The portion of the biopsy chosen for cryosections should optimally be about 0.5 cm in diameter and 1.0 cm in length. A variety of flash-freezing techniques have been described (14,15). One commonly used method is to orient the muscle for eventual cross-sectional histology (see Fig. 4.1), then immerse the biopsy in isopentane precooled to −160°C. All frozen tissue should be stored wrapped in foil inside an airtight container (e.g., polycon). Most laboratories use a −80°C freezer for specimen storage.
A standard histologic approach is to produce a hematoxylin and eosin (H&E) slide from each paraffin block and to cut serial frozen sections for H&E, Gomori trichrome, fiber typing by either adenosine triphosphatase (ATPase) or myosin heavy chain immunostaining, and nicotinamide adenine dinucleotide, reduced (NADH-TR). Cryosections for these standard stains are typically cut at a thickness of 10 μm. When needed, a wide range of histochemical, enzyme histochemical, immunoperoxidase, and immunofluorescence techniques can be applied to paraffin or frozen sections. Epon sections stained with toluidine blue with or without subsequent electron microscopy provide additional insight in selected cases. In a small subset of cases, ultrastructural features can be critical to reaching a specific diagnosis.
INTERPRETATION OF THE MUSCLE BIOPSY SPECIMEN
NORMAL MUSCLE
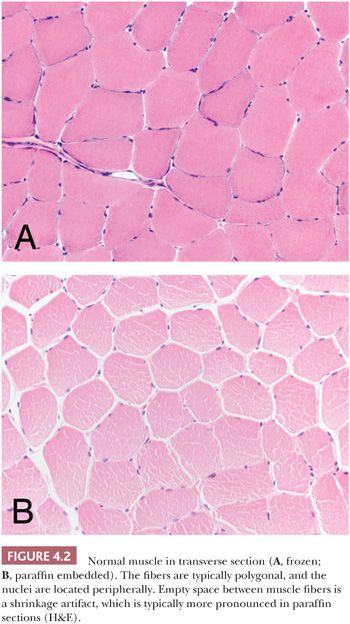
In the evaluation of the muscle biopsy specimen, familiarity with the normal structure is the basis for understanding the diversity of pathologic reactions that occur in neuromuscular disease. The reader is referred elsewhere for a more detailed discussion of the light microscopy, histochemistry, and electron microscopy of normal muscle (2–4). The myocyte is a multinucleated, syncytium-like cell with a shape resembling an elongated cylinder, although the normal adult fiber is not truly round but, instead, is polygonal or multifaceted in cross section (Fig. 4.2). The sarcolemmal nuclei are ordinarily located peripherally, often more than one per fiber in transverse sections. The diameter of the fibers depends on several factors. In general, powerful proximal muscles are made up of fibers with a mean diameter (50 to 70 μm) greater than that of smaller, distal, or ocular muscles (20 μm), which are devoted to finely coordinated activity. Fiber size is greater in males than in females, probably because of hormonal influences and the general propensity of males to engage in more strenuous physical activity. That exercise encourages fiber hypertrophy in both sexes is well established. Muscle fibers in children and the elderly are smaller than those in young, healthy adults.
In vertebrates, notably certain species of birds, one can distinguish between red (e.g., soleus) and white (e.g., pectoralis) muscles, with the color of red muscles being a consequence of their greater myoglobin content. Red muscle, with its larger mitochondrial population and higher capillary density, is adapted to aerobic respiration and is designed for postural or sustained activity. White muscle, which contains fewer mitochondria and abundant glycogen, is capable of anaerobic respiration; it is more suited to sudden and intermittent action. Although an entire muscle in lower animals may be composed of either red or white fibers, human muscle is constructed of both fiber types, which are arranged in a mixed mosaic pattern resembling a checkerboard. The location of the muscle in the body and its function determine the proportion of type 1 and type 2 fibers, but the average muscle has about twice as many type 2 fibers (60% to 65%) as type 1 fibers (35% to 40%) (16).
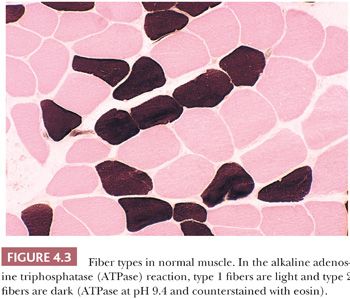
Fiber typing, the demonstration of the histochemical properties of muscle fibers within a biopsy specimen, is accomplished by carrying out enzyme histochemistry in frozen sections (Table 4.1). Fiber types are not evident in slides stained with H&E. The traditional approach is to perform myofibrillar ATPase reactions at acidic and alkaline pH. In the standard or alkaline ATPase reaction conducted at a pH of 9.4, type 1 fibers appear light (they can be seen better in sections counterstained with eosin) and type 2 fibers appear dark (Fig. 4.3). Fibers with intermediate staining properties are not seen in the alkaline reaction.
When the pH of the incubating solution is reduced to the acidic range (pH 4.2) in the so-called reverse ATPase reaction, type 1 fibers are very dark and type 2 fibers are very light. At a slightly less acidic pH 4.6, type 2A fibers are very light and the staining intensity of type 2B fibers is intermediate. Commercial antibodies to slow (type 1) and fast (type 2) myosin are now available. By means of automated immunoperoxidase staining, fiber typing with results similar to those obtained with ATPase reactions can be achieved in either cryosections or paraffin sections (3,17).

The oxidative stains, such as the NADH-TR reaction or specific mitochondrial enzyme histochemistry (e.g., succinic dehydrogenase or cytochrome C oxidase), reflect the concentration of mitochondria within the myofibers. The fiber staining is essentially the opposite of that seen in the alkaline ATPase reaction. Thus, intensely stained fibers are designated as type 1 (oxidative), and lighter fibers are designated as type 2. Typically, oxidative enzyme reactions further divide type 2 fibers into two subpopulations. Type 2B fibers are very lightly stained, whereas the staining intensity of type 2A fibers is intermediate between that of type 1 and that of 2B. The myofibril network is stained such that the sarcoplasm has a regular, fine granular appearance.
All muscle fibers contain phosphorylase as well as glycogen; these are more abundant in type 2 (glycolytic) fibers. Some laboratories take advantage of the histochemical reaction for phosphorylase, or the periodic acid-Schiff (PAS) stain, as a means of fiber typing, but our experience dictates that such staining is unpredictable, especially because much of the glycogen leaches out of the muscle sections during processing. We restrict use of the phosphorylase reaction to cases of possible enzyme deficiency (McArdle disease). Lipid vacuoles are more numerous in the sarcoplasm of type 1 fibers, yet fat stains such as oil red O seldom demonstrate an unequivocal difference between fiber types. Fat stains are more valuable when the diagnosis of lipid storage myopathy is suspected.
ARTIFACTS
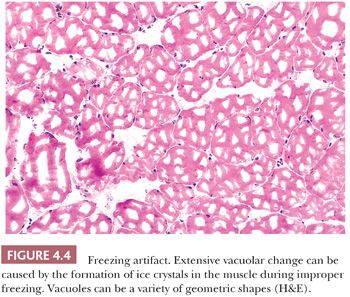
Among the most unavoidable and disturbing artifacts is the vacuolation produced by ice crystals that form during improper freezing of the muscle biopsy specimen (Fig. 4.4). It may also occur after improper transport or storage that allows thawing and refreezing of the sample. This type of artifact may simulate pathologic change, such as vacuolar myopathy, or it may distort pathologically altered fibers, thereby precluding proper interpretation. In contrast to pathologic vacuoles, the vacuoles associated with ice crystals are in the more slowly frozen or earlier thawing portion of the specimen, which is generally in the center or at the periphery of the tissue block, respectively. Vacuoles tend to affect every fiber in the region of artifact and may be arranged in a gradient according to size. Where the artifact is minimal, small vacuoles are numerous in each fiber, uniform in size, and arranged in a waffle pattern; they are located between the myofibrils. In poorly frozen regions, larger clear vacuoles are present. Muscle transported to the laboratory in saline can acquire artifactual separation of muscle fibers during freezing due to the expansion of water that accumulated in the interstitium.
