84 | The Practice of Genetics in Clinical Medicine |
APPLICATIONS OF MOLECULAR GENETICS IN CLINICAL MEDICINE
Genetic testing for inherited abnormalities associated with disease risk is increasingly used in the practice of clinical medicine. Germline alterations include chromosomal abnormalities (Chap. 83e), specific gene mutations with autosomal dominant or recessive patterns of transmission (Chap. 82), and single nucleotide polymorphisms with small relative risks associated with disease. Germline alterations are responsible for disorders beyond classic Mendelian conditions with genetic susceptibility to common adult-onset diseases such as asthma, hypertension, diabetes mellitus, macular degeneration, and many forms of cancer. For many of these diseases, there is a complex interplay of genes (often multiple) and environmental factors that affect lifetime risk, age of onset, disease severity, and treatment options.
The expansion of knowledge related to genetics is changing our understanding of pathophysiology and influencing our classification of diseases. Awareness of genetic etiology can have an impact on clinical management, including prevention and screening for or treatment of a range of diseases. Primary care physicians are relied upon to help patients navigate testing and treatment options. Consequently, they must understand the genetic basis for a large number of genetically influenced diseases, incorporate personal and family history to determine the risk for a specific mutation, and be positioned to provide counseling. Even if patients are seen by genetic specialists who assess genetic risk and coordinate testing, primary care providers should provide information to their patients regarding the indications, limitations, risks, and benefits of genetic counseling and testing. They must also be prepared to offer risk-based management following genetic risk assessment. Given the pace of genetics, this is an increasingly difficult task. The field of clinical genetics is rapidly moving from single gene testing to multigene panel testing, with techniques such as whole-exome and -genome sequencing on the horizon, increasing the complexity of test selection and interpretation, as well as patient education and medical decision making.
COMMON ADULT-ONSET GENETIC DISORDERS
INHERITANCE PATTERNS
Adult-onset hereditary diseases follow multiple patterns of inheritance. Some are autosomal dominant conditions. These include many common cancer susceptibility syndromes such as hereditary breast and ovarian cancer (due to germline BRCA1 and BRCA2 mutations) and Lynch syndrome (caused by germline mutations in the mismatch repair genes MLH1, MSH2, MSH6, and PMS2). In both of these examples, inherited mutations are associated with a high penetrance (lifetime risk) of cancer, although risk is not 100%. In other conditions, although there is autosomal dominant transmission, there is lower penetrance, thereby making the disorders more difficult to recognize. For example, germline mutations in CHEK2 increase the risk of breast cancer, but with a moderate lifetime risk in the range of 20–40%, as opposed to 50–70% for mutations in BRCA1 or BRCA2. Other adult-onset hereditary diseases are transmitted in an autosomal recessive fashion where two mutant alleles are necessary to cause disease. Examples include hemochromatosis and MYH-associated colon cancer. There are more pediatric-onset autosomal recessive disorders, such as lysosomal storage diseases and cystic fibrosis.
The genetic risk for many adult-onset disorders is multifactorial. Risk can be conferred by genetic factors at a number of loci, which individually have very small effects (usually with relative risks of <1.5). These risk loci (generally single nucleotide polymorphisms [SNPs]) combine with other genes and environmental factors in ways that are not well understood. SNP panels are available to assess risk of disease, but the optimal way of using this information in the clinical setting remains uncertain.
Many diseases have multiple patterns of inheritance, adding to the complexity of evaluating patients and families for these conditions. For example, colon cancer can be associated with a single germline mutation in a mismatch repair gene (Lynch syndrome, autosomal dominant), biallelic mutations in MYH (autosomal recessive), or multiple SNPs (polygenic). Many more individuals will have SNP risk alleles than germline mutations in high-penetrance genes, but cumulative lifetime risk of colon cancer related to the former is modest, whereas the risk related to the latter is significant. Personal and family histories provide important insights into the possible mode of inheritance.
FAMILY HISTORY
When two or more first-degree relatives are affected with asthma, cardiovascular disease, type 2 diabetes, breast cancer, colon cancer, or melanoma, the relative risk for disease among close relatives ranges from two- to fivefold, underscoring the importance of family history for these prevalent disorders. In most situations, the key to assessing the inherited risk for common adult-onset diseases is the collection and interpretation of a detailed personal and family medical history in conjunction with a directed physical examination.
Family history should be recorded in the form of a pedigree. Pedigrees should convey health-related data on first- and second-degree relatives. When such pedigrees suggest inherited disease, they should be expanded to include additional family members. The determination of risk for an asymptomatic individual will vary depending on the size of the pedigree, the number of unaffected relatives, the types of diagnoses, and the ages of disease onset. For example, a woman with two first-degree relatives with breast cancer is at greater risk for a specific Mendelian disorder if she has a total of 3 female first-degree relatives (with only 1 unaffected) than if she has a total of 10 female first-degree relatives (with 7 unaffected). Factors such as adoption and limited family structure (few women in a family) should to be taken into consideration in the interpretation of a pedigree. Additional considerations include young age of disease onset (e.g., a 30-year nonsmoking woman with a myocardial infarction), unusual diseases (e.g., male breast cancer or medullary thyroid cancer), and the finding of multiple potentially related diseases in an individual (e.g., a woman with a history of both colon and endometrial cancer). Some adult-onset diseases are more prevalent in certain ethnic groups. For instance, 2.5% of individuals of Ashkenazi Jewish ancestry carry one of three founder mutation in BRCA1 and BRCA2. Factor V Leiden mutations are much more common in Caucasians than in Africans or Asians.
Additional variables that should be documented are nonhereditary risk factors among those with disease (such as cigarette smoking and myocardial infarction; asbestos exposure and lung disease; and mantle radiation and breast cancer). Significant associated environmental exposures or lifestyle factors decrease the likelihood of a specific genetic disorder. In contrast, the absence of nonhereditary risk factors typically associated with a disease raises concern about a genetic association. A personal or family history of deep vein thrombosis in the absence of known environmental or medical risk factors suggests a hereditary thrombotic disorder. The physical examination may also provide important clues about the risk for a specific inherited disorder. A patient presenting with xanthomas at a young age should prompt consideration of familial hypercholesterolemia. The presence of trichilemmomas in a woman with breast cancer raises concern for Cowden syndrome, associated with PTEN mutations.
