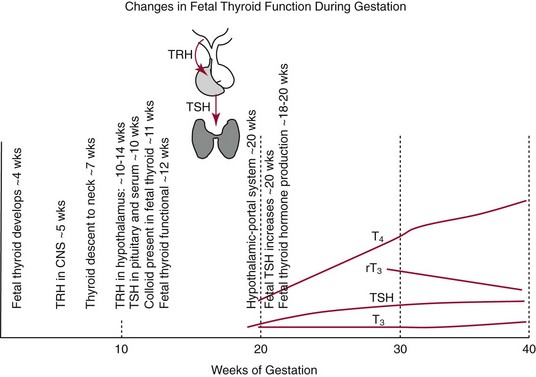
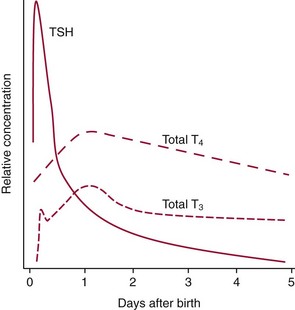
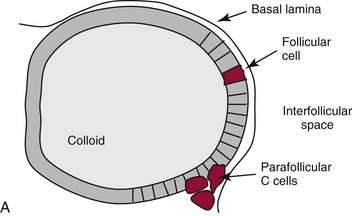
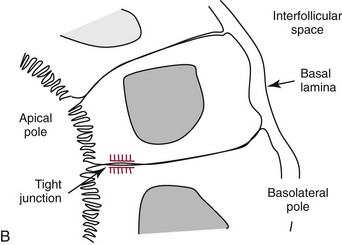
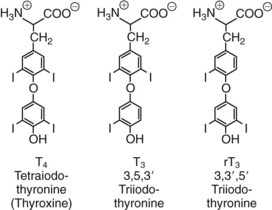
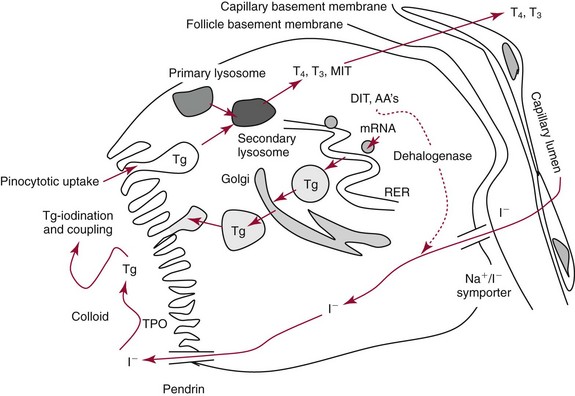
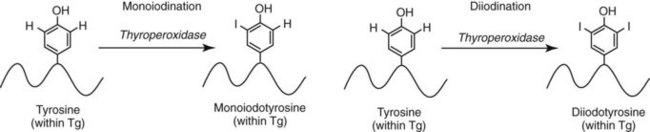
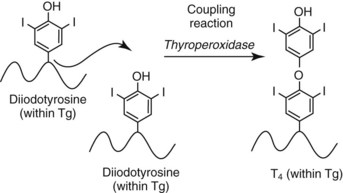
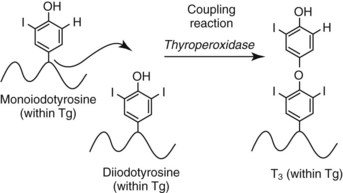
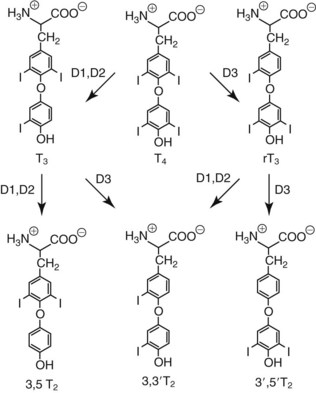
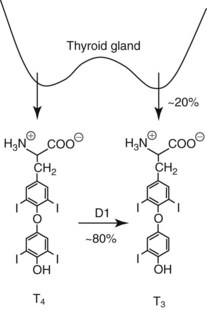
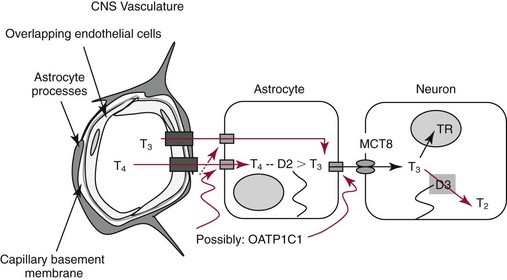
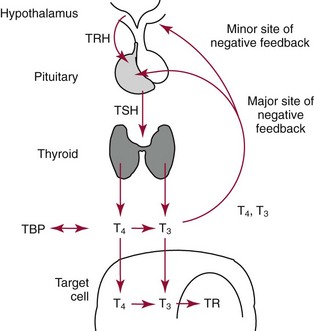
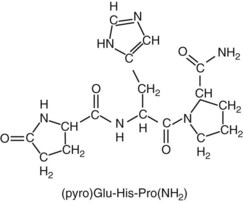
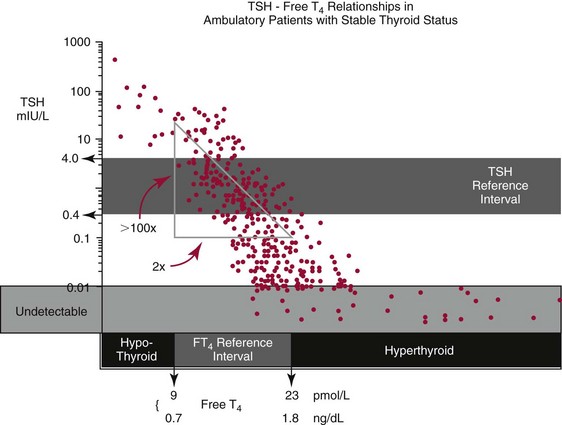
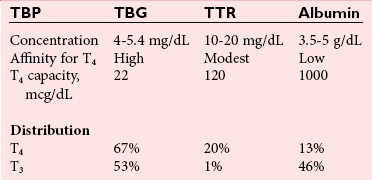
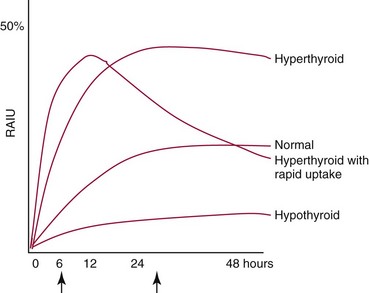
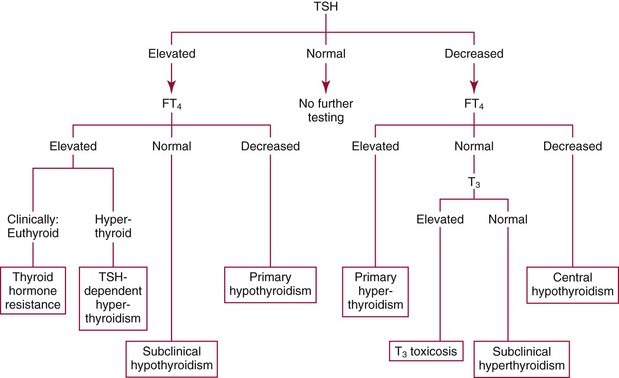
Chapter 55 The thyroid is a butterfly-shaped gland located in the front of the neck, just above the trachea. The fully developed thyroid gland in a human is composed of two lobes connected by a thin band of tissue, the isthmus, which gives the gland the appearance of a butterfly. The gland is closely attached to the trachea in the anterior aspect of the neck. Anatomically and embryologically, the thyroid gland is two glands in one: the thyroid follicular cells produce thyroid hormones and the parafollicular (or C) cells secrete calcitonin.21 The fetus is dependent on the transplacental passage of maternal thyroid hormone for the first half of gestation.26,29,65,225 Although only minute amounts of maternal thyroid hormone cross the placenta, maternal hypothyroidism in the first 20 weeks of gestation may adversely impact fetal central nervous system (CNS) development, leading to neuropsychologic impairment in infants and children.178 Worldwide, the most common cause of hypothyroidism during pregnancy is iodine deficiency. Conversely, in developed countries, where iodine fortification is widespread, the most common cause of primary hypothyroidism is Hashimoto thyroiditis (chronic autoimmune thyroiditis/inflammation of the thyroid gland). Hypothalamic thyrotropin-releasing hormone (TRH) stimulates thyrotropin [thyroid-stimulating hormone (TSH)], which in turn stimulates thyroid hormone synthesis. By 10 weeks, fetal thyroid follicles and thyroxine synthesis are demonstrable. By the mid second trimester, maturation of the hypothalamic-pituitary-thyroid axis occurs, so that by 20 weeks, the fetus is becoming responsible for its own production of thyroid hormone. Thyroxine-binding globulin (TBG) and thyroxine are first detectable in fetal serum at 8 to 10 weeks’ gestation and increase thereafter until they plateau at 35 to 37 weeks (Figure 55-1). Thyroxine [tetraiodothyronine (T4); see Table 55-1 for nomenclature and abbreviations) and TSH rise until birth, with T4 near ≈10 µg/dL and TSH reaching a concentration of 7 to 10 mIU/L. Beginning at ≈30 weeks, T4 is converted to triiodothyronine (T3) by increased activity of the type 1 deiodinase (D1), so that T3 concentrations rise, while reverse T3 (rT3) concentrations decline. At 30 weeks, T3 is less than 15 ng/dL; at term, T3 is ≈50 ng/dL. Within hours of birth, TSH, T4, and T3 rise rapidly (Figure 55-2).52 Cold stress is believed to be responsible for the massive TSH surge to concentrations of 70 to 100 mIU/L. By 2 to 3 days, the TSH concentration falls in most cases to less than 20 mIU/L. Total T4 peaks near ≈17 µg/dL. T4 then falls to adult concentrations by 1 to 2 months of age. The postbirth rise in T3 results from increased thyroid gland release in response to the rising TSH concentration and increased conversion of T4 to T3 due to maturation of the type 1 deiodinase enzyme (D1). Microscopically, the thyroid gland is composed of follicles or acini (Figure 55-3, A). The outside of the follicular unit/acinus is enveloped by a basement membrane. Along the inside of the basement membrane are squamous or cuboidal epithelial follicular cells. The height of the follicular cells reflects their biochemical activity: the greater their height, the more thyroid hormone synthetic activity is occurring. The basal pole of the follicular cell is oriented toward the basement membrane, and the apical pole is oriented toward the center of the follicle (Figure 55-3, B). In the center of the follicle, lined by the apical aspects of the follicular cells, is the lacuna, which contains colloid composed predominantly of thyroglobulin. Thyroid hormone is derived from the amino acid tyrosine. Thyronine is produced by substitution of a second phenol moiety for the phenolic hydrogen on tyrosine, producing a diphenyl ether having two phenol rings attached to one another through an ether linkage (Figure 55-4). Iodination of tyrosine to ultimately produce T4 and T3 involves the tyrosine residues on thyroglobulin.86 An isomer of T3 is reverse T3 (rT3), in which the alpha ring is monoiodinated and the beta ring is di-iodinated, producing 3,3′,5′-L-triiodothyronine. Reverse T3 is not biologically active. Almost all rT3 is formed by the extrathyroidal conversion of T4 to rT3, and approximately 80% of T3 is formed by extrathyroidal monodeiodination of T4 to T3. Thyroid hormone biosynthesis begins with the active transport of iodide (I−) into the thyroid gland via the Na+/I− (sodium-iodide) symporter (NIS, SLC5A; SLC = solute carrier; chromosome 19p12-13.2; Figure 