens (24), although a higher incidence is seen as incidental findings in autopsy (16,25). Coexistence of ACTH and PRL cell adenomas is most common (26), which is not surprising given the high incidence of incidental PRL cell adenomas in autopsy series. The distinction of two tumors in a single biopsy specimen is often difficult and may require both immunohistochemical and ultrastructural confirmation (24).
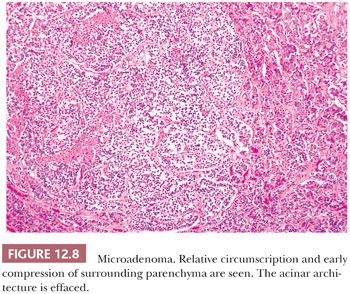
Endocrinologically functional tumors are often small, whereas silent or nonfunctioning tumors are large, coming to attention only as a result of mass effects. An accessible distinction of microadenomas (Fig. 12.8), which are defined by neuroimaging as tumors 1 cm in size or smaller, from macroadenomas (Fig. 12.9), which are tumors exceeding 1 cm, has long been in use (27). Giant adenomas, which are presently defined as adenomas greater than 4 cm in maximal dimension, are rare (28). Diffuse adenomas are ones that fill and expand the sella, often compressing the residual gland into a thin membrane. Larger or massive adenomas often efface the sellar floor, displace surrounding structures, and undergo suprasellar extension.
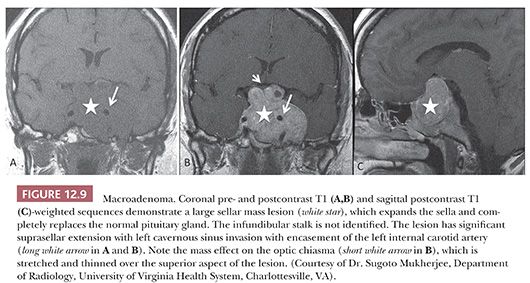
Suprasellar extension with elevation of the sellar diaphragm is common, as is the extension of tumor through its frequently incompetent diaphragmatic opening. The result of significant suprasellar extension is chiasmal compression with visual disturbance, typically bitemporal hemianopsia. Compression of the pituitary stalk may lead to disruption of the hypothalamic-pituitary trafficking of hormones leading to hypopituitarism and increased PRL release due to negative feedback, the so-called “pituitary stalk effect” (17,18). It is of note that pituitary adenomas, regardless of their size, are rarely associated with diabetes insipidus.
Massive suprasellar extension may result in the deep indentation of the brain in the region of the third ventricle. Extension into the middle, anterior, or, less often, posterior fossa may also be seen. Gross, operatively or radiographically apparent invasion of adjacent bony and soft tissues of the sellar region is not infrequently seen. In any case, it predisposes patients to tumor recurrence (29). Microscopic dural invasion, when systematically sought, is common, ranging from 65% to 95%, depending on tumor size (30,31). Documenting the presence of dural or bone invasion by microscopy is recommended for follow-up of the patient. Lateral growth into the cavernous sinus may occur through infrequent naturally occurring discontinuities in the fibrous membrane separating the sella from the sinus (32) or by way of invasion.
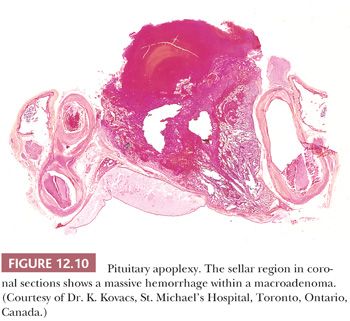
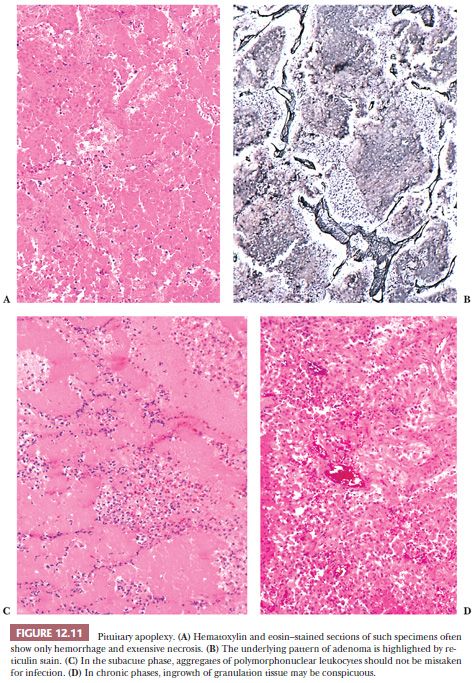
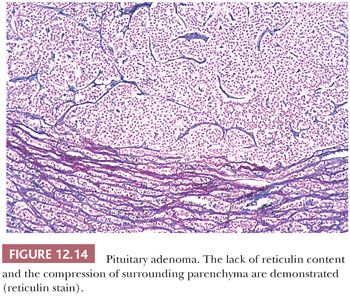
Pituitary apoplexy, which is defined as rapid enlargement of an adenoma by intratumoral hemorrhage and variable infarction, may be a surgical emergency (Fig. 12.10). Alternatively, the process may undergo subclinical evolution. Seen in approximately 10% of operated adenomas, it takes the form of hemorrhagic, necrotic, or cystic foci (33,34). Apoplexy affects all types of adenoma, but large, nonfunctioning tumors are particularly prone. The specimens usually consist of blood and necrotic tumor. Identification of the underlying tumor is aided by reticulin stains, which highlight the abnormal stromal pattern of the adenoma (Fig. 12.11).