Recall of family history is often inaccurate. This is especially so when the history is remote and families lose contact or separate geographically. It can be helpful to ask patients to fill out family history forms before or after their visits, because this provides them with an opportunity to contact relatives. Ideally, this information should be embedded in electronic health records and updated intermittently. Attempts should be made to confirm the illnesses reported in the family history before making important and, in certain circumstances, irreversible management decisions. This process is often labor intensive and ideally involves interviews of additional family members or reviewing medical records, autopsy reports, and death certificates.
Although many inherited disorders will be suggested by the clustering of relatives with the same or related conditions, it is important to note that disease penetrance is incomplete for most genetic disorders. As a result, the pedigree obtained in such families may not exhibit a clear Mendelian inheritance pattern, because not all family members carrying the disease-associated alleles will manifest clinical evidence of the condition. Furthermore, genes associated with some of these disorders often exhibit variable disease expression. For example, the breast cancer–associated gene BRCA2 can predispose to several different malignancies in the same family, including cancers of the breast, ovary, pancreas, skin, and prostate. For common diseases such as breast cancer, some family members without the susceptibility allele (or genotype) may develop breast cancer (or phenotype) sporadically. Such phenocopies represent another confounding variable in the pedigree analysis.
Some of the aforementioned features of the family history are illustrated in Fig. 84-1. In this example, the proband, a 36-year-old woman (IV-1), has a strong history of breast and ovarian cancer on the paternal side of her family. The early age of onset and the co-occurrence of breast and ovarian cancer in this family suggest the possibility of an inherited mutation in BRCA1 or BRCA2. It is unclear however, without genetic testing, whether her father harbors such a mutation and transmitted it to her. After appropriate genetic counseling of the proband and her family, the most informative and cost-effective approach to DNA analysis in this family is to test the cancer-affected 42-year-old living cousin for the presence of a BRCA1 or BRCA2 mutation. If a mutation is found, then it is possible to test for this particular alteration in other family members, if they so desire. In the example shown, if the proband’s father has a BRCA1 mutation, there is a 50:50 probability that the mutation was transmitted to her, and genetic testing can be used to establish the absence or presence of this alteration. In this same example, if a mutation is not detected in the cancer-affected cousin, testing would not be indicated for cancer-unaffected relatives.
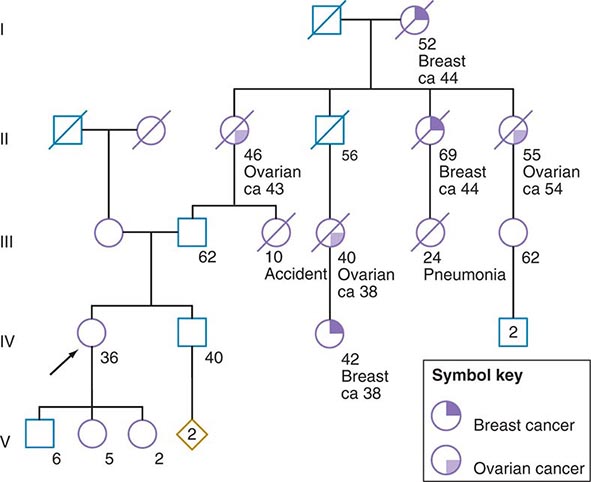
FIGURE 84-1 A 36-year-old woman (arrow) seeks consultation because of her family history of cancer. The patient expresses concern that the multiple cancers in her relatives imply an inherited predisposition to develop cancer. The family history is recorded, and records of the patient’s relatives confirm the reported diagnoses.
GENETIC TESTING FOR ADULT-ONSET DISORDERS
A critical first step before initiating genetic testing is to ensure that the correct clinical diagnosis has been made, whether it is based on family history, characteristic physical findings, pathology, or biochemical testing. Such careful clinical assessment can define the phenotype. In the traditional model of genetic testing, testing is directed initially toward the most probable genes (determined by the phenotype), which prevents unnecessary testing. Many disorders exhibit the feature of locus heterogeneity, which refers to the fact that mutations in different genes can cause phenotypically similar disorders. For example, osteogenesis imperfecta (Chap. 427), long QT syndrome (Chap. 277), muscular dystrophy (Chap. 462e), and hereditary predisposition to breast (Chap. 108) or colon (Chap. 110) cancer can each be caused by mutations in a number of distinct genes. The patterns of disease transmission, disease risk, clinical course, and treatment may differ significantly depending on the specific gene affected. Historically, the choice of which gene to test has been determined by unique clinical and family history features and the relative prevalence of candidate genetic disorders. However, rapid changes in genetic testing techniques, as discussed below, may impact this paradigm. It is now technically and financially feasible to sequence many genes (or even the whole exome) at one time. The incorporation of multiplex testing for germline mutations is rapidly evolving.
METHODOLOGIC APPROACHES TO GENETIC TESTING
Genetic testing is regulated and performed in much the same way as other specialized laboratory tests. In the United States, genetic testing laboratories are Clinical Laboratory Improvement Amendments (CLIA) approved to ensure that they meet quality and proficiency standards. A useful information source for various genetic tests is www.genetests.org. It should be noted that many tests need to be ordered through specialized laboratories.