55-5).20 Iodide transport is inhibited by lithium, which competes with sodium, but the NIS transports other anions besides iodide. In the follicular cells, the iodine concentration is 30- to 40-fold greater than the circulating concentration. When the thyroid gland is stimulated by TSH, even larger iodide concentration gradients can result. Antithyroid drugs such as propylthiouracil and methimazole inhibit iodination and coupling; this can lead to an 800-fold difference in the concentration of iodide in the thyroid gland versus the plasma. Once in the thyroid follicular cells, cytoplasmic iodide moves into the lacuna (colloid) via pendrin (SLC26A4 gene on chromosome 7q21-34) (see Figure 55-5).88 Pendrin is a passive iodide-transporting glycoprotein identified during studies of patients with Pendred syndrome. Pendred syndrome is an autosomal recessive disorder of dyshormonogenesis that is characterized by goiter and sensorineural deafness. Thyroglobulin (Tg) is necessary for thyroid hormone synthesis. Similar to other proteins, Tg is synthesized in follicular epithelial cells by ribosomes located on the endoplasmic reticulum (see Figure 55-5). Tg may represent up to 50% of protein synthesis in the thyroid gland and may account for 75% of glandular total protein. Although most Tg is secreted into the follicular lumen, a small amount of Tg is released from the follicular cells without transport into the colloid, and this Tg is not iodinated. Of consequence in autoimmune thyroid disease, iodination increases the immunogenicity of Tg. Increased dietary iodine exposure is associated with the development or expression of Hashimoto thyroiditis.112 Once in the lacuna, iodide is oxidized (“organified”) to an iodine radical by the thyroperoxidase (TPO) enzyme. Hydrogen peroxide (H2O2) serves as the terminal electron acceptor, forming H2O2−.185 Hydrogen peroxide is generated at the apical membrane by the action of DUOX1 and DUOX2 (DUOX was previously known as THOX, or thyroid oxidase). DUOX is a dual oxidase that has domains analogous to the domains found in nicotinamide adenine dinucleotide phosphate (NADPH) oxidoreductases. Hypothyroidism due to DUOX mutations has been reported.130 TPO catalyzes the monoiodination of Tg tyrosines to monoiodotyrosine (MIT) (Figure 55-6). Di-iodination of tyrosine forms di-iodotyrosine (DIT) (Figure 55-6). After Tg is iodinated to form MIT and DIT, TPO catalyzes the coupling reaction to produce T4 (Figure 55-7) and T3 (Figure 55-8). In the coupling reaction, T3 (bound to Tg) is formed from one DIT and one MIT residue with the transfer of one monoiodinated phenolic group to a DIT residue. T4 (bound to Tg) is formed from two DIT residues with the transfer of one di-iodinated phenolic group to another DIT residue. Tg, along with its T3 and T4 residues, remains in the colloid, providing a reservoir of thyroid hormone. With TSH stimulation, the apical pole of the follicular cell releases colloid into a vesicle by pinocytosis (see Figure 55-5). The follicular cell digests the intravesicular colloid containing Tg after fusion of the phagosome body with a primary lysosome. Fusion of the endocytosed Tg with the primary lysosome forms a secondary lysosome, in which digestion of Tg occurs, releasing T4, T3, MIT, DIT, and amino acids. By diffusion, the lipophilic T4 and T3 molecules exit the lysosome and cross the follicular cell plasma membrane to be captured in thyroid capillaries, where the vast majority of thyroid hormone is protein bound. Only 0.03% of total T4 is free (unbound and bioactive), and only 0.3% of total T3 is unbound and bioactive. Within the cytoplasm of the follicular cell, the released MIT and DIT are stripped of iodine by dehalogenase (Dhal) to produce free iodide ions, which can be recycled immediately for the synthesis of new thyroid hormone.64 Two dehalogenase genes have been described: Dhal1 and Dhal1b. Normally, only negligible amounts of MIT and DIT are released from the thyroid gland into the circulation. However, loss-of-function mutations in the dehalogenase enzyme potentially lead to iodine loss in the urine and increased concentrations of DIT and MIT in the circulation. The peripheral metabolism of thyroid hormones is very complex.191 Three homodimeric deiodinases are present: type 1 (D1), with inner and outer ring deiodinase activities found in liver, kidney, thyroid, and possibly the anterior pituitary gland (Figure 55-9); type 2 (D2), with outer ring deiodinase activity expressed in the CNS, anterior pituitary, brown fat, placenta, heart, skeletal muscle, and thyroid; and type 3 (D3), with inner ring deiodinase activity identified in CNS, liver, endometrium, and placenta. Approximately 40% of T4 is deiodinated to T3 by D1 or D2, and ≈45% is deiodinated to rT3 by D1 or D3. About 80% of circulating T3 comes from 5′-deiodination of T4; only ≈20% of T3 is released directly from the thyroid gland (Figure 55-9). Almost all circulating rT3 results from 5-deiodination of T4. By regulating the conversion of T4 to T3, the body, in part, regulates the metabolic rate. D1 converts T4 to rT3 (via 5-deiodination) and T4 to T3 (via 5′-deiodination) (Figure 55-10). The preferred substrates for D1 are sulfated T3 (converted to 3,3′-T2 via 5-deiodination) and rT3 (converted to 3,3′-T2 via 5′-deiodination). D1 activity is stimulated by thyroid hormone through increased gene transcription. Thus, increased D1 activity promotes peripheral conversion of T4 to T3 in hyperthyroidism. Therapeutically, D1 is inhibited by propylthiouracil, which is used to treat hyperthyroidism. In contrast to the effect of thyroid hormone on D1, D2 expression is suppressed by thyroid hormone. The Km for the D1-T4 complex is approximately 1000-fold greater than the corresponding Km for D2 or D3. Regulation of the T4→T3 and T4→ rT3 conversions is dependent on the tissue distribution of D1, D2, and D3. The free T3 (FT3)-to-T4 (FT4) ratio is affected by a genetic polymorphism in the D1 gene (DIO1; the single-nucleotide polymorphism rs2235544).143 Until recently it was believed that both T4 and T3 entered cells by passive diffusion across the plasma membrane. However, evidence has shown that thyroid hormones cross plasma membranes using specific transporters.74 One important thyroid hormone transporter is monocarboxylate transporter 8 (MCT8).118 The solute carrier family of monocarboxylate transporters has 14 members (MCT1 through MCT14); examples of other monocarboxylates transported by these carriers are lactate and pyruvate. Transport of thyroid hormone into neurons is especially critical for normal CNS development and function (Figure 55-11). OATP1C1 may be responsible for thyroid hormone uptake and release by astrocytes, although this has not yet been definitively demonstrated. Within the astrocyte, T4 is converted to T3 via D2 attached to the astrocyte membrane. T3 may exit the astrocyte via OATP1C1 to be taken up by neurons via MCT8. Within the neuron, T3 can interact with the intranuclear thyroid hormone receptor (TR), or can be converted to 3,3′-T2 via membrane-attached D3. T3 binds to specific intranuclear TRs to exert its hormonal effects. In humans (and various animals), thyroid hormone upregulates the expression of growth hormone,110 myelin basic protein, the alpha-myosin heavy chain, and a sarcoplasmic reticulum calcium ATPase, while downregulating the expression of the TSH beta chain and the beta-myosin heavy chain. In part, suppression of TSH beta chain synthesis contributes to negative feedback. Two types of TRs are known: TRα and TRβ (Table 55-2).218 TRα is encoded by the thyroid hormone receptor-alpha (THRA) gene on chromosome 17q11.2. Of its two expressed isoforms (TRα1 and TRα2), only TRα1 binds thyroid hormone. A TRα3 isoform produced by alternative splicing has also been reported. TRα1 is expressed throughout the body, but its mRNA is in highest concentrations in the heart, brain, liver, and kidneys. TABLE 55-2 Thyroid Hormone Receptor (TR) Sizes *Extended C-terminal region (does not bind T3). Thyroid Hormone Negative Feedback Control of TRH and TSH Thyroid hormone synthesis and secretion are controlled by a negative feedback system involving the hypothalamus, the pituitary, and the thyroid follicular cells (Figure 55-12).30 In contrast to other hypothalamic-anterior pituitary target organ systems that utilize a negative feedback system, however, the major site of thyroid hormone feedback is at the level of the anterior pituitary thyrotrophs—not at the level of the hypothalamus. Thyrotropin-releasing hormone (TRH; thyrotropin-releasing factor, thyroliberin, or protirelin) is encoded on chromosome 3, and the modified tripeptide L-pyroglutamyl-L-histidyl-L-prolinamide is secreted by the paraventricular nuclei (PVN) in the hypothalamus (Figure 55-13). Cyclization of the glutamate terminus is required for TRH bioactivity. The PVN are located in the anterior hypothalamus, rostral to the optic chiasm. TRH concentrations rise in thyroid hormone deficiency with TRH declining when thyroid hormone is in excess. TRH is delivered to the anterior pituitary gland via the hypothalamic-pituitary portal system. Thyrotrophs, the anterior pituitary cells that secrete thyroid-stimulating hormone (TSH; thyrotropin), express TRH receptors, which are G-protein–coupled receptors with seven transmembrane domains. When TRH binds to its receptor, the thyrotroph depolarizes, triggering calcium influx. Consequently, increased free cytosolic calcium activates the Ca2+-phosphatidylinositol cascade, effecting TSH release, synthesis, and glycosylation of alpha and beta TSH subunits. In relative terms, TRH has a greater effect on TSH glycosylation than hormone release.165 However, glycosylation of TSH is necessary for normal TSH bioactivity. When TRH is deficient, TSH may lack potency as the result of insufficient glycosylation, yet nonglycosylated TSH may retain much of its immunoreactivity. Therefore, immunoassays for TSH may not reflect the activity of the hormone when injury or disease results in a TRH deficiency. TRH modifies the sensitivity of the thyrotroph to thyroid hormone negative feedback; increased TRH makes the thyrotroph less sensitive to inhibition with decreasing TRH makes the thyrotroph more sensitive to negative feedback. The mechanism for TRH control involves reduced expression of TRs in the thyrotroph following TRH stimulation. When the thyrotroph is less sensitive to negative feedback from thyroid hormone, TSH release is potentiated. Therefore, TSH secretion rises in thyroid