THE ROLE OF THE PATHOLOGIST
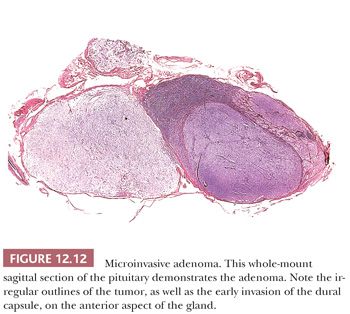
The assessment of pituitary adenomas begins at the time of surgery. Although smears or frozen sections may be performed to identify the tumor, systematic assessment of resection margins is an unrealistic endeavor. Total surgical excision may be complicated by factors such as irregular tumor shape, lack of cleavage planes and a capsule, and grossly inapparent foci of dural invasion (Fig. 12.12). Furthermore, because of lack of stroma, the soft consistency of adenomas obscures landmarks and may even promote contamination of the operative field.
Although pituitary adenomas are the most common tumors in the sellar region, a spectrum of lesions enters into their differential diagnosis. The number of diagnostic possibilities is markedly reduced by attention to such factors as the presence or absence of sellar enlargement and clinical and biochemical data regarding hormone secretion and hyperfunction. A working relationship between the pathologist and surgeon is essential, not only to ensure the procurement of an adequate specimen but also to maximize clinicopathologic correlation.
SPECIMEN HANDLING
Fresh tissues should be promptly transported from the operating room on a moist Telfa pad; delayed fixation and drying artifact must be avoided. Smears and touch preparations are preferable to frozen sections in the assessment of adenomas. Smears best demonstrate cytologic detail (Fig. 12.13). Cytologic methods obviate the mechanical and freezing artifacts that affect permanent sections and immunohistochemical preparations. Once the tissues are frozen, they often show nonspecific or reduced immunoreactivity. Furthermore, they are useless for ultrastructural study.
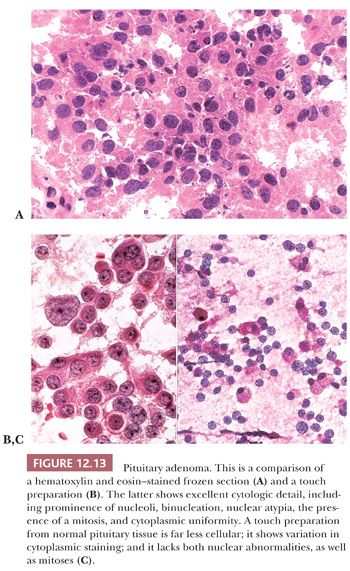
The specimen should be formalin fixed only after a minute portion, at minimum a single 1-mm fragment, has been placed in glutaraldehyde for possible ultrastructural study. With an adequate sample, consideration can be given to freezing a portion for biochemical or genetic studies. When diagnostic tissue is scant or no lesion is identified on cytologic or frozen section assessment, the entire specimen should be step sectioned to obtain H&E and unstained slides.
In addition to the preparation of an H&E-stained slide, consecutive microsections should be cut for reticulin stains (Fig. 12.14), as well as immunostains for pituitary hormones. Performance of a full hormone battery, including GH, PRL, ACTH, LH, FSH, TSH, and α-subunit of glycoproteins, is preferable for better classification of the tumors, but immunostains can be selectively applied, depending on the clinical setting and size of the specimen. This is particularly true in the setting of Cushing disease, in which serial immunostains for ACTH may be more useful in confirming the presence of a small, often fragmented pituitary adenoma rather than the entire hormonal panel. In large institutions with advanced endocrine and neurosurgical departments, the full spectrum of antibodies for pituitary hormones is usually applied to all but the adenomas of Cushing disease. As stated earlier, electron microscopy plays an important, albeit defined, role in the assessment of adenomas.
CLASSIFICATION OF PITUITARY ADENOMAS
In broad terms, the World Health Organization (WHO) and Systematized Nomenclature of Medicine (SNOMED) classify adenohypophyseal tumors as typical and atypical adenomas (WHO 8272/0 and 8272/1), as well as carcinomas (WHO 8272/3) (35).