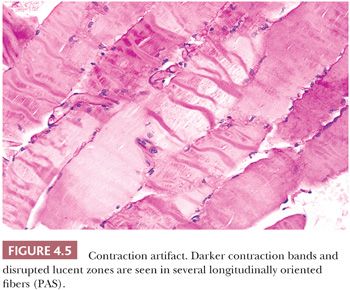
With severe contraction artifact in longitudinal sections, dark contraction bands alternate with lucent, disrupted zones within the fiber (Fig. 4.5). The latter appear as jagged cracks in the sarcoplasm in transversely oriented sections. Contraction artifact is most noticeable at the edges of the specimen. This artifact is commonly seen in unclamped specimens, when plastic disposable clamps that only partially prevent contraction are used or when the muscle is injected with local anesthetic. Contraction artifact renders muscle particularly unfit for electron microscopy. The orderly structural landmarks are obliterated by myofibrillar fragmentation and disorientation.
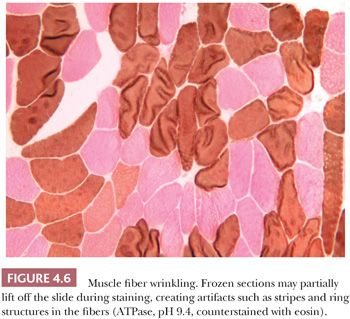
A frequent artifact occurs in frozen sections when the section lifts partially off the slide. The staining intensity of the fibers varies, and a variety of striped or ring structures result from curling or wrinkling of the fibers (Fig. 4.6).
GENERAL PATHOLOGIC REACTIONS OF MUSCLE
When evaluating muscle biopsies, the surgical pathologist is likely to come across one or more of the features described in the next few paragraphs. Very few of the pathologic changes that may be observed in the muscle biopsy specimen are totally specific for a single disease, although each has diagnostic significance because it is restricted to one disease or a limited number of diseases. The presence of each pathologic change described in the following sections should engender a differential diagnosis in the mind of the observer. Arrival at a precise diagnosis involves correlation not only with the clinical information and laboratory test results but also integrating the various pathologic findings to arrive at a pattern or signature of the disease process. All of the pathologic changes discussed here are light microscopic features, and only some are appreciated in greater detail ultrastructurally. Where appropriate, a description or illustration of the fine structure accompanies the description of the light microscopic features.
Variations in Muscle Fiber Size: Smallness, Atrophy, and Hypertrophy
Undoubtedly, the most common abnormality encountered in muscle biopsy pathology is variation in fiber size. Diameters range widely with the age, sex, and physical activity of the patient. At birth, muscle fiber diameters average 10 to 15 μm, whereas in adults, the normal range is from 40 to 80 μm (3). One of the most demanding challenges for the surgical pathologist is interpretation of hypertrophy and abnormal smallness due to developmental abnormalities or atrophy when abnormal fiber size is the predominant pathologic finding. The usefulness of histochemistry can be considerable in this situation. Because the muscle fiber depends on neural and other influences for survival, the disruption of these influences results in the atrophy of the fiber. The most common form of atrophy in neuromuscular disease is that caused by denervation. In addition, the maintenance of muscle fiber integrity requires regular activity. Disuse, as may occur with prolonged bed rest, can lead to muscle atrophy. Finally, a reduction in fiber volume may be a complication of aging, ischemia, or poor nutrition.
Hypertrophy, by comparison, is principally a consequence of an increase in muscular work, either during exercise or as a compensatory reaction of normal, intact fibers to the atrophy of others in their midst. In an evaluation of fiber size, the measurement of cell diameters may be fruitful. A quick evaluation of the range of diameters can be performed using the pointer arrow after measuring the arrow at various magnifications with a glass slide or eyepiece micrometer. Morphometric analysis of the muscle specimen may be indicated in the event that changes in fiber diameters are minimal and subtle. Morphometry can be performed manually with an eyepiece micrometer or electronically with a computer-assisted image analyzer (18). To obtain statistically significant morphometric data, the lesser diameter of each muscle fiber should be measured, and the minimum number of fibers in the sample should be 200. Histograms of each fiber type can be generated from ATPase- or myosin heavy chain immunoperoxidase-stained sections.
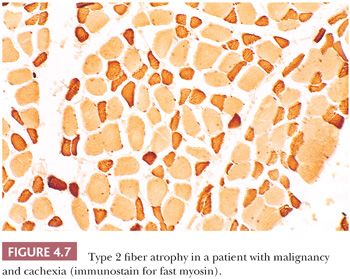
The atrophic or hypertrophic process may be selective, involving only one fiber type, or nonselective (19). Selective atrophy of type 1 fibers is seen most commonly in myotonic dystrophy but is also seen in distal myopathy, nemaline myopathy, centronuclear myopathy, and congenital fiber–type disproportion (Table 4.2). Type 2 fiber atrophy (Fig. 4.7) is observed in myasthenia gravis, acute denervation, disuse, and systemic malignancy (20). In our experience, more than half of such cases are attributable to corticosteroid therapy (Table 4.3). Hypertrophy restricted to type 1 fibers is relatively specific for infantile spinal muscular atrophy (SMAI), although such hypertrophy may develop in normal athletes who undergo endurance training. Type 2 fiber hypertrophy has been reported in runners, notably sprinters, and in congenital fiber–type disproportion. Non-fiber type–selective alterations in fiber size are somewhat uninformative from a diagnostic standpoint. Denervation accounts for a large proportion of cases of nonselective atrophy but certainly not all of them. Hypertrophy involving both major fiber types is frequently noted in muscular dystrophy, inclusion body myositis, myotonia congenita, and acromegaly.


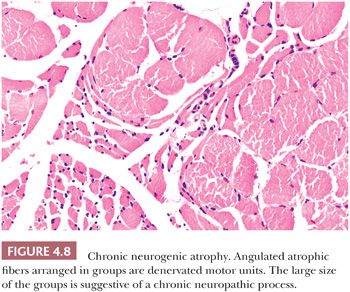
The pattern of atrophy may be diagnostic in some atrophic diseases. Grouped atrophy, which is recognized as a clustering of five or more small angular fibers, is essentially pathognomonic for chronic neurogenic disorders (Fig. 4.8). Panfascicular atrophy, an extreme version of grouped atrophy wherein the vast majority of fibers in each fascicle are severely atrophic, is a distinctive feature of SMAI. Perifascicular atrophy typifies dermatomyositis, in which fiber atrophy is limited mainly to the periphery of the fascicles. Unfortunately, in many muscle biopsy specimens, atrophic fibers are randomly situated in the section. This pattern of atrophy is totally nonspecific.