hormone deficiency and declines in thyroid hormone excess. An inverse logarithmic relationship exists between TSH and FT4 concentrations (Figure 55-14); a 50% decline in FT4 concentration results in a 100-fold increase in TSH. TSH is a 30 kDa heterodimeric glycoprotein that shares a subunit with luteinizing hormone (LH), follicular stimulating hormone (FSH), and human chorionic gonadotropin (hCG).174 All four hormones contain a 14.7 kDa alpha subunit (gene location: chromosome 6q21.1-q23). Each of these hormones has a unique beta subunit that is responsible for the specific activity of the hormone. The 15.6 kDa TSH beta chain is encoded by a three-exon gene located on chromosome 1p. The alpha chain contains two oligosaccharides, and the TSH beta subunit contains one oligosaccharide modification. The effects of TSH on the thyroid follicular cell are mediated through TSH receptor (TSHR)–G-protein–adenyl cyclase–coupled synthesis of intracellular cyclic adenosine monophosphate (cAMP).197 At supraphysiologic concentrations (100× normal), TSH will signal through the inositol-phosphate diacylglycerol cascade, activating phospholipase C (PLC) and raising intracellular calcium concentrations, with subsequent stimulation of H2O2 generation and iodide efflux into the follicular lumen. Both T4 and T3 are highly bound to plasma proteins in circulation. Protein-bound T4 and T3 serve as thyroid hormone reservoirs within plasma. In the thyroid gland, Tg serves as a reservoir. The unbound fraction of circulating thyroid hormone is the biologically active form, so FT4 concentrations correlate more closely with the clinical status of the patient than do total T4 concentrations.15,142 However, elevations in FT3 usually correlate with the clinical status of the patient, low FT3 concentrations do not always equate with clinical hypothyroidism, as evidenced in the sick euthyroid syndrome (see later). Similarly, elevated free concentrations of T4 or T3 do not always correlate with hyperthyroidism because of the possibility of peripheral thyroid hormone resistance syndromes and rare MCT8 loss-of-function mutations. Alterations in the concentrations of thyroid hormone–binding proteins can profoundly affect the total concentrations of T4 and T3 without significant changes in the free hormone concentrations. T4 binds to thyroxine-binding globulin (TBG; a 54 kDa α1-globulin), thyroxine-binding prealbumin (TBPA, or transthyretin; 54 kDa), and albumin. T3 binds almost exclusively to TBG and albumin; TBPA binds only negligible amounts of T3 (Table 55-3).176 On serum protein electrophoresis, TBPA migrates in the prealbumin region (hence, its name). TBG (chromosome Xq11-23) is a 395 amino acid acidic glycoprotein with one iodothyronine-binding site. TBG has 4 heterosaccharide sidechains with 5 to 9 sialic acid moieties. As the degree of sialylation increases (an effect of estrogen), the half-life of TBG increases, thus raising TBG concentrations. Increased concentrations of TBG raise total T4, yet FT4 remains within the euthyroid interval as the result of negative feedback (see Figure 55-12). Similarly, decreased TBG concentrations lower total T4 while maintaining normal FT4. Causes of elevations and depressions in thyroid hormone–binding protein (TBP) are shown in Box 55-1. In congenital TBG excess (an X-linked dominant condition), male hemizygotes display threefold to fivefold elevations in TBG concentration, while female heterozygotes typically have twofold to threefold elevations in TBG. Abnormal forms of albumin (familial dysalbuminemic hyperthyroxinemia) and TBPA (familial euthyroid thyroid excess) alter the concentrations of total T4, yet do not affect FT4, total T3, or FT3. These conditions are discussed later, along with antithyroid hormone autoantibodies that raise total thyroid hormone concentrations. Isolated euthyroid hypertriiodothyroninemia (with all other thyroid parameters normal) caused by a rare form of dysalbuminemia has been reported. Thyroid gland location, anatomy, and function is also assessed radiographically. For example, thyroidal uptake of radioactive iodine (I-123 or I-131) or technetium pertechnetate (99mTc-pertechnetate; TcO4−) is assessed over time as an index of thyroid function.116 The degree of uptake of an exogenously administered radioactive tracer versus time reflects the activity of the thyroid gland (Figure 55-15). Radioactive iodine or technetium pertechnetate can be used. The radioactive iodine uptake (RAIU) is typically expressed as the counts measured (via scanning of the thyroid gland) divided by the total counts administered. The reference interval for the RAIU is usually 5 to 25%, which means that 5 to 25% of administered radioactive tracers are present in the thyroid gland at the time of the scan (usually at 24 hours). The perchlorate discharge test is used to detect defects in thyroid gland iodide oxidation or iodination of Tg. Radioactive iodine is administered prior to perchlorate, and any radioiodine still in the follicular cells that has not yet been incorporated into colloidal Tg is released.227 Perchlorate does not block iodination of Tg. If the NIS has transported radioiodide into the thyroid gland, but the iodide is not yet incorporated into Tg after perchlorate