Genetic testing is performed largely by DNA sequence analysis for mutations, although genotype can also be deduced through the study of RNA or protein (e.g., apolipoprotein E, hemoglobin S, and immunohistochemistry). For example, universal screening for Lynch syndrome via immunohistochemical analysis of colorectal cancers for absence of expression of mismatch repair proteins is under way at multiple hospitals throughout the United States. The determination of DNA sequence alterations relies heavily on the use of polymerase chain reaction (PCR), which allows rapid amplification and analysis of the gene of interest. In addition, PCR enables genetic testing on minimal amounts of DNA extracted from a wide range of tissue sources including leukocytes, mucosal epithelial cells (obtained via saliva or buccal swabs), and archival tissues. Amplified DNA can be analyzed directly by DNA sequencing, or it can be hybridized to DNA chips or blots to detect the presence of normal and altered DNA sequences. Direct DNA sequencing is frequently used for determination of hereditary disease susceptibility and prenatal diagnosis. Analyses of large alterations of the genome are possible using cytogenetics, fluorescent in situ hybridization (FISH), Southern blotting, or multiplex ligation-dependent probe amplification (MLPA) (Chap. 83e).
Massively parallel sequencing (also called next-generation sequencing) is significantly altering the approach to genetic testing for adult-onset hereditary susceptibility disorder. This technology encompasses several high-throughput approaches to DNA sequencing, all of which can reliably sequence many genes at one time. Technically, this involves the use of amplified DNA templates in a flow cell, a very different process than traditional Sanger sequencing which is time-consuming and expensive.
Multiplex panels for inherited susceptibility are commercially available and include testing of a number of genes that have been associated with the condition of interest. For example, panels are available for Brugada syndrome, hypertrophic cardiomyopathy, and Charcot-Marie-Tooth neuropathy. For many syndromes, this type of panel testing may make sense. However, in other situations, the utility of panel testing is less certain. Currently available breast cancer susceptibility panels contain six genes or more. Many of the genes included in the larger panels are associated with only a modest risk of breast cancer, and the clinical application is uncertain. An additional problem of sequencing many genes (rather than the genes for which there is most suspicion) is the identification of one or more variants of uncertain significance (VUS), discussed below.
Whole-exome sequencing (WES) is also now commercially available, although largely used in individuals with syndromes unexplained by traditional genetic testing. As cost declines, WES may be more widely used. Whole-genome sequencing is also commercially available. Although it may be quite feasible to sequence the entire genome, there are many issues in doing so, including the daunting task of analyzing the vast amount of data generated. Other issues include: (1) the optimal way in which to obtain informed consent, (2) interpretation of frequent sequence variation of uncertain significance, (3) interpretation of alterations in genes with unclear relevance to specific human pathology, and (4) management of unexpected but clinically significant genetic findings.
Testing strategies are evolving as a result of these new genetic testing platforms. As the cost of multiple gene panels and WES continue to fall, and as interpretation of such test results improve, there may be a shift from sequential single-gene (or a few genes) testing to multigene testing. For example, presently, a 30-year-old woman with breast cancer but no family history of cancer and no syndromic features would undergo BRCA1/2 testing. If negative, she would subsequently be offered TP53 testing. Notably, a reasonable number of individuals offered TP53 testing for Li-Fraumeni syndrome decline because mutations are associated with extremely high cancer risks (including childhood cancers) in multiple organs and there are no proven interventions to mitigate risk. Without features consistent with Cowden syndrome, the woman would not be routinely offered PTEN testing or testing for CHEK2, ATM, BRIP, BARD, NBN, and PALB2. However, it is now possible to synchronously analyze all of the aforementioned genes, for a nominally higher cost than BRCA1/2 testing alone. Concerns about such panels include appropriate consent strategies related to unexpected findings, VUS, and unclear clinical utility of testing moderate-penetrance genes. Thus, changes from the traditional model of single-gene genetic testing should be done with caution (Fig. 84-2).
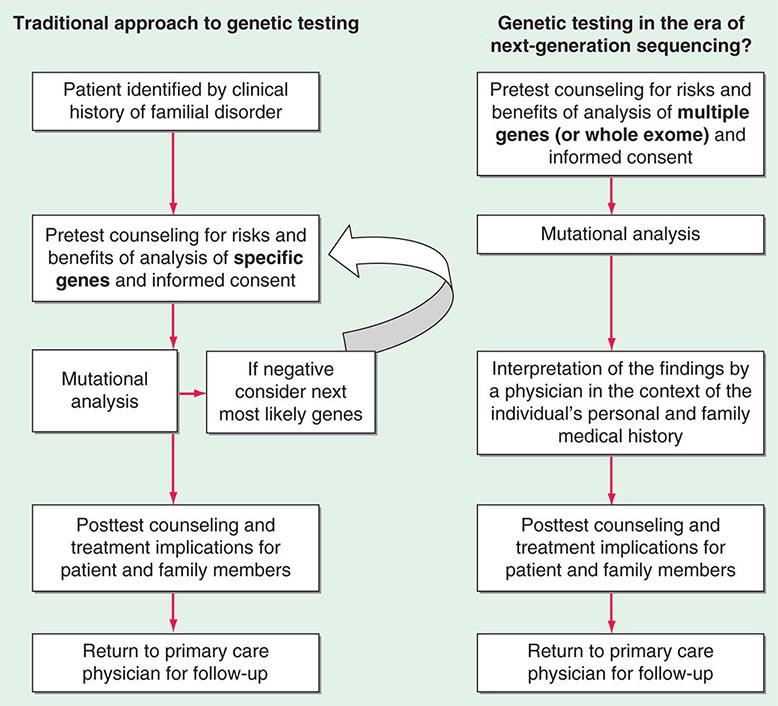
FIGURE 84-2 Approach to genetic testing.
Limitations to the accuracy and interpretation of genetic testing exist. In addition to technical errors, genetics tests are sometimes designed to detect only the most common mutations. In addition, genetic testing has evolved over time. For example, it was not possible to obtain commercially available comprehensive large genomic rearrangement testing for BRCA1 and BRCA2 until 2006. Therefore, a negative result must be qualified by the possibility that the individual may have a mutation that was not included in the test. In addition, a negative result does not mean that there is not a mutation in some other gene that causes a similar inherited disorder. A negative result, unless there is known mutation in the family, is typically classified as uninformative.