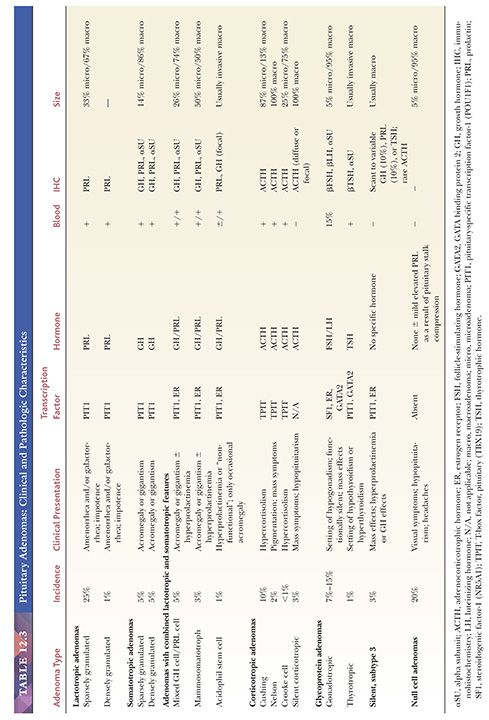
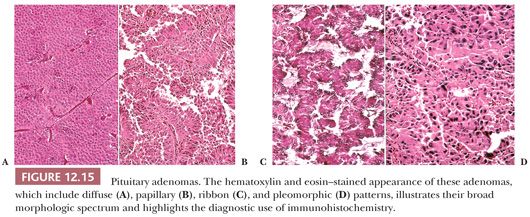
Morphologically, adenomas may show a variety of growth patterns including diffuse, papillary, and trabecular arrangements similar to other neuroendocrine tumors, which can be present in any adenoma type (Fig. 12.15). Their recognition, although of no prognostic significance, is worthwhile because a spectrum of lesions enters into the differential diagnosis of sellar region pathology. Cytologically, adenomatous cells may be acidophilic, basophilic, or chromophobic; however, these tintorial characteristics do not identify specific adenoma types. Note that all adenomas are synaptophysin immunoreactive but not all stain for chromogranin. Adenomas are focally immunoreactive for cytokeratin, in particular CAM5.2, and epithelial membrane antigen (EMA) (36). Electron microscopy has revealed much about the nature of normal pituitary cells (37) and their respective tumors. At present, although less used in clinical practice (38), ultrastructural analysis continues to play a valuable role in the diagnosis of unusual adenoma types and the expanding spectrum of rare sellar region tumors (Table 12.4) (37–39).
The classification of adenomas and their clinicopathologic and immunohistochemical features are summarized in Table 12.3. The major adenoma subgroups are discussed in turn in the following sections.
PITUITARY ADENOMA SUBTYPES
PROLACTIN CELL ADENOMA
Nearly half of the newly clinically diagnosed pituitary adenomas are PRL-producing tumors (40). The so-called prolactinomas or lactotropic tumors are composed entirely of PRL cells. Although the majority of prolactinomas are medically treated by dopamine agonists, a significant number of the patients undergo surgical resection due to several clinical issues (17,18).
Prolactinomas are the second most frequently occurring adenoma in MEN1 after somatotroph adenomas (22,41). On the other hand, isolated familial prolactinomas are rare (42). Whereas estrogens cause prolactinomas in experimental animals and pregnancy may be associated with enlargement of some, these agents alone are generally incapable of inducing prolactinomas in humans (43–45).
Microadenomas generally occur in reproductive-age women who exhibit exquisite sensitivity to PRL excess by manifesting with amenorrhea, galactorrhea, or both. In men and postmenopausal women, prolactinomas may appear to be clinically nonfunctional, growing to macroadenoma dimensions and exhibiting invasion (46–48). The basis for this relative aggressiveness appears to be related to differences in tumor invasion and proliferative activity (47,48). Approximately 50% of prolactinomas treated by surgery are macroadenomas and a third of the cases are grossly or radiographically invasive at initial surgery (48). Not surprisingly, the frequency of invasion increases with tumor size. Serum PRL levels are uniformly elevated in patients with prolactinomas, although the levels range from little more than normal (~20 ng/mL) to extremely high (≥2000 ng/mL). In general, PRL levels correlate with the tumor size (17,18). As noted earlier, mild increases of PRL (<150 ng/mL) may be a result of “stalk section effect” and are not diagnostic of adenoma (17,18).