Nuclei
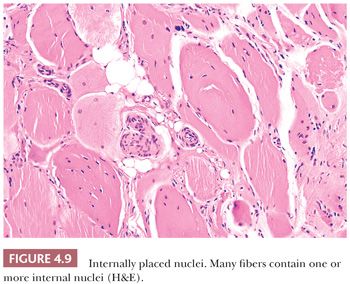
One of the more frequent and easily recognized abnormalities observed in muscle biopsies is the internalization of muscle fiber nuclei. According to quantitative studies, the nuclei in cross sections of normal muscle are peripheral (subsarcolemmal) in 97% to 99% of fibers. In specimens from patients with neuromuscular disease, the counts of internal nuclei are commonly elevated to 5% to 10% of fibers. Although the sarcoplasmic integrity appears undisturbed, such fibers are often mildly or moderately altered in size (Fig. 4.9). This inconstancy of nuclear position is of no specific diagnostic import, and it is a reaction to a variety of diverse injuries. However, if most of the fibers contain randomly distributed internal nuclei, the diagnosis is more likely a myopathic condition (Table 4.4). The virtually pathognomonic criterion of centronuclear myopathy is the presence of a single central or paracentral nucleus within most muscle fibers. As opposed to nuclear internalization in the absence of injury to the sarcoplasm, after myonecrosis, the nuclei may no longer be subsarcolemmal. They commonly migrate internally in degenerating and regenerating fibers, no matter what the cause. Severely atrophic fibers also contain multiple nuclei that seem to remain intact, forming pyknotic nuclear clusters as the sarcoplasm is progressively diminished in volume. Most often seen in neuropathic disorders or end-stage muscle, these nuclear clusters can be mistaken for lymphocytic inflammation.

Fiber Splitting
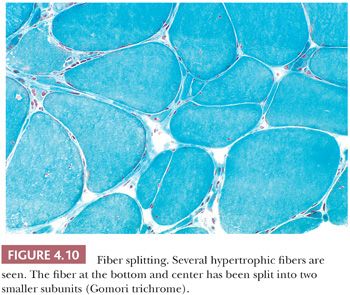
Muscle fibers that have become hypertrophic with multiple internalized nuclei commonly split into smaller subunits of two or more smaller fibers that appear to be mature myocytes with intact sarcoplasm. Before splitting is concluded, the fiber assumes a segmented appearance as slitlike spaces form invaginations between individual segments (Fig. 4.10). Within each space, the extensions of the plasma membrane remain continuous around the dividing portions of the cell. Fiber splitting is typically more conspicuous in chronic necrotizing myopathies (e.g., muscular dystrophies or inflammatory myopathies) (21). Internalization of capillaries sometimes accompanies the fiber splitting process (see panel A of Fig. 4.36).
Fiber Shape
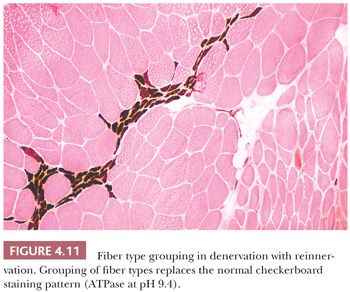
Assessing fiber shape is typically more accurate in frozen sections than in paraffin sections. In contrast to the normal polygonal contour of the myofiber viewed in the transverse plane, rounded fibers favor a myopathic process, particularly muscular dystrophy or congenital myopathy. In neuropathic diseases (excluding SMAI where small fibers are rounded), the atrophic fibers are angular (or ensiform) and grouped (Fig. 4.11). These fibers appear flattened and narrow with tapered or pointed ends. Grouping is the result of simultaneous atrophy of entire motor units. Angulated atrophic fibers are also found in myasthenia gravis, disuse, myotonic dystrophy, and steroid myopathy.
Inflammation
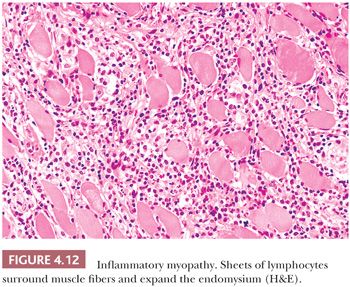
Interstitial inflammatory infiltrates are most frequently encountered in immunologically mediated or idiopathic inflammatory myopathies (Table 4.5). Most important among these are polymyositis (PM), dermatomyositis (DM), and inclusion body myositis, which make up nearly a third of all cases evaluated at the University of Buffalo. The inflammatory cells in PM invade the endomysium, sometimes enveloping necrotic fibers. Sheets of inflammatory cells expand the endomysial spaces in acute, more severe disease (Fig. 4.12). In DM, they surround intramuscular blood vessels with minimal infiltration of the vascular walls. The inflammatory cells are mononuclear in type, chiefly consisting of mature lymphocytes. Plasma cells are a minor part of the inflammatory response. Rare eosinophils are seen, and neutrophils are absent.
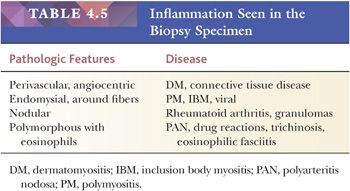
An inflammatory myopathy histologically identical to PM and DM may accompany any of the systemic connective tissue diseases. However, crucial differences among several of these collagen vascular disorders are diagnostically reliable. Nodular infiltrates composed largely of plasma cells are highly suggestive of rheumatoid arthritis. Polyarteritis nodosa and systemic lupus erythematosus, on the other hand, are typically associated with a vasculitis. Polyarteritis nodosa shows an affinity for larger vessels, especially arteries, within the epimysium and perimysium. The entire wall of the vessel may be infiltrated by inflammatory cells, which usually include conspicuous eosinophils. In systemic lupus erythematosus, the affected vessels are smaller in caliber, and they exhibit fibrinoid necrosis. Areas of vascular injury attract neutrophils, and the remnant of their breakdown, nuclear dust, is also seen. With the use of immunohistochemical techniques, deposits of immunoglobulin and complement can be demonstrated at sites of vascular injury in both conditions.
Granulomatous inflammation may be indicative of sarcoidosis or idiopathic granulomatous myositis, a chronic progressive myopathy of middle-aged women without systemic manifestations (22,23). The granulomas in both are sharply circumscribed and nonnecrotizing. They invade and expand the interstitium, and they are composed of lymphocytes, epithelioid histiocytes, and multinucleated giant cells. Trichinella infestation may elicit a granulomatous response in muscle that usually contains numerous eosinophils. Other microbial agents, including bacteria, fungi, and viruses, may cause inflammatory myopathy, but a muscle biopsy is not routinely a part of the diagnostic investigation of these infections.
Fiber Necrosis (Myonecrosis) and Regeneration
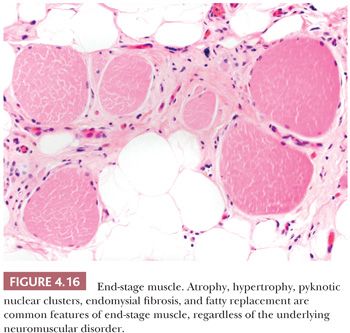
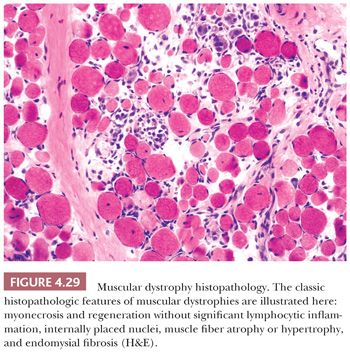
The initial sign of necrosis in light microscopic sections is an alteration in the tinctorial properties of the muscle fiber that is perhaps best appreciated in H&E stains. The acutely necrotic fiber first stains more intensely eosinophilic and then pales to a wan shade of pink. Simultaneously, there is a loss of striations in the sarcoplasm. The myocyte nuclei may become fragmented and eventually are no longer visible. As the process of myonecrosis proceeds, the sarcoplasm is evacuated by macrophages that eventually occupy the entire space within the original myocyte basement membrane (Fig. 4.13; also see Fig. 4.29 later in this chapter). Often, even before the total removal of the necrotic sarcoplasmic debris, the phase of regeneration has supervened; therefore, both regenerative and phagocytic activity may be seen in the same fiber. Necrosis may be a part of the pathologic response in many muscle diseases, but it is prevalent in muscular dystrophies (especially Duchenne and related limb-girdle muscular dystrophies) and the inflammatory myopathies (Table 4.6).