VUS are another limitation to genetic testing. A VUS (also termed unclassified variant) is a sequence variation in a gene where the effect of the alteration on the function of the protein is not known. Many of these variants are single nucleotide substitutions (also called missense mutations) that result in a single amino acid change. Although many VUSs will ultimately be reclassified as benign polymorphisms, some will prove to be functionally important. As more genes are sequenced (for example, in a multiplex panel or through WES), the percentage of individuals found to have a VUS increases significantly. The finding of a VUS is difficult for patients and providers alike and complicates decisions regarding medical management.
Clinical utility is an important consideration because genetic testing for susceptibility to chronic diseases is increasingly integrated into the practice of medicine. In some situations, there is clear clinical utility to genetic testing with significant evidence-based changes in medical management decisions based on results. However, in many cases, the discovery of disease-associated genes has outpaced studies that assess how such information should be used in the clinical management of the patient and family. This is particularly true for moderate- and low-penetrance gene mutations. Therefore, predictive genetic testing should be approached with caution and only offered to patients who have been adequately counseled and have provided informed consent.
Predictive genetic testing falls into two distinct categories. Presymptomatic testing applies to diseases where a specific genetic alteration is associated with a near 100% likelihood of developing disease. In contrast, predisposition testing predicts a risk for disease that is less than 100%. For example, presymptomatic testing is available for those at risk for Huntington’s disease; whereas, predisposition testing is considered for those at risk for hereditary colon cancer. It is important to note that for the majority of adult-onset disorders, testing is only predictive. Test results cannot reveal with confidence whether, when, or how the disease will manifest itself. For example, not everyone with the apolipoprotein E4 allele will develop Alzheimer’s disease, and individuals without this genetic marker can still develop the disorder.
The optimal testing strategy for a family is to initiate testing in an affected family member first. Identification of a mutation can direct the testing of other at-risk family members (whether symptomatic or not). In the absence of additional familial or environmental risk factors, individuals who test negative for the mutation found in the affected family member can be informed that they are at general population risk for that particular disease. Furthermore, they can be reassured that they are not at risk for passing the mutation on to their children. On the other hand, asymptomatic family members who test positive for the known mutation must be informed that they are at increased risk for disease development and for transmitting the alteration to their children.
Pretest counseling and education are important, as is an assessment of the patient’s ability to understand and cope with test results. Genetic testing has implications for entire families, and thus individuals interested in pursuing genetic testing must consider how test results might impact their relationships with relatives, partners, spouses, and children. In families with a known genetic mutation, those who test positive must consider the impact of their carrier status on their present and future lifestyles; those who test negative may manifest survivor guilt. Parents who are found to have a disease-associated mutation often express considerable anxiety and despair as they address the issue of risk to their children. In addition, some individuals consider options such as preimplantation genetic diagnosis in their reproductive decision making.
When a condition does not manifest until adulthood, clinicians and parents are faced with the question of whether at-risk children should be offered genetic testing and, if so, at what age. Although the matter is debated, several professional organizations have cautioned that genetic testing for adult-onset disorders should not be offered to children. Many of these conditions have no known interventions in childhood to prevent disease; consequently, such information can pose significant psychosocial risk to the child. In addition, there is concern that testing during childhood violates a child’s right to make an informed decision regarding testing upon reaching adulthood. On the other hand, testing should be offered in childhood for disorders that may manifest early in life, especially when management options are available. For example, children with multiple endocrine neoplasia 2 (MEN 2) may develop medullary thyroid cancer early in life and should be considered for prophylactic thyroidectomy (Chap. 408). Similarly, children with familial adenomatous polyposis (FAP) due to a mutation in APC may develop polyps in their teens with progression to invasive cancer in the twenties, and therefore, colonoscopy screening is started between the ages of 10 and 15 years (Chap. 110).
INFORMED CONSENT
Informed consent for genetic testing begins with education and counseling. The patient should understand the risks, benefits, and limitations of genetic testing, as well as the potential implications of test results. Informed consent should include a written document, drafted clearly and concisely in a language and format that is understandable to the patient. Because molecular genetic testing of an asymptomatic individual often allows prediction of future risk, the patient should understand all potential long-term medical, psychological, and social implications of testing. There have long been concerns about the potential for genetic discrimination. The Genetic Information Nondiscrimination Act (GINA) was passed in 2008 and provides some protections related to job and health insurance discrimination. It is important to explore with patients the potential impact of genetic test results on future health as well as disability and life insurance coverage. Patients should understand that alternatives remain available if they decide not to pursue genetic testing, including the option of delaying testing to a later date. The option of DNA banking should be presented so that samples are readily available for future use by family members, if needed.
FOLLOW-UP CARE AFTER TESTING
Depending on the nature of the genetic disorder, posttest interventions may include: (1) cautious surveillance and awareness; (2) specific medical interventions such as enhanced screening, chemoprevention, or risk-reducing surgery; (3) risk avoidance; and (4) referral to support services. For example, patients with known deleterious mutations in BRCA1 or BRCA2 are strongly encouraged to undergo risk-reducing salpingo-oophorectomy and are offered intensive breast cancer screening as well as the option of risk-reducing mastectomy. In addition, such women may wish to take chemoprevention with tamoxifen, raloxifene, or exemestane. Those with more limited medical management and prevention options, such as patients with Huntington’s disease, should be offered continued follow-up and supportive services, including physical and occupational therapy and social services or support groups as indicated. Specific interventions will change as research continues to enhance our understanding of the medical management of these genetic conditions and more is learned about the functions of the gene products involved.
Individuals who test negative for a mutation in a disease-associated gene identified in an affected family member must be reminded that they may still be at risk for the disease. This is of particular importance for common diseases such as diabetes mellitus, cancer, and coronary artery disease. For example, a woman who finds that she does not carry the disease-associated mutation in BRCA2 previously discovered in the family should be reminded that she still requires the same breast cancer screening recommended for the general population.