administration, a supranormal amount of radioiodine is released from the thyroid gland (an increase in the perchlorate discharge of radioiodine occurs). Because the signs and symptoms of thyroid dysfunction are extremely variable, thyroid function studies are often measured in clinical practice.36,41 An enlarged thyroid gland (goiter) is typically evaluated by measurement of TSH and thyroid hormones, and on the basis of (1) history, (2) physical examination, and (3) laboratory results; patients may be classified as (1) euthyroid, (2) hypothyroid, or (3) hyperthyroid.95,125,153 Patients presenting with a thyroid mass are typically euthyroid. Suspicious masses are typically followed by ultrasound-guided fine-needle aspiration of the mass with cytologic examination. A thorough discussion of all forms of thyroid cancer is beyond the scope of this chapter, although the role of Tg measurements will be examined as a tumor marker for differentiated thyroid cancers.67 If clinical findings do not suggest an abnormality in the hypothalamic-pituitary thyroid axis, laboratory evaluation is begun with measurement of TSH (Figure 55-16).4,95A If TSH is within the reference interval, thyroid dysfunction generally can be excluded. If TSH is below the reference interval, measurement of FT4 is indicated. If TSH is depressed and FT4 is elevated, the biochemical diagnosis of primary hyperthyroidism is established. Although total T3 can be measured at this point (and is expected to be greatly elevated), this value should not change the diagnosis or the initial therapy. If TSH is depressed and FT4 is within the reference interval, T3 should be measured in search of T3 toxicosis. The authors favor total T3 measurements over FT3 measurements because total T3 measurements are both more accurate and less expensive than FT3 measurements. If TSH is depressed and both FT4 and T3 are normal on more than one occasion, subclinical hyperthyroidism is diagnosed. This assumes that causes of TSH suppression such as high-dose glucocorticoids and dopamine have been excluded. If TSH is depressed, FT4 is normal, and T3 is elevated, the diagnosis of T3 toxicosis is established.50 If TSH is elevated and FT4 is within the reference interval on more than one occasion in an asymptomatic patient, subclinical hypothyroidism is diagnosed (assuming that heterophilic antibodies have been excluded as a cause of TSH elevation).8,145 Elevated TSH and FT4 raise the possibility of central hyperthyroidism or thyroid hormone resistance. These disorders are differentiated clinically: patients with thyroid hormone resistance usually are euthyroid or, at worst, mildly hypothyroid, however, those with true hyperthyroidism clinically manifest thyrotoxicosis (see later). Because of the log-linear relationship of TSH to FT4, FT4 usually is normal as long as TSH is between 0.5 and 10 mcIU/mL. Hypothyroidism is defined as a deficiency in thyroid hormone secretion and action that produces a variety of clinical signs and symptoms of hypometabolism.73,84,198 This common disorder occurs in 2 to 15% of the population, more commonly in women than in men. The risk of developing hypothyroidism increases with age.39 Clinical symptoms suggesting hypothyroidism include (1) mental dullness (including mental retardation in children with untreated or undertreated congenital hypothyroidism), (2) somnolence, (3) increased sleeping, (4) lethargy, (5) easy fatigability, (6) hoarseness or deepening of the voice, (7) hair loss, (8) weight gain, (9) cold intolerance, (10) menstrual irregularities, (11) infertility, (12) growth failure, (13) delayed puberty in adolescents, (14) constipation, (15) muscle weakness or cramps, and (16) depressed affect or frank clinical depression.171 Physical signs compatible with hypothyroidism include (1) bradycardia, (2) decreased pulse pressure, (3) cool and/or dry skin, (4) puffy eyes, (5) loss of the outer lateral eyebrows, (6) delayed relaxation phase of reflexes (“hung-up” reflexes), (7) myopathy, (8) carotenemia, (9) occasional galactorrhea, and (10) radiologic evidence of delayed bone age in children. In cases of severe hypothyroidism, congestive heart failure or coma may develop.90 In children with untreated congenital hypothyroidism, severe growth failure and mental retardation ensue. The development of biological and biochemical processes is delayed in cases of congenital hypothyroidism. For example, affected infants often have prolonged jaundice as the result of immaturity of UDP-glucuronyl transferase. A rare (and controversial) manifestation of Hashimoto thyroiditis is encephalopathy.216 However, no definitive clinical test is available to diagnose encephalopathy resulting from Hashimoto thyroiditis. The diagnosis is considered when encephalopathy of unknown origin is associated with autoantibodies against the thyroid gland. The causes of primary hypothyroidism are classified as endogenous or exogenous (Box 55-2).38 Endogenous disorders are conditions that develop within the patient such as autoimmune thyroid gland dysfunction, inborn errors, and developmental abnormalities. Exogenous disorders are conditions that originate outside the patient such as iodine deficiency or excess, goitrogen or drug effects, and postsurgical hypothyroidism or