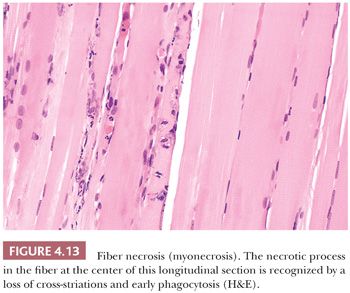

Hyaline fibers are pathologically rounded and enlarged. They are more deeply stained than normal fibers, whether in paraffin or frozen sections stained with H&E, trichrome, PAS, or histochemical methods (Fig. 4.14). The sarcoplasm is smudged and homogeneous. The hyaline fiber is a bona fide pathologic change in tissue unaffected by contraction artifact, and in many instances, it represents an early stage of cell necrosis (24). Serial sections through the same fiber may reveal zones of unequivocal necrosis and phagocytosis adjacent to hyalinization. Hyaline fibers are most commonly encountered in Duchenne muscular dystrophy, and they are most numerous in this condition.
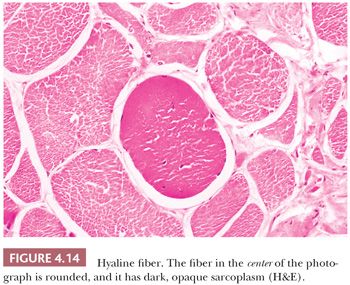
Fiber necrosis (myonecrosis) generally acts as a stimulus for subsequent regeneration. Hence, the presence of regenerating fibers in a biopsy specimen, even in the absence of necrotic fibers, is a likely indicator of previous necrosis. The regeneration of fibers is believed to arise primarily from the proliferation of satellite cells and generation and fusion of myoblasts (25). Regenerating fibers are most readily visualized in H&E sections by the basophilia of their sarcoplasm. The nuclei are typically increased in number, are larger than normal, have vesicular chromatin and prominent nucleoli, and often are internally placed (Fig. 4.15; also see Fig. 4.29). Ultrastructurally, the regenerating fibers are replete with ribosomes, which explains the sarcoplasmic basophilia at the light microscopic level (26).
Fibrosis and Fatty Infiltration
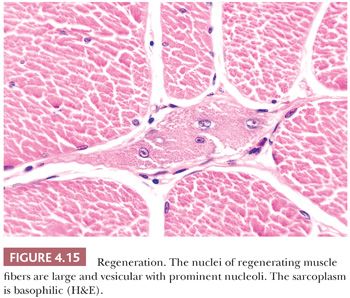
Endomysial fibrosis is a common pathologic component of chronic necrotizing myopathies such as muscular dystrophies and myositis. The fibrosis is simply a manifestation of inflammation and repair as can be seen in most tissues. However, the factors that provoke end-stage interstitial fibrosis and fatty replacement of muscle have not been adequately explained by research investigations even though they are the bequest of chronic neuromuscular disease of both myopathic and neurogenic origin. Even the most seasoned pathologist experiences difficulty in interpreting the biopsy specimen with marked fibrosis or fatty infiltration because, at this juncture in the natural history of the disease, the active pathologic process has probably subsided and the opportunity of discovering specific pathologic changes is irretrievably lost (Fig. 4.16). Because end-stage muscle is unlikely to provide information relevant to the patient’s diagnosis, the biopsy of a severely involved muscle should be discouraged.
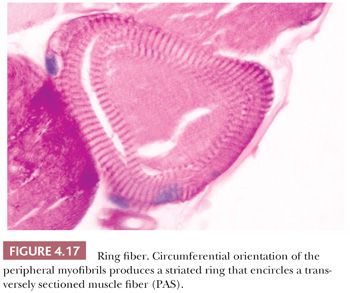
Ring Fibers
The ring is formed by a peripheral bundle of myofibrils that is directed circumferentially, encircling the inner portion of the myofiber, which otherwise appears normal in disposition and structure. In cross sections of muscle, the striated annulation is oriented in the transverse plane rather than in the longitudinal axis of the fiber (Fig. 4.17). The striations are easily visible in phosphotungstic acid hematoxylin (PTAH) or PAS-stained sections, resin sections, and ultrastructurally; with electron microscopy, the abnormally oriented myofibrils usually exhibit a normal architecture except for the hypercontraction of the sarcomeres (27). Although ring fibers have been reported in a variety of diseases, they are most consistently observed in limb-girdle dystrophy and myotonic dystrophy. Large numbers of ring fibers especially favor the latter diagnosis.
Inclusion Bodies
Inclusions may be located within the nuclei or within the sarcoplasm. Intranuclear inclusions suggest the diagnosis of oculopharyngeal dystrophy or inclusion body myositis. Sarcoplasmic inclusions suggest the diagnosis of myofibrillar myopathy or inclusion body myositis. In oculopharyngeal dystrophy, intranuclear inclusions are difficult to detect by light microscopy. Electron microscopic examination of the muscle tissue is likely to reveal nuclear inclusions composed of 8.5-nm unbranched tubular structures.
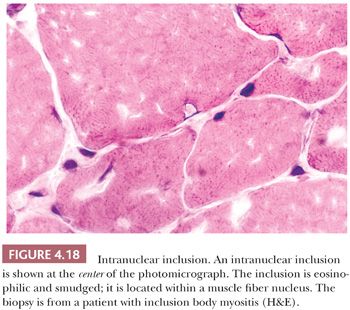
Nuclear inclusions in inclusion body myositis are faintly pink to wine colored in H&E stains, and they often fill the nucleus, leaving a rim of peripheral chromatin (Fig. 4.18). Pink or red inclusions may also be seen associated with the sarcoplasmic rimmed vacuoles (see Fig. 4.28). By electron microscopy, these inclusions, which consist of membranous whorls and bundles of filaments measuring 15 to 18 nm in diameter, may be present in few fibers, and they are extremely difficult to identify. The filaments are considered to be β-amyloid fibrils (see “Inclusion Body Myositis” section). Because the filaments are sparse, Congo red stains tend to be negative despite contrary claims in the literature.
In the myofibrillar myopathies, some congenital myopathies, and miscellaneous other neuromuscular disorders, the inclusion bodies represent protein aggregates within the cytoplasm of affected muscle fibers. They may contain intermediate filaments such as desmin, which form poorly defined, smudged areas within the fibers. In hyaline body myopathy, the inclusions, which contain myosin filaments, are subsarcolemmal, well-defined, and slightly different in texture from the surrounding sarcoplasm. The protein composition of nonspecific cytoplasmic inclusion bodies has not been defined.