GENETIC COUNSELING AND EDUCATION
Genetic counseling should be distinguished from genetic testing and screening, although genetic counselors are often involved in issues related to testing. Genetic counseling refers to a communication process that deals with human problems associated with the occurrence of risk of a genetic disorder in a family. Genetic risk assessment is complex and often involves elements of uncertainty. Counseling, therefore, includes genetic education as well as psychosocial counseling. Genetic counseling can be useful in a wide range of situations (Table 84-1). The role of the genetic counselor includes the following:
1. Gather and document a detailed family history.
2. Educate patients about general genetic principles related to disease risk, both for themselves and for others in the family.
3. Assess and enhance the patient’s ability to cope with the genetic information offered.
4. Discuss how nongenetic factors may relate to the ultimate expression of disease.
5. Address medical management issues.
6. Assist in determining the role of genetic testing for the individual and the family.
7. Ensure the patient is aware of the indications, process, risks, benefits, and limitations of the various genetic testing options.
8. Assist the patient, family, and referring physician in the interpretation of the test results.
9. Refer the patient and other at-risk family members for additional medical and support services, if necessary.
INDICATIONS FOR GENETIC COUNSELING |
Genetic counseling is generally offered in a nondirective manner, wherein patients learn to understand how their values factor into a particular medical decision. Nondirective counseling is particularly appropriate when there are no data demonstrating a clear benefit associated with a particular intervention or when an intervention is considered experimental. For example, nondirective genetic counseling is used when a person is deciding whether to undergo genetic testing for Huntington’s disease. At this time, there is no clear benefit (in terms of medical outcome) to an at-risk individual undergoing genetic testing for this disease because its course cannot be altered by therapeutic interventions. However, testing can have an important impact on the individual’s perception of advanced care planning and his or her interpersonal relationships and plans for childbearing. Therefore, the decision to pursue testing rests on the individual’s belief system and values. On the other hand, a more directive approach is appropriate when a condition can be treated. In a family with FAP, colon cancer screening and prophylactic colectomy should be recommended for known APC mutation carriers. The counselor and clinician following this family must ensure that the at-risk family members have access to the resources necessary to adhere to these recommendations.
Genetic education is central to an individual’s ability to make an informed decision regarding testing options and treatment. An adequate knowledge of patterns of inheritance will allow patients to understand the probability of disease risk for themselves and other family members. It is also important to impart the concepts of disease penetrance and expression. For most complex adult-onset genetic disorders, asymptomatic patients should be advised that a positive test result does not always translate into future disease development. In addition, the role of nongenetic factors, such as environmental exposures and lifestyle, must be discussed in the context of multifactorial disease risk and disease prevention. Finally, patients should understand the natural history of the disease as well as the potential options for intervention, including screening, prevention, and in certain circumstances, pharmacologic treatment or prophylactic surgery.
THERAPEUTIC INTERVENTIONS BASED ON GENETIC RISK FOR DISEASE
Specific treatments are available for a number of genetic disorders. Strategies for the development of therapeutic interventions have a long history in childhood metabolic diseases; however, these principles have been applied in the diagnosis and management of adult-onset diseases as well (Table 84-2). Hereditary hemochromatosis is usually caused by mutations in HFE (although other genes have been less commonly associated) and manifests as a syndrome of iron overload, which can lead to liver disease, skin pigmentation, diabetes mellitus, arthropathy, impotence in males, and cardiac issues (Chap. 428). When identified early, the disorder can be managed effectively with therapeutic phlebotomy. Therefore, when the diagnosis of hemochromatosis has been made in a proband, it is important to counsel and offer testing to other family members in order to minimize the impact of the disorder.
EXAMPLE OF GENETIC TESTING AND POSSIBLE INTERVENTIONS |
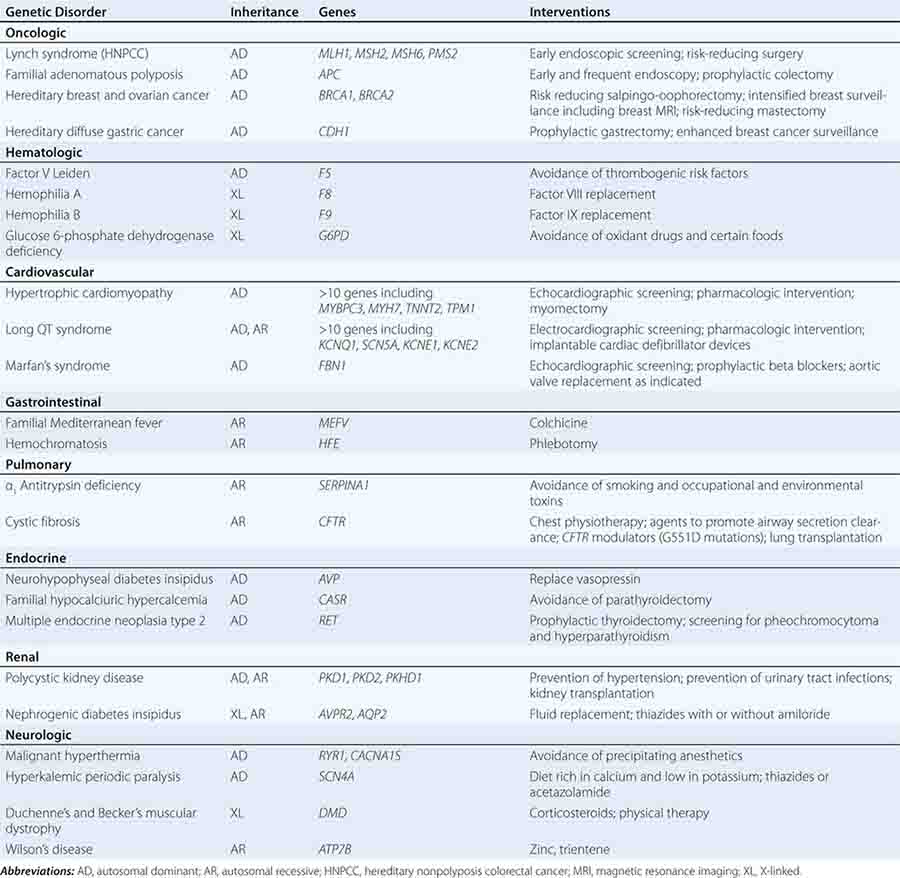
Preventative measures and therapeutic interventions are not restricted to metabolic disorders. Identification of familial forms of long QT syndrome, associated with ventricular arrhythmias, allows early electrocardiographic testing and the use of prophylactic antiarrhythmic therapy, overdrive pacemakers, or defibrillators. Individuals with familial hypertrophic cardiomyopathy can be screened by ultrasound, treated with beta blockers or other drugs, and counseled about the importance of avoiding strenuous exercise and dehydration. Those with Marfan’s syndrome can be treated with beta blockers or angiotensin II receptor blockers and monitored for the development of aortic aneurysms.