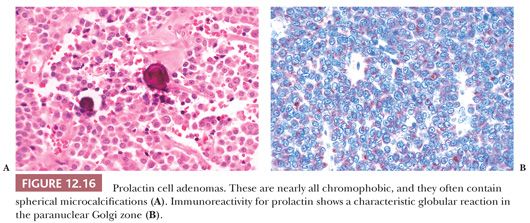
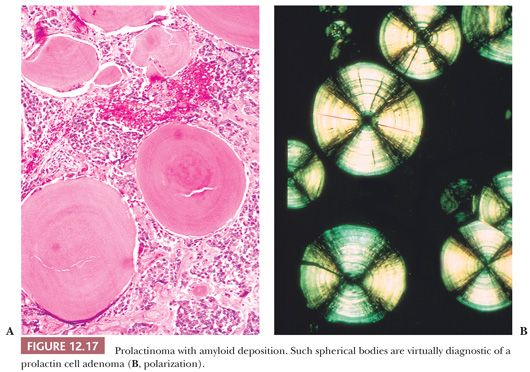
Nearly all prolactinomas are sparsely granulated and thus chromophobic (Fig. 12.16). Densely granulated (eosinophilic) examples are rare. Approximately 10% to 20% of prolactinomas feature psammomatous microcalcification (49). The latter is usually scant but may be so abundant that it forms a “pituitary stone.” The presence of microcalcifications in an adenoma strongly suggests the diagnosis of prolactinoma, as does the rare finding of spherical amyloid bodies (Fig. 12.17) (50). Immunoreactivity for PRL is typically strong but is paranuclear in location (Fig. 12.16). The ultrastructural features of PRL cell adenoma are distinctive (Table 12.4) and include abundant rough endoplasmic reticulum, as well as “misplaced exocytosis” or granule extrusion between neoplastic cells (37,38).
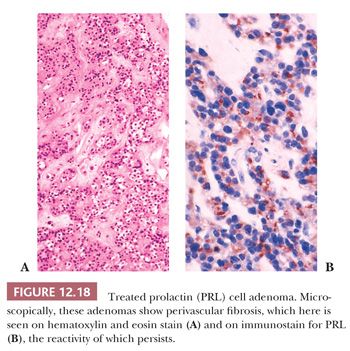
Given the efficacy of dopamine agonist therapy, the frequency of prolactinomas in surgical series has dramatically decreased from 30% to 10%. These agents produce atrophy of tumoral cells with resultant tumor shrinkage or arrest; the effect is reversible with cessation of therapy and thus not curative. The morphologic effects of such dopamine agonists as bromocriptine have been well characterized (51,52). They include diminution of cell size, condensation of nuclei, reduction in synthetic and secretory organelles, cessation of granule extrusion, and interstitial collagen deposition. Necrosis plays no significant part. Perhaps because of uneven D2 receptor loss, these changes may not affect all tumor cells. When administered long-term, they may induce dense tumoral fibrosis (Fig. 12.18).
Small tumor size and normalization of PRL levels after surgical or medical therapy of prolactinoma are favorable prognostic features. Increased age, male sex, large tumors, and invasion are negative predictors for surgical outcomes (48). Recurrence has been associated with invasion (48,53). The main differential diagnosis of prolactinoma is the acidophil stem cell adenoma (39) (see “Acidophil Stem Cell Adenoma” section). Key distinguishing features include its often large size in the presence of low PRL levels, minor immunoreactivity for both PRL and GH, and distinctive ultrastructural features.
GROWTH HORMONE–PRODUCING ADENOMAS
Growth Hormone Cell Adenoma
Two clinical disorders are associated with GH-producing adenomas: gigantism, which begins in childhood or adolescence, and acromegaly, a far more common disease affecting adults. These effects are largely mediated by insulin-like growth factor-1 (IGF-1) produced by the liver and more reliably elevated than GH levels.
Although GH cell adenomas are associated with clinical or immunohistologic evidence of GH production, only a minority of adenomas produces GH alone (54,55). The vast majority of adenomas produce both GH and PRL or are plurihormonal, also expressing TSH and/or α-subunit. Acromegaly can also be the result of somatotroph hyperplasia caused by ectopic production of growth hormone–releasing hormone (GHRH) by a variety of endocrine tumors and in the McCune-Albright syndrome (21,22). As previously commented, GH adenomas also occur in Carney complex and familial isolated pituitary adenoma (22).