hypothyroidism following radioactive iodine treatment. Excluding the newborn period, autoimmune thyroid disease (AITD) is the most common cause of thyroid disease and primary hypothyroidism.111,148,203 Hashimoto thyroiditis (chronic lymphocytic thyroiditis) leads to destruction of the thyroid follicular cells through a cell-mediated autoimmune process.102 Histologically, the gland is infiltrated with lymphocytes and plasma cells to the extent that secondary lymphoid follicles develop within the thyroid gland that are similar to the secondary follicles observed in normal lymph nodes. Initially, the gland is usually enlarged. Over time, with destruction of the gland, the gland can atrophy or become firm. In the rare condition of Riedel thyroiditis (Riedel disease or struma, ligneous struma, ligneous thyroiditis, chronic fibrous thyroiditis), the thyroid gland becomes fibrotic with possible attachment to adjacent structures that can produce, for example, tracheal compression.144 Subacute (viral) thyroiditis may also cause Riedel thyroiditis.31 Not all cases of Riedel thyroiditis are presaged by Hashimoto thyroiditis. The diagnosis of Hashimoto thyroiditis is supported by recognition of autoantibodies against TPO or Tg.113 Ninety percent of patients with chronic lymphocytic thyroiditis (the histologic description of Hashimoto thyroiditis) have antithyroperoxidase autoantibodies (TPOA), antithyroid microsomal autoantibodies (TMA), and/or antithyroglobulin autoantibodies (TgA), making these autoantibodies excellent markers for Hashimoto thyroiditis.182 TPOA testing was initially pursued by testing for TMA (see section on thyroid autoantibody testing). TMA testing is being replaced by specific testing for TPOA using TPO as the antigen in the immunoassay. TPOA/TMA positivity is more common at the time of diagnosis than TgA positivity with TgA usually appearing later in the disease process. TgA overall are less common than TMA or TPOA. Ultrasound has limited value and is not routinely used to detect Hashimoto thyroiditis. In many families, AITD appears to be inherited in an autosomal dominant pattern although some family members develop Hashimoto thyroiditis and other family members develop Graves’ disease. Several genetic loci have been associated with susceptibility to Hashimoto thyroiditis (or AITD): HLA-DR, CTLA-4, CD40, FCRL3, Tg, the TSH receptor, PTPN22, and the IL-2 receptor. However, no single Mendelian locus explains the apparent autosomal dominant pattern of inheritance of AITD.66 It is most appropriate to describe AITD as polygenic and multifactorial, indicating that both environment and multiple genes provide susceptibility to these very common disorders. Nonendocrine and endocrine autoimmune disorders can occur with increased frequency in people with AITD. Chronic lymphocytic gastritis causing pernicious anemia may accompany AITD, particularly in older patients. AITD also occurs commonly in association with type 1 diabetes and pernicious anemia.61,91,104 Another, less common, association is AITD and Addison disease.82 AITD can occur as part of autoimmune polyglandular syndrome type 2 (APS 2) and, more rarely, APS 1.161,175 APS 2 affects women more often than men with onset in childhood or early adulthood and is diagnosed when Addison disease (or adrenal autoantibodies) occurs together with AITD and/or type 1 diabetes. In contrast to the polygenic nature of APS 2, mutations in AIRE (the autoimmune regulator gene that is a transcription factor; gene location: chromosome 21q22.3) produce autoimmune polyglandular syndrome type 1 (APS 1), which is inherited in an autosomal recessive pattern, thus affecting males and females equally.18,93 Another variant of AITD is postpartum thyroiditis (PPT).192 PPT develops presumably as a consequence of a decline in the natural immunosuppression of pregnancy following delivery. PPT follows ≈5% of all pregnancies and ≈10% of pregnancies in women with type 1 diabetes. The clinical phenotype of PPT is transient hypothyroidism, hyperthyroidism, or both (one following the other) with a return to the euthyroid state by approximately 1 year postpartum. Women with thyroid autoantibodies are at highest risk for PPT. Later in life, permanent clinical evidence of AITD can develop in women afflicted with PPT. Thyroid autoimmunity, even in the absence of hypothyroidism, may impair fertility in women and increase the risk of spontaneous abortion in those women who do become pregnant.154 Therefore an indication for thyroid autoantibody testing in the absence of hypothyroidism would be female infertility or recurrent miscarriage. Convincing data suggest that T4 treatment of pregnant women who are positive for thyroid autoantibodies (especially TPOA) leads to a higher frequency of miscarriage (13.8%) than is seen in pregnant women who lack TPOA (2.4% rate of miscarriage), and that T4 treatment of the TPOA-positive group reduces the rate of miscarriage to approximately 3.5%.136 Inborn errors in thyroid hormone biosynthesis (dyshormonogenesis) are rare causes of primary hypothyroidism. These defects usually present early in life and can appear in newborns as a goiter. In very rare cases, asphyxia from tracheal compression has been reported.213 Biochemical defects include iodine