The field of pharmacogenetics identifies genes that alter drug metabolism or confer susceptibility to toxic drug reactions. Pharmacogenetics seeks to individualize drug therapy in an attempt to improve treatment outcomes and reduce toxicity. Examples include thiopurine methyltransferase (TPMT) deficiency, dihydropyrimidine dehydrogenase deficiency, malignant hyperthermia, and glucose-6-phosphate deficiency. Despite successes in this area, it is not always clear how to incorporate pharmacogenetics into clinical care. For example, although there is an association with CYP2C6 and VKORC1 genotypes and warfarin dosing, there is no evidence that incorporating genotyping into clinical practice improves patient outcomes.
The identification of germline abnormalities that increase the risk of specific types of cancer is rapidly changing clinical management. Identifying family members with mutations that predispose to FAP or Lynch syndrome leads to recommendations of early cancer screening and prophylactic surgery, as well as consideration of chemoprevention and attention to healthy lifestyle habits. Similar principles apply to familial forms of melanoma as well as cancers of the breast, ovary, and thyroid. In addition to increased screening and prophylactic surgery, the identification of germline mutations associated with cancer may also lead to the development of targeted therapeutics, for example, the ongoing development of PARP inhibitors in those with BRCA-associated cancers.
Although the role of genetic testing in the clinical setting continues to evolve, such testing holds the promise of allowing early and more targeted interventions that can reduce morbidity and mortality. Rapid technologic advances are changing the ways in which genetic testing is performed. As genetic testing becomes less expensive and technically easier to perform, it is anticipated that there will be an expansion of its use. This will present challenges, but also opportunities. It is critical that physicians and other health care professionals keep current with advances in genetic medicine in order to facilitate appropriate referral for genetic counseling and judicious use of genetic testing, as well as to provide state-of-the-art, evidence-based care for affected or at-risk patients and their relatives.
85e | Mitochondrial DNA and Heritable Traits and Diseases |
Mitochondria are cytoplasmic organelles whose major function is to generate ATP by the process of oxidative phosphorylation under aerobic conditions. This process is mediated by the respiratory electron transport chain (ETC) multiprotein enzyme complexes I–V and the two electron carriers, coenzyme Q (CoQ) and cytochrome c. Other cellular processes to which mitochondria make a major contribution include apoptosis (programmed cell death) and additional cell type–specific functions (Table 85e-1). The efficiency of the mitochondrial ETC in ATP production is a major determinant of overall body energy balance and thermogenesis. In addition, mitochondria are the predominant source of reactive oxygen species (ROS), whose rate of production also relates to the coupling of ATP production to oxygen consumption. Given the centrality of oxidative phosphorylation to the normal activities of almost all cells, it is not surprising that mitochondrial dysfunction can affect almost any organ system (Fig. 85e-1). Thus, physicians in many disciplines might encounter patients with mitochondrial diseases and should be aware of their existence and characteristics.
FUNCTIONS OF MITOCHONDRIA |
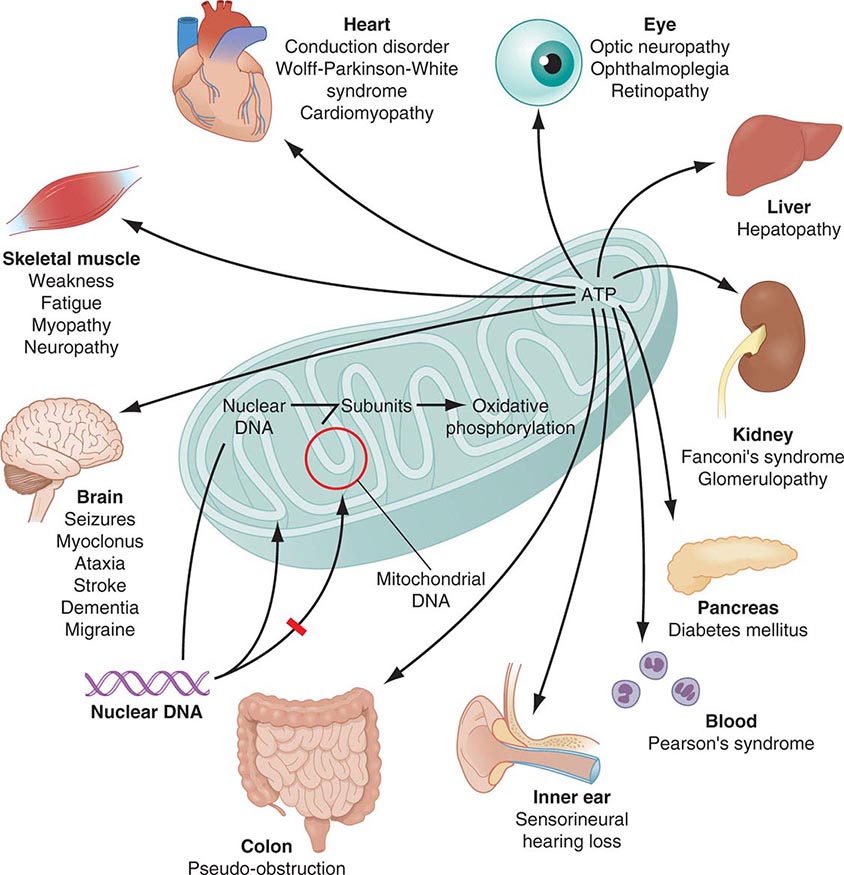
FIGURE 85e-1 Dual genetic control and multiple organ system manifestations of mitochondrial disease. (Reproduced with permission from DR Johns: Mitochondrial DNA and disease. N Engl J Med 333:638, 1995.)