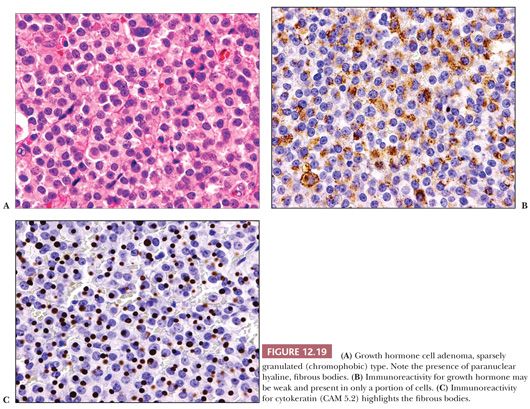
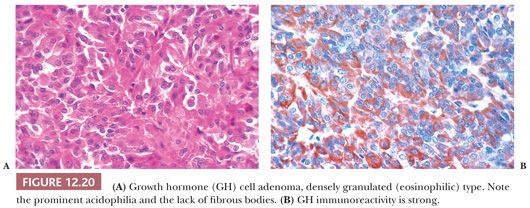
The pathology of GH-producing or somatotroph adenomas has been extensively studied (56–58). Two variants of GH cell adenomas are described: the densely granulated (eosinophilic) and the sparsely granulated (chromophobic) adenomas. Densely granulated adenomas are more frequent than sparsely granulated GH cell adenoma (59). The histologic patterns of GH cell adenomas are often nonspecific and diffuse, but nuclear pleomorphism and multinucleation are most often seen in sparsely granulated (chromophobic) tumors. The latter often show only scant GH immunoreactivity (Fig. 12.19). In contrast, densely granulated eosinophilic tumors stain strongly (Fig. 12.20). A characteristic of sparsely granulated adenomas is the presence of paranuclear eosinophilic, low–molecular-weight keratin containing “fibrous bodies” (Fig. 12.19C). At the ultrastructural level, these consist of intermediate filament whorls, often enmeshing organelles (Table 12.4). Sparsely granulated GH cells do not occur in the normal pituitary. In contrast, the cells of densely granulated tumors resemble normal GH cells, their eosinophilia being a result of abundant larger secretory granules. Such tumors lack fibrous bodies. Although the endocrinologic features and secretory activity of densely granulated and sparsely granulated variants of GH cell adenoma are similar, sparsely granulated tumors are more aggressive (59). These tumors are mostly macroadenomas, tend to arise in younger patients than densely granulated adenomas (44 vs. 50 years), and have higher invasive incidence (55,59). On rare occasion, GH adenomas are associated with gangliocytomas (60). Clinically nonfunctional or “silent GH cell adenomas” are rare (61).
Medical treatment of GH-producing adenomas involves the use of long-acting somatostatin analogs, such as octreotide. Morphologically, their effect varies but includes mild to moderate interstitial fibrosis (62). Tumor shrinkage is minor despite reduction in GH levels (62).
Mixed Growth Hormone Cell–Prolactin Cell Adenoma
As has already been noted, most GH-producing adenomas also secrete PRL. Indeed, nearly 40% of acromegalic patients have hyperprolactinemia (55). Because any number of lesions in the sellar region may be associated with hyperprolactinemia as a result of stalk section effect, a diagnosis of GH adenoma with a PRL-producing component cannot be made without immunohistochemical confirmation.
The predominant clinical feature of the mixed GH cell–PRL cell adenoma is acromegaly. The effects of concomitant GH and PRL elevation are not always apparent. Such tumors, which are variably acidophilic, are of interest because they are composed of two distinct, albeit related, cell types—somatotrophs and lactotrophs (Fig. 12.21) (37,38). They represent approximately 5% of pituitary adenomas. Both GH and PRL immunoreactivity are strong. Distinguishing them ultrastructurally from the acidophil stem cell adenoma is important but generally easy. The latter, an aggressive form of GH- and PRL-producing adenoma, shows much less hormone reactivity and masquerades as a prolactinoma, with GH elevation and acromegaly being infrequent (see the following section) (39).
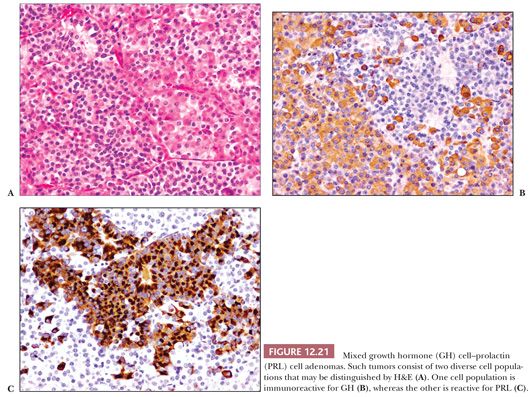
Mammosomatotroph Cell Adenoma
This unusual GH- and PRL-producing tumor represents only 1% of pituitary adenomas and is the most common tumor underlying gigantism (63). It is of interest because this well-differentiated, eosinophilic tumor is composed of a single cell type sharing ultrastructural features of both GH and PRL cells. Key features include densely granulated cells resembling somatotrophs but exhibiting both large (up to 1500 nm) granules and misplaced exocytosis, a feature of lactotrophs (37,38,64). Immunoreactivity for both hormones is strong. Again, making a distinction from acidophil stem cell adenoma is essential (Table 12.4).
Acidophil Stem Cell Adenoma
This unique form of pituitary adenoma is uncommon and comprises less than 1% of all adenomas and 5% of GH-producing tumors. Most patients present with hyperprolactinemia with minor symptomatology related to GH hypersecretion (39). The adenoma is composed of immature cells, perhaps stem cells, of the acidophil (GH and PRL cell) line. Small numbers of acidophil stem cells are found in the normal pituitary. On light microscopy, acidophil stem cell adenomas are chromophobic or somewhat oncocytic in appearance (Fig. 12.22). In general, acidophil stem cell adenoma shows greater immunoreactivity for PRL than for GH, which may be very focal or lacking. Scattered fibrous bodies, immunoreactive for cytokeratin, may be seen. Ultrastructurally, the tumor consists of a single, rather poorly differentiated cell type showing features intermediate between GH cells (fibrous bodies) and PRL cells (misplaced exocytosis) (Table 12.4) (39). Secretory granules are sparse and small. An often conspicuous feature is the presence of giant mitochondria, which may be apparent on light microscopy as cytoplasmic vacuoles, some approaching the size of the nucleus (Fig. 12.22). Electron microscopy is required to establish a firm diagnosis of acidophil stem cell adenoma (Table 12.4).