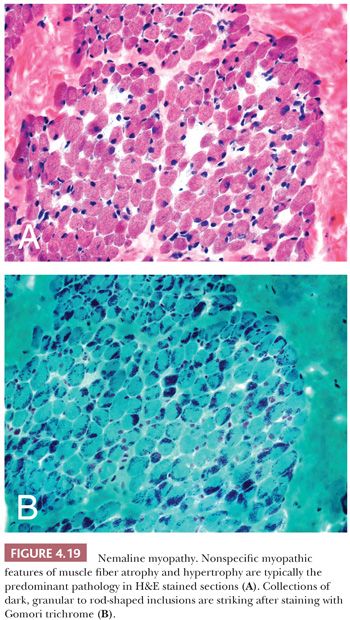
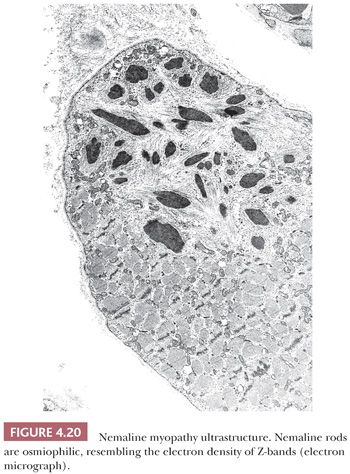
In contrast, nemaline rods are composed primarily of proteins normally found in Z-bands. Rods were first recognized in nemaline myopathy, a congenital and nonprogressive muscle disease of childhood (28). The name nemaline is derived from the Greek root nema (“thread” or “threadlike”) to emphasize the pathologic marker of this disorder. The threads or rods tend to cluster beneath the sarcolemma. Rods easily escape detection in H&E sections but are often readily apparent with Gomori trichrome stains of frozen sections (Fig. 4.19) or in toluidine blue–stained resin sections. Ultrastructurally, the rods are osmiophilic oblong or rectangular structures of varying dimensions, typically less than 5 μm (Fig. 4.20). Their latticelike appearance resembles that of the normal Z-band (29), likely a consequence of a protein composition that is similar to Z-bands. Since the original description of nemaline myopathy, it has become increasingly evident that rods are not unique to a single disease entity. Occasional rods in a small number of fibers have been reported in muscle specimens of muscular dystrophy and PM, for example. One cannot be comfortable with predicting the diagnosis of nemaline myopathy unless, in the appropriate clinical setting, many fibers in the specimen contain rods and they are numerous within each fiber.
Mottled Fibers and Lobulated Fibers
The peculiar, uneven staining reaction of mottled or moth-eaten fibers is satisfactorily demonstrated only in oxidative enzyme preparations. Zones of weak enzyme activity with irregular and poorly delimited borders are randomly dispersed in the sarcoplasm (Fig. 4.21). Ultrastructurally, mottled areas reveal a lack of mitochondria and the destruction of the myofilaments. The fact that the ultrastructural integrity of much of the cell sarcoplasm between the zones of mottling is preserved upholds the notion that the mottled fiber has the capacity for recovery and that it represents a form of reversible injury. Moth-eaten change is a nonspecific abnormality.
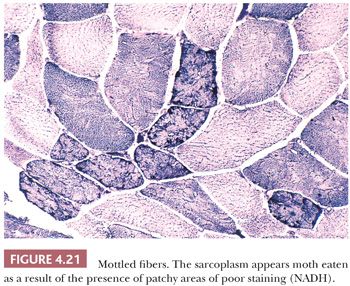
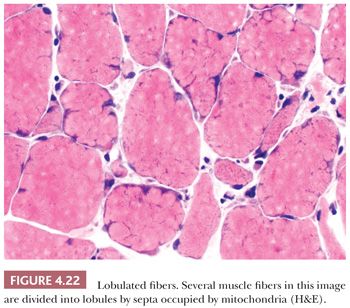
Lobulated fibers have a superficial resemblance to mottled fibers (see preceding paragraph) and fibers containing minicores (see next paragraph). Unlike moth-eaten changes and cores, lobulations are easily recognized on H&E and Gomori trichrome (Fig. 4.22) as well as oxidative stains. The peripheral cytoplasm of muscle fibers is divided by thin triangular clusters of mitochondria that extend a short distance centrally from the sarcolemma. Although generally considered to be a feature of myopathic disease, lobulated fibers do not have disease specificity.
Cores and Targets
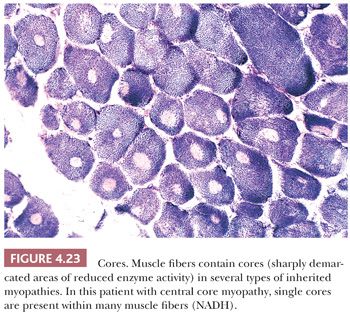
Oxidative enzyme reactions are the ideal technique for identifying cores that appear as sharply demarcated regions of depleted or absent mitochondria (Fig. 4.23). Large cores are often also evident with H&E and Gomori trichrome stains. Ultrastructurally and in resin sections, distinguishing between structured and unstructured cores is possible (30). The cross-banding pattern is evident in the structured core, whereas cross-striations are absent from the unstructured core, which perhaps represents a later stage in core development. Cores cannot be regarded as a specific pathologic finding because they occur in a variety of diseases. However, the muscle fibers of several inherited myopathies contain cores, and one nosologic group, the core myopathies, is defined by them. As a nonspecific change, cores are present in less than 10% of fibers, whereas they are numerous and are located more often in type 1 fibers in central core disease. Although cores are single and centrally placed within the fiber in classic central core disease, they may be multiple and eccentric in other conditions.
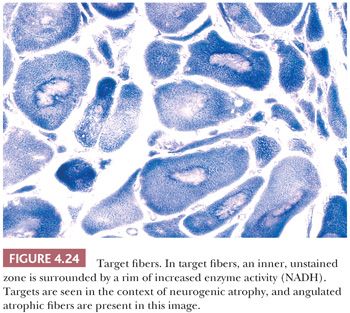
For diagnostic purposes, target fibers are considered pathognomonic for neurogenic atrophy (31), but unfortunately, they are identified in less than 25% of cases. Despite many similarities, targets and cores can often be distinguished. Of greatest significance is the three-zone structure of the target. The central zone, which resembles the unstructured core, is surrounded by an intermediate zone, which is darkly stained in oxidative enzyme reactions (Fig. 4.24). This rim, which is not a feature of cores, sharply contrasts with the third zone, the outer normal portion of the fiber. The term targetoid refers to target-like, sharply demarcated regions that lack the intermediate zone or rim of increased oxidative enzyme activity. Targetoid change and cores essentially are morphologically identical, but the term targetoid is typically used in neuropathic disease and cores in myopathic disease.
Mitochondrial Abnormalities
Abnormalities of mitochondria occur in a wide variety of diseases (32–34). They may be generalized disorders, or they may be limited to skeletal muscle. The mitochondrial abnormalities are similar in all of these diseases, and they are often recognized by the presence of ragged red fibers. Classic ragged red fibers are readily identified by the Gomori trichrome, in which intensely red, subsarcolemmal protrusions from the cell surface give the margins of involved fibers an irregular, ragged appearance (Fig. 4.25). Such fibers are surrounded by prominent, sometimes dilated capillaries that appear to indent them and to be increased in number. Ragged red fibers are equally impressive in oxidative enzyme reactions, especially succinate dehydrogenase reactions, in which large collections of mitochondria are seen as dark, coarsely granular deposits that not only may be subsarcolemmal but may also diffuse throughout the fiber.
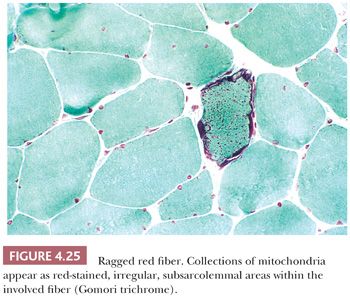
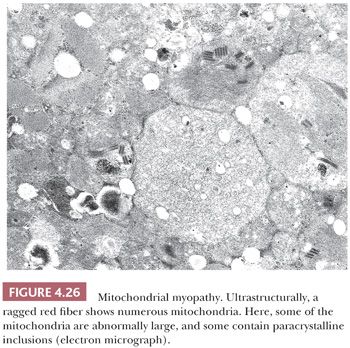
Electron microscopy shows that ragged red fibers contain accumulations of mitochondria that may be enlarged and abnormal in shape. Their cristae may be excessively numerous and disorganized, or they may be concentrically arranged. Among the commonly encountered matrix inclusions are glycogen aggregates, clear vacuoles, floccular densities, myelin figures, and paracrystalline structures having a square or rectangular conformation and resembling a parking lot or grid (Fig. 4.26). These inclusions contain mitochondrial creatine kinase and perhaps other proteins.