The integrated activity of an estimated 1500 gene products is required for normal mitochondrial biogenesis, function, and integrity. Almost all of these are encoded by nuclear genes and thus follow the rules and patterns of nuclear genomic inheritance (Chap. 84). These nuclear-encoded proteins are synthesized in the cell cytoplasm and imported to their location of activity within the mitochondria through a complex biochemical process. In addition, the mitochondria contain their own small genome consisting of numerous copies (polyploidy) per mitochondrion of a circular, double-strand mitochondrial DNA (mtDNA) molecule comprising 16,569 nucleotides. This mtDNA sequence (also known as the “mitogenome”) might represent the remnants of endosymbiotic prokaryotes from which mitochondria are thought to have originated. The mtDNA sequence contains a total of 37 genes, of which 13 encode mitochondrial protein components of the ETC (Fig. 85e-2). The remaining 22 tRNA- and 2 rRNA-encoding genes are dedicated to the process of translating the 13 mtDNA-encoded proteins. This dual nuclear and mitochondrial genetic control of mitochondrial function results in unique and diagnostically challenging patterns of inheritance. The current chapter focuses on heritable traits and diseases related to the mtDNA component of the dual genetic control of mitochondrial function. The reader is referred to Chaps. 84 and 462e for consideration of mitochondrial disease originating from mutations in the nuclear genome. The latter include (1) disorders due to mutations in nuclear genes directly encoding structural components or assembly factors of the oxidative phosphorylation complexes, (2) disorders due to mutations in nuclear genes encoding proteins indirectly related to oxidative phosphorylation, and (3) mtDNA depletion syndromes (MDS) characterized by a reduction of mtDNA copy number in affected tissues without mutations or rearrangements in the mtDNA.
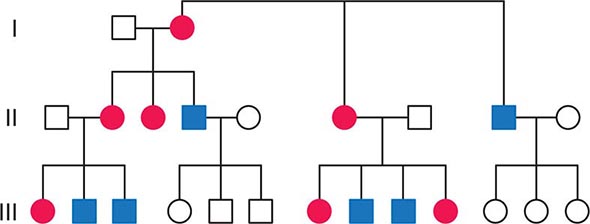
FIGURE 85e-2 Maternal inheritance of mitochondrial DNA (mtDNA) disorders and heritable traits. Affected women (filled circles) transmit the trait to their children. Affected men (filled squares) do not transmit the trait to any of their offspring.
MITOCHONDRIAL DNA STRUCTURE AND FUNCTION
As a result of its circular structure and extranuclear location, the replication and transcription mechanisms of mtDNA differ from the corresponding mechanisms in the nuclear genome, whose nucleosomal packaging and structure are more complex. Because each cell contains many copies of mtDNA, and because the number of mitochondria can vary during the lifetime of each cell, mtDNA copy number is not directly coordinated with the cell cycle. Thus, vast differences in mtDNA copy number are observed between different cell types and tissues and during the lifetime of a cell. Another important feature of the mtDNA replication process is a reduced stringency of proofreading and replication error correction, leading to a greater degree of sequence variation compared to the nuclear genome. Some of these sequence variants are silent polymorphisms that do not have the potential for a phenotypic or pathogenic effect, whereas others may be considered pathogenic mutations.
With respect to transcription, initiation can occur on both strands and proceeds through the production of an intronless polycistronic precursor RNA, which is then processed to produce the 13 individual mRNA and 24 individual tRNA and rRNA products. The 37 mtDNA genes comprise fully 93% of the 16,569 nucleotides of the mtDNA in what is known as the coding region. The control region consists of ~1.1 kilobases (kb) of noncoding DNA, which is thought to have an important role in replication and transcription initiation.
MATERNAL INHERITANCE AND LACK OF RECOMBINATION
In contrast to homologous pair recombination that takes place in the nucleus, mtDNA molecules do not undergo recombination, such that mutational events represent the only source of mtDNA genetic diversification. Moreover, with very rare exceptions, it is only the maternal DNA that is transmitted to the offspring. The fertilized oocyte degrades mtDNA carried from the sperm in a complex process involving the ubiquitin proteasome system. Thus, although mothers transmit their mtDNA to both their sons and daughters, only the daughters are able to transmit the inherited mtDNA to future generations. Accordingly, mtDNA sequence variation and associated phenotypic traits and diseases are inherited exclusively along maternal lines.
As noted below, because of the complex relationship between mtDNA mutations and disease expression, sometimes this maternal inheritance is difficult to recognize at the clinical or pedigree level. However, evidence of paternal transmission can almost certainly rule out an mtDNA genetic origin of phenotypic variation or disease; conversely, a disease affecting both sexes without evidence of paternal transmission strongly suggests a heritable mtDNA disorder (Fig. 85e-2).