transport defects from loss-of-function mutations in the NIS (no increase in the perchlorate discharge); defects in thyroperoxidase, DUOX2,130 and pendrin (with increased perchlorate discharge); thyroglobulin deficiency; and iodotyrosine dehalogenase mutations (potentially causing iodine deficiency through loss of MIT, DIT, and iodine in the urine).129 Developmental causes of primary hypothyroidism involve aplasia or hypoplasia of the thyroid gland and ectopic and lingual thyroid glands.202 These disorders account for ≈75% of cases of congenital hypothyroidism. Other causes of congenital hypothyroidism include thyroid dyshormonogenesis (10% of cases; see earlier), hypothalamic or pituitary abnormalities (5% of cases). Ten percent of cases of congenital hypothyroidism are transient. Rare causes of congenital hypothyroidism involve loss-of-function mutations in the pituitary TRH receptor,23 the TSH beta chain,128 and the TSH receptor.19,58 Mutations in anterior pituitary transcription factors (HESX1, LHX3, LHX4, PROP1, PIT1) can lead to TSH deficiency. Causes of transient congenital hypothyroidism include (1) severe maternal iodine deficiency, (2) acute iodine exposure, (3) transplacental passage of thionamides taken for treatment of maternal hyperthyroidism, (4) transplacental transfer of TSH receptor blocking antibodies, (5) hypothyroxinemia of prematurity, and (6) heterozygosity for a DUOX2 mutation. Worldwide, the most common cause of goiter is iodine deficiency. In iodine-deficient areas, especially mountainous areas, iodine deficiency produces “endemic” goiter. Frank hypothyroidism, however, is far less common because with iodine deficiency, more T3 is synthesized than T4, and within the thyroid gland, more T4 is converted to T3 to maintain the euthyroid state overall. Endemic goiters can develop nodularity (with or without hemorrhage into the nodule); in such cases, neoplasia must be excluded. Large goiters can produce dysphagia, obstruction of the trachea, or compression of the recurrent laryngeal nerves. A rare cause of iodine deficiency is nephrotic syndrome, with increased urinary loss of iodine often in the form of thyroid hormone.53,114 Although it is logical that iodine deficiency would produce hypothyroidism because iodine is a necessary component in the synthesis of thyroid hormone, it is ironic that excess iodine can interfere with normal thyroid gland function.106 However, excessive iodine does suppress thyroid gland function. High doses of iodine [super-saturated potassium iodide (SSKI) or Lugol’s solution] are routinely used before planned surgical thyroidectomy for Graves’ disease, to inhibit further thyroid hormone release and decrease the vascularity of the gland to reduce surgical blood loss. Various drugs affect thyroid gland function (Table 55-4).63 Collectively known as thionamides or thioureas, propylthiouracil (PTU), methimazole, and carbimazole inhibit the oxidation of iodide and the subsequent binding of iodine to tyrosine residues in Tg. Other drugs with thionamide-like activity include ethionamide, aminoglutethimide, phenylbutazone, and para-aminosalicylic acid. Evidence suggests an immunosuppressive effect of thionamides on thyroid autoimmunity. TABLE 55-4 Effects of Some Drugs on Tests of Thyroid Function ↓, Reduced serum concentration; ↑, increased serum concentration; Data from Smallridge RD. Chapter 33. Thyroid function tests. In: Becker KL, ed. Principles and practice of endocrinology and metabolism, 7th edition. Philadelphia, Pa: JB Lippincott, 1995:299-306; Stockigt JR. Thyroid hormone changes in critical illness: the sick euthyroid “syndrome.” Diagn Endo Metab 1997;15:39-46. PTU, methimazole, and carbimazole are commonly used to treat hyperthyroidism. (Note: Carbimazole is not available in the United States.) When large doses of PTU are used, the drug decreases peripheral conversion of T4 to T3 through inhibition of the D1 deiodinase. Because of this effect (in addition to the direct suppressive effect of PTU on the thyroid gland), some endocrinologists argue that PTU should be chosen over methimazole for the treatment of hyperthyroidism. However, recognition of serious or even fatal liver disease in a small (but significant) number of children treated with PTU is increasing.100 Therefore, many pediatric endocrinologists discourage the routine use of PTU for the treatment of hyperthyroidism in children. Reduced granulocyte counts and subsequent infections are serious but uncommon side effects of thionamides. Therefore the white blood cell count and the differential count should be monitored during such therapy. A variety of other drugs have been known to cause hypothyroidism. For example, lithium, which is used in the treatment of bipolar disorder (manic-depressive illness), can induce hypothyroidism.85 The action of lithium appears similar to that of high-dose iodine, inhibiting thyroid hormone release and organification of iodine. Prolonged use of nitroprusside, a drug used to treat acute-onset, severe hypertension or severe heart failure by inducing preload and afterload reduction, may lead to hypothyroidism. Cyanide (CN−) released from nitroprusside is metabolically converted to thiocyanate (SCN−), which inhibits iodide uptake by the thyroid