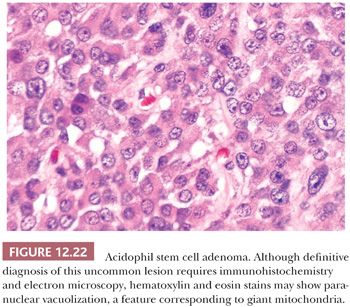
Acidophil stem cell adenomas are invasive macroadenomas (39). The diagnosis is of clinical importance because acidophil stem cell adenomas may be clinically mistaken for prolactinomas. Unlike the latter, they fail to respond to dopamine agonist therapy.
Plurihormonal Growth Hormone–Producing Adenomas
In this minority of acromegaly-associated adenomas, the tumor produces not only GH but also PRL and one or more glycoprotein hormones, usually TSH and/or α-subunit (63). Immunoreactivity for LH or FSH is exceptional (65). Hypersecretion of PRL may be clinically or biochemically apparent, but TSH secretion is rarely expressed, even at the biochemical level. Most plurihormonal tumors are macroadenomas. A majority shows radiographic or gross operative invasion. This group of tumors is of particular pathologic interest because they may consist of one, two, or three ultrastructurally distinct cell types, including GH, PRL, and/or TSH cells (Table 12.4).
Extrapituitary Disease and Growth Hormone Excess
This topic was briefly alluded to earlier. Acromegaly in association with an extrasellar neoplasm is usually a result of ectopic secretion of GHRH by a neuroendocrine neoplasm, such as bronchial carcinoid tumor, pancreatic endocrine tumor, or pheochromocytoma (66–69). In such cases, the pituitary is enlarged and shows GH cell hyperplasia and not an adenoma (67).
CORTICOTROPH CELL ADENOMAS
Adenomas that produce ACTH fall into two major groups: endocrinologically active tumors associated with either Cushing disease or Nelson syndrome and clinically nonfunctioning or silent corticotropic adenomas.
CUSHING DISEASE
Although Cushing disease may be rarely caused by corticotroph cell hyperplasia, the vast majority of cases are caused by an ACTH-producing adenoma. The incidence of the disease is 1 to 10 cases per million per year. Although the great majority of ACTH adenomas in the setting of Cushing disease occur sporadically, Cushing disease is rarely associated with an inherited condition including MEN1 and familial isolated pituitary adenomas (70).
ACTH-producing tumors represent nearly 15% of all adenomas. Affected patients vary greatly in age (peak incidence is 30 to 40 years), and there is a 5 to 10 times higher incidence of Cushing disease in women than men (18). The clinical diagnosis of Cushing disease may be challenging and relies on the combination of clinical, laboratory, and neuroimaging data, which are beyond the scope of this chapter.
Most ACTH cell tumors (87%) are microadenomas (mean size, 5 mm), but some measure no more than 1 to 2 mm. At surgery, only approximately 12% to 15% of ACTH cell adenomas are found to be invasive (32,71), a factor clearly underlying persistent disease and recurrence. Invasion has been directly linked to tumor size (72). Postoperative remission is achieved in 90% of microadenomas but in only 65% of macroadenomas. Thus, tumor size and invasiveness have great influence on surgical success (72).
Most functional ACTH adenomas arise in the midline of the pituitary, the so-called mucoid wedge. Occasional examples are posteriorly situated and must be distinguished from clinically insignificant foci of basophil invasion of the posterior pituitary. Of double adenomas in surgical specimens, the combination of an ACTH cell adenoma and a prolactinoma is best known (24,26).
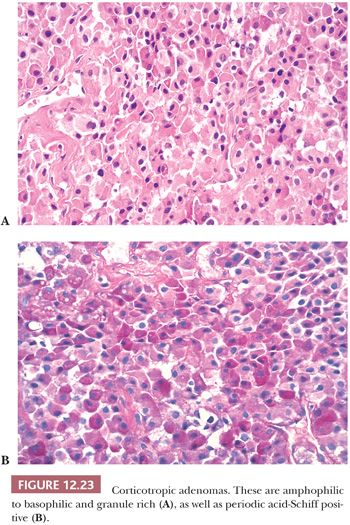
On light microscopy, ACTH adenomas are amphophilic or basophilic and often strongly PAS positive (Fig. 12.23). Immunohistochemistry shows not only the presence of ACTH but also the presence of other hormones derived from the proopiomelanocortin (POMC) precursor molecule, including β-lipotropin, endorphin, enkephalins, and melanocyte-stimulating hormone (MSH); however, these hormones are of little clinical value. ACTH adenomas express the transcription factor TPIT (Table 12.3). The ultrastructural features of ACTH adenomas are distinctive and include an abundance of secretory granules ranging in configuration from round to heart- or teardrop-shaped and with varying electron density, as well as perinuclear cytoplasmic intermediate filament bundles composed of keratin (Table 12.3). Their presence explains the variable cytokeratin immunoreactivity of corticotropic adenomas. Corticotroph adenomas may rarely be associated with gangliocytomas (60).