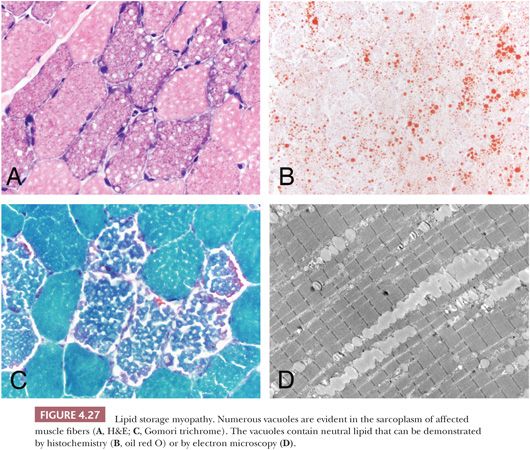
Vacuolar Change
Vacuoles may contain abnormal quantities of glycogen or lipid, or they may be composed of clustered lysosomes/autophagosomes (Table 4.7). Hence, routine microscopic sections with vacuolar change of an unexplained nature should be stained with PAS and oil red O or other suitable fat stains. We prefer to use resin sections, in which the osmiophilic fat deposits are more clearly demonstrated than they are in routine fat stains (Fig. 4.27). An abundance of lipid within myofibers is a relatively specific finding that is usually indicative of a lipid storage disease or mitochondrial myopathy, with the latter frequently being recognized by the presence of ragged red fibers. Although deposits of glycogen raise the specter of carbohydrate storage disease or other disorders affecting glycogen metabolism, such as hypothyroidism (35), they can be an incidental, fortuitous finding in virtually any disorder. Especially when only one or two fibers in the specimen are involved and the vacuoles are crescentlike, subsarcolemmal, and visible primarily in PAS stains, they are probably of no clinical significance.

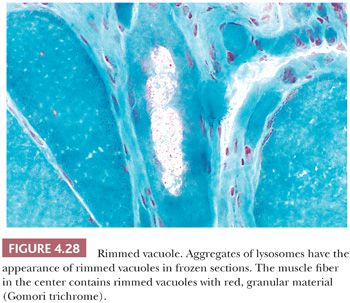
The so-called rimmed vacuole has received attention as a diagnostic criterion of oculopharyngeal dystrophy (36), distal myopathy (37), and inclusion body myositis (38). These vacuoles may be sharply demarcated and may contain granular material (Fig. 4.28). Granular material typically rims the vacuoles, appearing basophilic in H&E stains and red in Gomori trichrome stains of frozen sections. Ultrastructurally, this type of vacuole is not always enclosed by a membrane, and it contains lysosomal membranous profiles with or without tubulofilamentous material. The rimmed vacuole is believed to be derived from the autophagic vacuole, which classically is membrane bound and is a repository for the debris of autodigestion. In at least two genetic diseases, autophagic vacuoles are numerous (39,40). These vacuoles are rimmed by membrane that expresses proteins typically found in the sarcolemma. Enzyme histochemistry or immunostaining for these sarcolemmal proteins demonstrate strikingly numerous cytoplasmic vacuoles in Danon disease and X-linked myopathy with excessive autophagy (XMEA) (see Fig. 4.36).
Changes in Fiber Type Distribution
The normal checkerboard staining paradigm that is so familiar in histoenzymatic reactions may fall victim to certain pathologic conditions. Predominance of one fiber population is emblematic of the congenital myopathies. Type 1 fiber predominance is found in central core disease, nemaline myopathy, centronuclear myopathy, and some cases of congenital fiber–type disproportion. In chronic denervation, the normal checkerboard pattern is replaced by large groups of fibers with identical histochemical properties. This fiber type grouping (Fig. 4.11) is a result of reinnervation of the denervated muscle. Normal anatomic and physiologic principles help to explain type grouping. The muscle fibers in each motor unit are of a single histochemical type. The neuron, through its axon and intramuscular branches (nerve twigs), which innervate the individual fibers, governs the fiber type in the motor unit. Reinnervation of a type 1 fiber by a type 2 nerve twig has been shown experimentally to convert the fiber from type 1 to type 2. The opposite occurs with a type 2 fiber and a type 1 twig. Thus, when neighboring denervated fibers of differing histochemical types undergo reinnervation by collateral sprouts from a single axon that has remained viable, all of the fibers are converted to one histochemical type, and fiber type grouping results.
NEUROMUSCULAR DISEASES
The muscle biopsy is only one facet of the overall diagnostic evaluation of the patient. It must not be interpreted in a vacuum without a consideration of the clinical history, physical examination findings, and the results of all pertinent laboratory tests. This section provides the background needed to interpret the biopsy findings in the context of the clinical setting. Those neuromuscular diseases that are most often encountered and in which the muscle biopsy significantly contributes to the diagnosis are emphasized. Based on more than 25 years of experience at the University of Buffalo and the University of Iowa, the most common neuromuscular diseases evaluated by muscle biopsy are either inflammatory or neurogenic. Despite best efforts, a specific diagnosis cannot be reached in some biopsies, so the pathology reports must be descriptive without a definitive interpretation.
MUSCULAR DYSTROPHIES
The rather meaningless term dystrophy, which literally means “deficient nutrition,” was popularized toward the close of the nineteenth century, when the pathogenesis of the muscular dystrophies was totally mysterious. The muscular dystrophies, which share certain clinical and pathologic attributes, have long been considered part of a common rubric. In general, the initial symptoms are manifested during childhood or young adulthood; however, late adult onset cases do occur. The cardinal symptom is muscular weakness that is steadily and unremittingly progressive. By definition, all forms of muscular dystrophy are genetic (Table 4.8); some are inherited, whereas others are de novo mutations. The histopathologic criteria of muscle fiber necrosis (myonecrosis), regeneration, and endomysial fibrosis are common to all patients (Fig. 4.29), but the severity of dystrophic changes varies among genotypes, among allelic variants of the same genotype, among muscles within individual patients, within a single biopsy, and over time within individual patients. Specific genetic diagnoses can be suggested by immunostaining, but definitive diagnoses rely on molecular genetic testing.