gland. Fortunately, nitroprusside only rarely causes hypothyroidism. Amiodarone is an antiarrhythmic drug that contains two iodine atoms per molecule and can induce hypothyroidism or hyperthyroidism.22,204 Various recombinant DNA-derived biologicals (e.g., interferon-alpha, interleukin-2) used to treat chronic viral hepatitis or cancer have been found to cause thyroid dysfunction (hypothyroidism or hyperthyroidism).124 Surgical removal of the thyroid gland will produce hypothyroidism. External irradiation of the thyroid gland (e.g., treatment of lymphoma or Hodgkin disease) or ingestion of radioactive iodine can ablate the gland, causing hypothyroidism.123 Administration of radioactive iodine to pregnant women can lead to ablation of the fetal thyroid gland, causing congenital hypothyroidism. Only rarely do viral or bacterial infections of the thyroid gland occur. Even more rarely do these entities damage the thyroid gland sufficiently to produce hypothyroidism. Viral infection of the thyroid gland is termed subacute thyroiditis (de Quervain, granulomatous, or giant cell thyroiditis) and can produce generalized thyroid gland tenderness. Bacterial infection of the thyroid gland is termed acute thyroiditis and can produce a thyroid abscess.3 In a patient with clinical evidence of hypothyroidism, supported by laboratory findings of a low FT4, if TSH is not elevated to the extent predicted (by FT4), or if TSH is within or below the reference interval, central hypothyroidism should be considered.92,229 Isolated pituitary TSH deficiency is rare, as most patients with secondary hypothyroidism also have other anterior pituitary hormone deficiencies (panhypopituitarism). Causes of central hypothyroidism (hypothalamic or pituitary disease) include (1) tumor, (2) hemorrhage, (3) trauma, (4) malformation, (5) postinfectious damage, and (6) postsurgical damage. Also, several rare autosomal recessive or dominant hereditary disorders involving transcription factor mutations can cause hypopituitarism and TSH deficiency: examples include paired-like homeodomain transcription factor-1 (Pit-1) and PROphet of Pit-1 (PROP1) mutations.131 Subclinical hypothyroidism is defined by a persistent elevation in TSH (6 to 12 weeks or longer) in the setting of FT4 concentrations that are repeatedly found within the reference interval.49 Other conditions wherein TSH is elevated but FT4 is normal encompass recent reinstitution of thyroid hormone replacement therapy (FT4 returns to normal before TSH declines), poor compliance with treatment in primary hypothyroidism, recovery from nonthyroidal illness, positively interfering heterophilic antibodies [e.g., human antimouse antibodies (HAMA)] in double-antibody immunoassays, and thyroid hormone resistance. Controversy is ongoing over the appropriate upper limit of the reference interval for TSH. Basing the TSH reference interval on the central 95% of the general, healthy population results in an upper limit of normal between 4.0 and 5.0 mIU/L for many TSH assays. If patients with thyroid autoantibodies such as TPOA, a family history of thyroid disease, or an abnormal thyroid gland ultrasound are excluded from the reference population, the upper limit of the reference interval declines to approximately 2.5 to 3.0 mIU/L. Therefore, an upper limit of 2.5 to 3.0 mIU/L for TSH may be a truer definition of normal, because the reference population mostly excludes individuals with endogenous thyroid disease or propensity to develop thyroid disease. However, it is unclear how patients with TSH concentrations between 2.5/3.0 mIU/L and 4.0/5.0 mIU/L should be managed. It has been argued that lowering the upper limit of the reference interval will provide no clear patient benefit and inevitably will lead to confusion and possible overtreatment of subclinical hypothyroidism. Overtreatment carries the risk of predisposing the patient to osteoporosis and atrial fibrillation. As noted previously, the value of treatment when TSH is between 4.0/5.0 and 9.9 mIU/L is currently debated. Another component of the TSH reference interval debate is that according to National Health and Nutrition Examination Survey (NHANES) data,9,134A,209 TSH rises as populations age. For example, in healthy adults age 80 and older, the upper limit of the reference interval for TSH is 7.5 mIU/L.
The Thyroid
Pathophysiology and Thyroid Function Testing
Thyroid Gland: Structural and Functional Ontogeny
Thyroid Gland Anatomy
Biochemistry
Amino Acids
kDa
TRα1
410
47
TRα2
490*
55
TRβ1
461
53
TRβ2
514†
58
Physiology
Hypothalamic Physiology
Anterior Pituitary Thyrotroph Physiology
Thyroid Follicular Cell Physiology
Thyroid Hormones in The Circulation
Radiographic Thyroid Testing
Perchlorate Discharge
Clinical Conditions
Hypothyroidism
Primary Hypothyroidism
Autoimmune Hypothyroidism
Inborn Errors in Thyroid Hormone Biosynthesis
Developmental Hypothyroidism Disorders
Hypothyroidism Caused by Iodine Deficiency or Excess
Drug-Induced Hypothyroidism

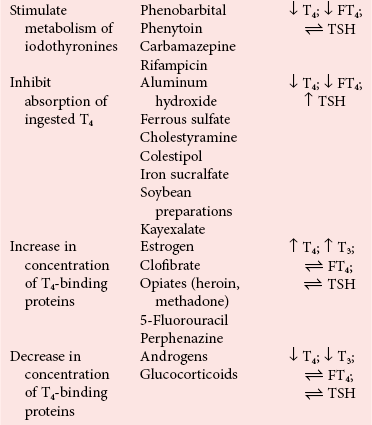
 , no change.
, no change.
Surgical and Radiation-Induced Hypothyroidism
Viral or Bacterial Thyroiditis
Central Hypothyroidism
Subclinical Hypothyroidism
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Basicmedical Key
Fastest Basicmedical Insight Engine