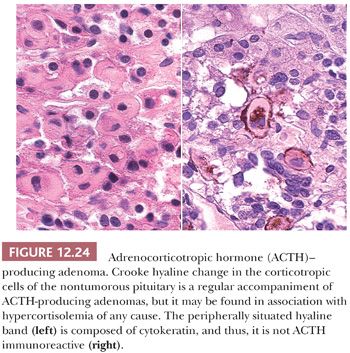
In Cushing disease, the extratumoral pituitary typically shows Crooke hyaline change, which is the result of massive perinuclear accumulation of cytokeratin filaments (Fig. 12.24). The change results from glucocorticoid feedback on the pituitary and presumably acts as a physical barrier to granule secretion. On occasion, ACTH cell adenomas per se show extensive Crooke change (73,74). These so-called Crooke cell adenomas are aggressive tumors with high rates of invasion (75).
Nelson Syndrome
The lesion underlying this disorder, now decreasing in frequency, has long been said to involve a small ACTH cell adenoma, one undetectable by radiographic means and thus prompting an adrenalectomy. Continued tumor growth is then furthered by lack of inhibitory feedback effects of glucocorticoids (Nelson syndrome). However, with the advances of diagnostic imaging techniques, tumors in this clinical scenario are now rarely seen. An additional mechanism involves adrenalectomy after incomplete resection of an invasive ACTH adenoma. In any case, the tumors of Nelson syndrome are often aggressive and most become or are invasive macroadenomas (71).
The adenomas of Nelson syndrome can be either amphophilic or basophilic and are variably PAS positive. Ultrastructurally, perinuclear cytoplasmic microfilament bundles are sparse or absent. Crooke hyaline change is lacking in extratumoral ACTH cells. Malignant transformation of Nelson adenomas accounts for a significant proportion of pituitary carcinomas (see “Pituitary Carcinoma” section later in this chapter) (76).
Silent Corticotroph Cell Adenomas
Some patients with ACTH immunoreactive tumors present without evidence of Cushing disease or hormone excess. These “silent” variants of corticotropic adenomas occur in two forms: either basophilic (densely granulated; subtype 1) or chromophobic (sparsely granulated; subtype 2) (37,38). Both tumors are unassociated with serum ACTH elevation or clinical signs of glucocorticoid excess. Some have suggested that silent corticotroph adenomas are incapable of hormone secretion, whereas others have found that they elaborate abnormal forms of high–molecular-weight ACTH, ACTH-related peptides (77), or endorphins (78). Silent corticotroph cell adenomas may arise from intermediate lobe corticotrophs, a cell population physiologically distinct from ACTH cells of the anterior lobe (11).
Histologically, silent corticotroph cell adenomas type 1 are in general composed of basophilic, PAS-positive cells; demonstrate strong ACTH immunoreactivity (Fig. 12.25); and contain abundant cytokeratin filaments. Type 2 tumors, however, are chromophobic or slightly basophilic tumors, resembling null cell adenomas, stain only weakly and focally for ACTH, and lack cytokeratin filaments. The ultrastructure of these two adenoma subtypes differs (Table 12.4). Type 1 tumors resemble regular Cushing adenomas, whereas type 2 tumors possess far fewer and smaller granules and lack intermediate filaments.
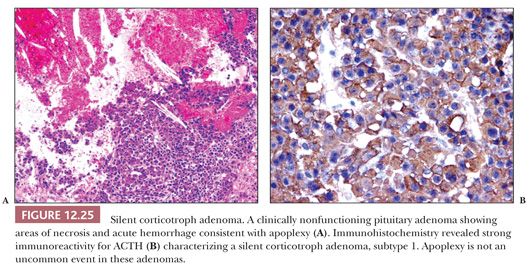
Because silent corticotroph adenomas are endocrinologically inactive, most grow to macroadenoma proportions and are invasive. In approximately 35% of cases, these tumors undergo spontaneous infarction (pituitary apoplexy) (79). The distinction of silent corticotroph cell adenoma from other nonfunctioning adenomas including gonadotroph and null cell adenomas is important due to the increased recurrence rate of these tumors in comparison with ordinary nonfunctioning adenomas (80). Only rarely these silent adenomas may become functional tumor with clinical symptoms of Cushing disease (81,82).