Dystrophinopathy (Duchenne and Becker Muscular Dystrophies)
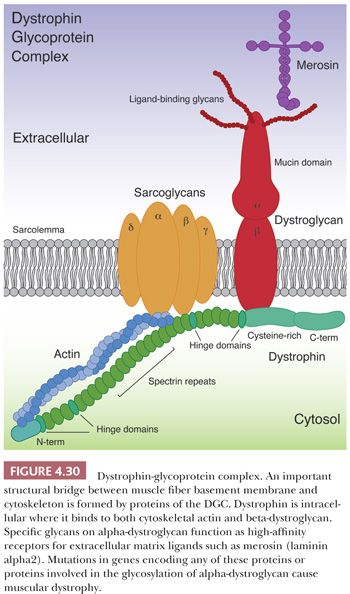
Duchenne muscular dystrophy (DMD) is the most common form of muscular dystrophy and also among the most severe. The dystrophin gene (DMD) that is responsible for DMD is located on the short arm of the X chromosome at Xp21 (41). With a 14-kb coding sequence within 2.5 to 3.0 Mb of DNA, it is the largest known human gene. Approximately two-thirds of DMD cases are caused by large deletions or duplications that are easily detected by genomic hybridization (42). Complete sequencing of coding exons and exon/intron boundaries discovers all but about 2% of the remaining DMD mutations (43). The identification of mutations permits an accurate diagnosis, including carrier and prenatal detection. Patients with DMD are greatly deficient in full-length dystrophin, a 427-kd, rod-shaped protein that is found predominantly in skeletal and cardiac muscle (44,45). Dystrophin is localized just beneath the sarcolemmal membrane and is a key component of the dystrophin-glycoprotein complex (DGC; Fig. 4.30). The DGC is presumed to promote sarcolemmal stability, particularly during muscle contraction, by forming a structural alignment between cytoskeletal actin inside and extracellular matrix proteins (e.g., merosin [laminin α2 or laminin 211], perlecan, and agrin) within the basement membrane (46). Other key members of the DGC are dystroglycans and sarcoglycans (see Fig. 4.30 and sections on limb-girdle and congenital muscular dystrophies).
As an X-linked recessively inherited disorder, DMD almost exclusively affects boys who are neurologically intact at birth. However, by the time the child attempts to stand or walk, the first signs of overt disease are noticeable. A subtle awkwardness gradually gives way to limb-girdle pattern weakness, sparing the muscles of facial expression and swallowing. A paradoxical enlargement of selected muscles that also are weak and flabby is characteristic of DMD. This pseudohypertrophy, which is associated with fatty infiltration and reactive fibrosis, is especially apparent in the calves and buttocks. Mild intellectual disability that cannot be explained on the basis of physical incapacitation is considered intrinsic to the disease. Reduced expression of dystrophin isoforms normally found in brain may underlie the cognitive abnormalities. Death is often hastened by an insidious cardiomyopathy leading to sinus tachycardia, cardiac arrhythmias, and congestive heart failure. An extremely high level of serum creatine kinase is an early indicator of this form of dystrophy, and it may precede severe pathologic alterations in muscle.
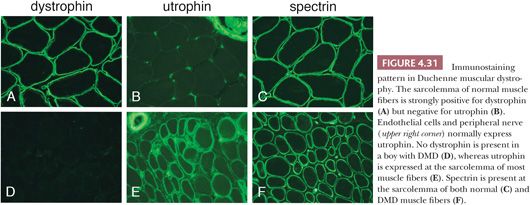
In theory, advances in molecular genetic diagnostic techniques and education of clinicians should eliminate the need for muscle biopsies to diagnose the vast majority of DMD patients. However, muscle biopsies continue to be done for a variety of reasons, including an inability to pay for genetic testing, failure to consider DMD in the differential, or finding DMD sequence variants of uncertain significance. If a muscle biopsy is obtained, myonecrosis and regeneration, endomysial fibrosis, and increased internally placed nuclei are striking, often exceeding that expected from the severity of clinical disease. Necrotic and regenerating fibers are frequently seen in clusters. Immunostains are readily available for detecting dystrophin (47,48). Whereas circumferential staining for dystrophin is robust in normal muscle and non-dystrophinopathy forms of muscular dystrophy, specimens from DMD patients are virtually dystrophin negative (Fig. 4.31). Because the amount of dystrophin expression depends on the specific DMD mutation, some patients with clinical severity most consistent with DMD can have weakly positive dystrophin immunostaining. Normal-intensity dystrophin immunostaining can sometimes be seen in some cells of a DMD muscle: revertant muscle fibers that result from spontaneous exon skipping or Schwann cells expressing their normal Dp116 dystrophin isoform. An important component of the immunostaining signature for DMD is the sarcolemmal expression of utrophin, a developmentally regulated muscle protein with structural similarity to dystrophin (Fig. 4.31). The upregulation of utrophin is thought to be partially compensatory for the loss of dystrophin. All nonnecrotic muscle fibers in DMD biopsies express spectrin.
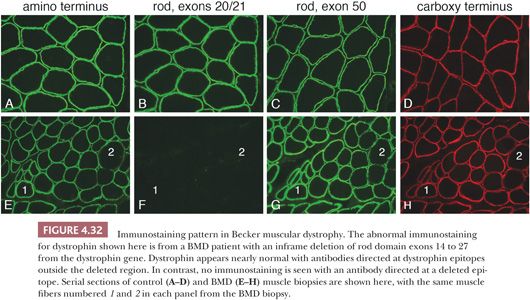
Becker muscular dystrophy (BMD) is the milder allelic form of dystrophinopathy seen in affected males. The symptoms in Becker dystrophy are less severe than in DMD, and the rate of progression is slower. The histopathologic changes are similar to, but less severe than, those of DMD. The mutations responsible for BMD are a mixture of inframe deletions, duplications, splice site mutations, and missense mutations leading to pathologic amino acid substitutions (43). The amount of dystrophin expressed in BMD muscle may be reduced or may be sufficient in quantity but abnormal in molecular size or structure (49,50). Thus, immunostains are positive for dystrophin, but the pattern of abnormal staining varies widely. It is important to use anti-dystrophin antibodies that each recognize a different epitope of the full length protein. A common approach uses antibodies DYS1, DYS2, and DYS3 (epitopes in the rod domain, at the C-terminus, and near the N-terminus, respectively) that will identify BMD patients with significantly reduced amounts of dystrophin. However, this panel of antibodies will miss some patients with inframe deletions who produce nearly normal amounts of truncated dystrophin. Alternative antibodies are readily available that bind to dystrophin epitopes within hot spots for inframe deletions (43). Using this approach, immunostaining diagnosis can be obvious (Fig. 4.32). Utrophin is often, but not always, upregulated in BMD muscle.
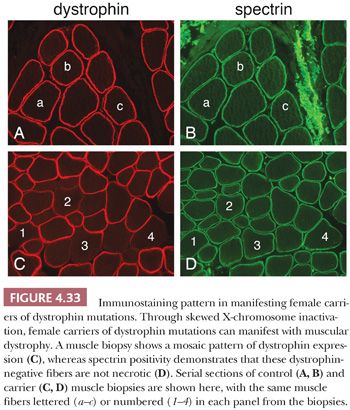
Manifesting female carriers of DMD mutations comprise a minor proportion of dystrophinopathy patients. However, their clinical similarities to other forms of muscular dystrophy often lead to a muscle biopsy before genetic testing is done. The histopathology is typically dystrophic, but immunostaining for dystrophin reveals a patchwork of negative and normal muscle fibers (Fig. 4.33). Skewed X-chromosome inactivation is thought to underlie the pathology that leads to clinical muscular dystrophy manifestations.
Limb-Girdle Muscular Dystrophies
Limb-girdle muscular dystrophy (LGMD) is not a single disease entity but rather a collection of dystrophies tenuously bound together by a common thread—the involvement of proximal axial muscles (51). These dystrophies are genetically very heterogeneous with at least 6 autosomal dominant (LGMD type 1) and at least 15 autosomal recessive (LGMD type 2) forms (Table 4.8). The age of onset ranges from early childhood (overlapping with the congenital muscular dystrophies described later) to late adulthood, and progression of disease varies widely. Many of the LGMD genes encode proteins associated with the sarcolemma (e.g., caveolin-3, dysferlin, and sarcoglycans) or critical for posttranslational O-mannosylation of α-dystroglycan, a sarcolemmal protein critical for binding to extracellular matrix proteins. Other LGMD proteins are part of the sarcomeric apparatus (e.g., titin) or nuclear envelope (e.g., lamin A/C) or have as yet uncertain localization and function (e.g., anoctamin 5). The prevalence of LGMD subtypes varies widely among ethnic groups and geographic regions, yet the most common subtypes in patients of European ancestry are calpainopathy (LGMD 2A), dysferlinopathy (LGMD 2B), sarcoglycanopathies (LGMD 2C-2F), dystroglycanopathies (e.g., LGMD 2I), and anoctaminopathy (LGMD 2L).