Extrapituitary Disease and Adrenocorticotropin Hormone Excess
In 10% to 25% of cases, Cushing syndrome is the result of ectopic ACTH production by a neuroendocrine neoplasm, most often a bronchial carcinoid tumor or a small cell carcinoma. In such instances, ACTH levels are markedly elevated, and the adrenal glands undergo massive cortical hyperplasia. The pituitary, which is normal in size, shows only Crooke hyaline change. Ectopic production of corticotropin-releasing hormone (CRH) by neuroendocrine tumors has also been reported. In such instances, the pituitary shows marked ACTH cell hyperplasia (83).
THYROTROPH CELL ADENOMA
Thyrotropic tumors are the least common of the pituitary adenomas. The majority of these adenomas occurs in adults and affects females (84,85). Patients may present clinically with mild hyperthyroidism, although patients with euthyroidism and slight hypothyroidism have also been reported (85). TSH-producing adenomas are seen in the setting of MEN1 and Carney complex, some as plurihormonal lesions (86,87). In the setting of primary hypothyroidism, combined TSH and PRL cell hyperplasia causes pituitary enlargement, mimicking adenoma (see the following texts) (88). Most TSH adenomas are invasive (75%) and aggressive (84). Thus, medical therapy and/or radiotherapy are often required.
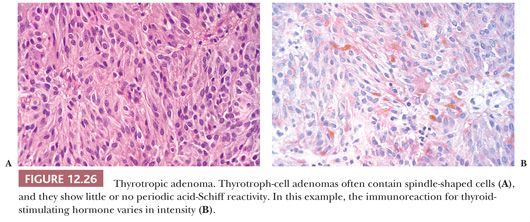
Most TSH adenomas are invasive macroadenomas and unusually sclerotic in texture. Histologically, the majority of these adenomas are chromophobic tumors composed of angulated or spindled cells. In addition to containing TSH (Fig. 12.26), most are also immunoreactive for α-subunit. Ultrastructurally, their cells resemble normal thyrotrophs, featuring processes containing microtubules, prominence of lysosomes, and sparse, minute secretory granules (Table 12.4).
GONADOTROPH CELL ADENOMA
Tumors producing βLH and βFSH are relatively common and represent the majority of nonfunctioning adenomas. Gonadotroph derived, they are well characterized in both morphologic (89) and clinical (90,91) terms. Most such adenomas occur in older adults, with men being preferentially affected. The great majority of these adenomas are clinically nonfunctioning tumors. Therefore, some investigators designate these adenomas as “silent gonadotroph adenomas” (92). In fact, gonadotroph adenoma cells produce only small amounts of intact gonadotropins or their individual βLH, βFSH, and α-subunits. Therefore, hormone level elevation may not be apparent. Hormonal excess is best seen in men because physiologic elevation of gonadotropins in women is normal after menopause (93).
Gonadotroph cell adenomas are large macroadenomas, often with suprasellar extension, and they cause pressure-related neurologic and visual symptoms (72%), as well as hypopituitarism (67%) (91). Radiographic or grossly apparent invasiveness is noted in only 20% to 30% of cases (71,91). Pituitary apoplexy is a recognized complication. Not surprisingly, occasional examples are incidental findings.
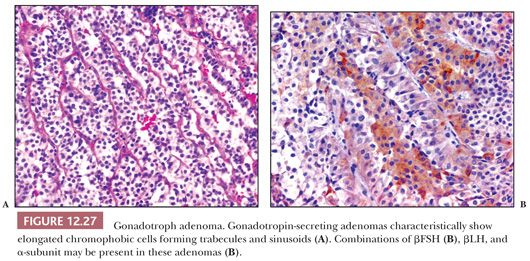
Unlike normal gonadotrophs, the adenoma cells are polygonal to elongate. Although a diffuse pattern of the tumor cells may be seen, sinusoidal, perivascular pseudorosettes or even papillae are prominent. The cells are mostly chromophobic or oncocytic (Fig. 12.27) and contain only scant, peripherally situated, PAS-positive granules. Immunostains show variable reactivity for βFSH and/or βLH and α-subunit. Most commonly, βFSH is stronger than βLH immunoreactivity. Chromogranin and synaptophysin are strongly reactive. Steroidogenic factor 1 (SF1), the transcription factor associated with gonadotrophs, is also positive (94). Ultrastructurally, gonadotroph cell adenomas feature polar cells with process formation and small numbers of minute secretory granules disposed beneath the plasmalemma, features not seen in normal gonadotroph cells (89). Gonadotroph cell adenomas in men also differ from those in women. The latter exhibits the characteristic “honeycomb transformation” of Golgi complexes (Table 12.4) (95). Because gonadotroph cell adenomas share many features of null cell adenoma, including frequent oncocytic change and scant LH/FSH immunoreactivity (89), their distinction may not be possible on immunostains alone.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