A large number of LGMD cases result from abnormalities of the DGC (Fig. 4.30). Mutations in any of the four DGC-associated sarcoglycan genes result in partial or total sarcoglycan deficiency, which can be detected by immunostaining for α-sarcoglycan and γ-sarcoglycan (52,53). Most sarcoglycanopathies are LGMD 2C, 2D, or 2E. Proteins involved in the O-mannosylation of α-dystroglycan make up the remaining DGC-associated LGMDs (54). An abnormality in immunostaining for α-dystroglycan can be detected using antibodies specific to the O-mannose glycoepitope. As a group, these LGMDs are termed dystroglycanopathies; phenotypically, they are the mild end of a spectrum of disease related by pathogenesis and genetics to a heterogeneous group of congenital muscular dystrophies described in the following section. The most prevalent of the LGMD-dystroglycanopathies is LGMD 2I caused by mutations in FKRP. A single point mutation that leads to the amino acid substitution L276I represents approximately 80% of all mutant alleles. This point mutation is particularly prevalent in northern Europeans and North American Hutterites (55). At the other end of the spectrum, only a single patient has been described with mutations in dystroglycan itself (DAG1). Like dystrophinopathy, some patients with sarcoglycanopathy or dystroglycanopathy may have severe cardiomyopathy (56,57).
Another of the more prevalent LGMD subtypes is 2A, which is caused by a mutations in the CAPN3 gene coding for calpain-3, a calcium-activated protease. This cytoplasmic, perhaps sarcomere-associated, protein is not readily amenable to evaluation by immunostaining. If other common LGMD proteins appear normally expressed by immunostaining, it may be useful to evaluate calpain-3 by Western blotting prior to sequencing CAPN3 (58,59).
Dysferlin and anoctamin 5 are also among the more prevalent LGMD subtypes, with dysferlinopathy particularly prevalent in southern Europe (60,61). In addition to LGMD presentations, mutations in each of these genes (DYSF and ANO5, respectively) can alternatively cause a distal myopathy phenotype, Miyoshi myopathy. Whether the presentation is limb-girdle or distal, many patients with either genotype over time progress to have both proximal and distal weakness. Dysferlinopathy (LGMD 2B) is amenable to detection by immunostaining. Absence or near absence of immunostaining, especially when verified by Western blotting, is highly predictive of DYSF mutations (62). However, a wide range of abnormal expression patterns seen in most muscular dystrophies and many other types of myopathy can make dysferlin immunostaining difficult to interpret (53). Antibodies are not yet available for anoctamin 5.
The remaining LGMD subtypes are much less common. Only caveolinopathy (LGMD 1C) is amenable to diagnostic immunostaining (53). Two of the dominant LGMD subtypes (1A and 1E) overlap with myofibrillar myopathies due to mutations in MYOT and DES, respectively (63,64). Thus, biopsies from these patients may have myofibrillar inclusions detectable by routine histologic evaluation. One of the recessive LGMD subtypes (2J) can be seen in families with a dominant distal myopathy (tibial muscular dystrophy or Udd myopathy) seen predominantly in Finland (65). Mutations in titin (TTN) and lamin A/C (LMNA) can cause a wide variety of other clinical phenotypes, including cardiomyopathy (66,67).
Congenital Muscular Dystrophies
The defining characteristic of congenital muscular dystrophies (CMDs) is presentation prior to 1 year of age. Otherwise, clinical and pathologic phenotypes are exceedingly heterogeneous (68,69). More than 20 genes are known. For the dystroglycanopathy group, more than 15 genes have been discovered that contribute to the O-mannosylation of α-dystroglycan (70–72). Except for the CMD associated with LMNA mutations, the CMDs are autosomal recessive. Clinical phenotypes can include brain and eye abnormalities. The brain involvement in merosin-deficient CMD (MDC1A) is typically limited to abnormal white matter signal on magnetic resonance imaging (MRI) (73). Much more severe phenotypes are found in the dystroglycanopathy group: Walker-Warburg syndrome, muscle-eye-brain disease, and Fukuyama CMD (70,73).
The vast majority of CMD proteins are related to skeletal muscle extracellular matrix: laminin α2 (merosin), collagen VI, and α-dystroglycan (dystroglycanopathies). Each of these latter CMDs can be diagnosed by immunostaining. The use of multiple antibodies increases specificity and sensitivity of this diagnostic approach. For example, partial merosin deficiency is more readily detected by using antibodies to more than one epitope. The specificity for dystroglycanopathy is improved by evaluating dystrophin and β-dystroglycan in addition to α-dystroglycan. For collagen VI CMD (Ullrich CMD), a dual-label immunofluorescence method (anti-collagen VI in combination with either anti-perlecan or anti-laminin) is recommended to detect the failure of secreted collagen VI to localize in muscle fiber basement membranes (74,75).
Muscle biopsy evaluation in some patients with CMD caused by LMNA mutations can easily be misinterpreted as myositis (76). As there is no immunodiagnostic method to detect lamin A/C abnormalities, the correct diagnosis relies on familiarity with the typical clinical phenotype and the lymphocytic inflammatory changes commonly present in the muscle.
Myofibrillar Myopathies
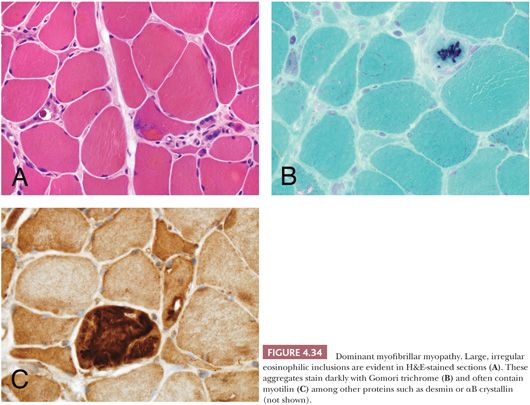
This heterogeneous group of autosomal dominant myopathies are sometimes included with nemaline myopathy and inclusion body myopathies under the terminology protein aggregate myopathies (77–79). The defining morphologic characteristic is the aggregation of multiple proteins (often including desmin, dystrophin, αB-crystallin, and myotilin) within the cytoplasm of muscle fibers (79–81). These aggregates are typically large, irregular inclusions easily seen on sections stained with H&E or Gomori trichrome (Fig. 4.34). They fail to stain with NADH or ATPase enzyme histochemistry. Although immunostaining can help confirm that myofibrillar inclusions are present, it usually does not predict the genotype of the patient. The myofibrillar myopathies are most often adult-onset, slowly progressive conditions with either proximal or distal presentations; LGMD 1A (MYOT) and 1E (DES) are allelic disorders. Affected patients may also have dysphagia and cardiac muscle involvement; cardiomyopathy is particularly prevalent in desminopathy (82). A severe congenital, recessive myopathy due to CRYAB mutations has been described in which the patients have extreme muscle rigidity and fatal respiratory failure (83,84). Under the electron microscope, myofibrillar inclusions are composed of granular and filamentous, electron dense material that does not help to predict the genotype. They often are arranged in register with the Z-bands. In the recessive CRYAB patients, the inclusions have a dappled appearance (Fig. 4.35).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