MULTIPLE COPY NUMBER (POLYPLOIDY), HIGH MUTATION RATE, HETEROPLASMY, AND MITOTIC SEGREGATION
Each aerobic cell in the body has multiple mitochondria, often numbering many hundreds or more in cells with extensive energy production requirements. Furthermore, the number of copies of mtDNA within each mitochondrion varies from several to hundreds; this is true of both somatic as well as germ cells, including oocytes in females. In the case of somatic cells, this means that the impact of most newly acquired somatic mutations is likely to be very small in terms of total cellular or organ system function; however, because of the manyfold higher mutation rate during mtDNA replication, numerous different mutations may accumulate with aging of the organism. It has been proposed that the total cumulative burden of acquired somatic mtDNA mutations with age may result in an overall perturbation of mitochondrial function, contributing to age-related reduction in the efficiency of oxidative phosphorylation and increased production of damaging ROS. The accumulation of such acquired somatic mtDNA mutations with aging may contribute to age-related diseases, such as metabolic syndrome and diabetes, cancer, and neurodegenerative and cardiovascular disease in any given individual. However, somatic mutations are not carried forward to the next generation, and the hereditary impact of mtDNA mutagenesis requires separate consideration of events in the female germline.
The multiple mtDNA copy number within each cell, including the maternal germ cells, results in the phenomenon of heteroplasmy, in contrast to much greater uniformity (homoplasy) of somatic nuclear DNA sequence. Heteroplasmy for a given mtDNA sequence variant or mutation arises as a result of the coexistence within a cell, tissue, or individual of mtDNA molecules bearing more than one version of the sequence variant (Fig. 85e-3). The importance of the heteroplasmy phenomena to the understanding of mtDNA-related mitochondrial diseases is critical. The coexistence of mutant and nonmutant mtDNA and the variation of the mutant load among individuals from the same maternal sibship, and across organs and tissues within the same individual, play a pivotal role in the manifestation and severity of disease and are crucial to understanding the complexity of inheritance of mtDNA disorders. At the level of the oocyte, the percentage of mtDNA molecules bearing each version of the polymorphic sequence variant or mutation depends on stochastic events related to partitioning of mtDNA molecules during the process of oogenesis itself. Thus, oocytes differ from each other in the degree of heteroplasmy for that sequence variant or mutation. In turn, the heteroplasmic state is carried forward to the zygote and to the organism as a whole, to varying degrees, depending on mitotic segregation of mtDNA molecules during organ system development and maintenance. For this reason, in vitro fertilization, followed by preimplantation genetic diagnosis (PGD), is not as predictive of the genetic health of the offspring in the case of mtDNA mutations as in the case of the nuclear genome. Similarly, the impact of somatic mtDNA mutations acquired during development and subsequently also shows an enormous spectrum of variability.
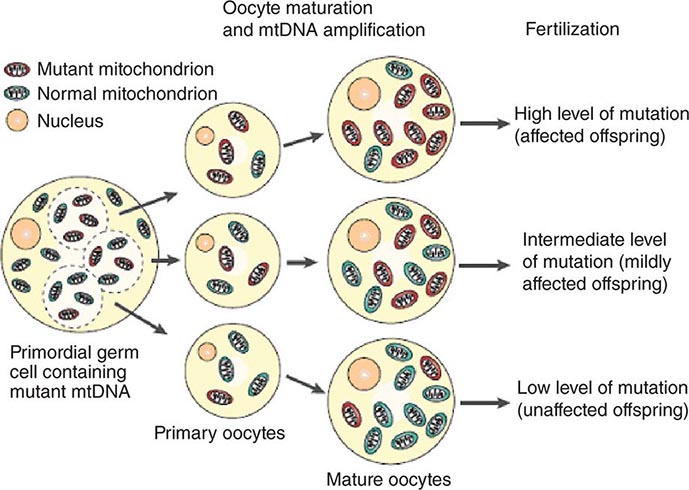
FIGURE 85e-3 Heteroplasmy and the mitochondrial genetic bottleneck. During the production of primary oocytes, a selected number of mitochondrial DNA (mtDNA) molecules are transferred into each oocyte. Oocyte maturation is associated with the rapid replication of this mtDNA population. This restriction-amplification event can lead to a random shift of mtDNA mutational load between generations and is responsible for the variable levels of mutated mtDNA observed in affected offspring from mothers with pathogenic mtDNA mutations. Mitochondria that contain mutated mtDNA are shown in red, and those with normal mtDNA are shown in green. (Reproduced with permission from R Taylor, D Turnbull: Mitochondrial DNA mutations in human disease. Nat Rev Genetics 6:389, 2005.)
Mitotic segregation refers to the unequal distribution of wild-type and mutant versions of mtDNA molecules during all cell divisions that occur during prenatal development and subsequently throughout the lifetime of an individual. The phenotypic effect or disease impact will, thus, be a function not only of the inherent disruptive effect (pathogenicity) on the mtDNA-encoded gene (coding region mutations) or integrity of the mtDNA molecule (control region mutations), but also of its distribution among the multiple copies of mtDNA in the various mitochondria, cells, and tissues of the affected individual. Thus, one consequence can be the generation of a bottleneck due to the marked decline in given sets of mtDNA variants, consequent to such mitotic segregation. Heterogeneity arises from differences in the degree of heteroplasmy among oocytes of the affected female, together with subsequent mitotic segregation of the pathogenic mutation during tissue and organ development, and throughout the lifetime of the individual offspring. The actual expression of disease might then depend on a threshold percentage of mitochondria whose function is disrupted by mtDNA mutations. This in turn confounds hereditary transmission patterns and hence genetic diagnosis of pathogenic heteroplasmic mutations. Generally, if the proportion of mutant mtDNA is less than 60%, the individual is unlikely to be affected, whereas proportions exceeding 90% cause clinical disease.
HOMOPLASMIC VARIANTS AND HUMAN MTDNA PHYLOGENY
In contrast to classic mtDNA diseases, most of which begin in childhood and are the result of heteroplasmic mutations as noted above, during the course of human evolution, certain mtDNA sequence variants have drifted to a state of homoplasmy, wherein all of the mtDNA molecules in the organism contain the new sequence variant. This arises due to a “bottleneck” effect followed by genetic drift during the very process of oogenesis itself (Fig. 85e-3). In other words, during certain stages of oogenesis, the mtDNA copy number becomes so substantially reduced that the particular mtDNA species bearing the novel or derived sequence variant may become the increasingly predominant, and eventually exclusive, version of the mtDNA for that particular nucleotide site. All of the offspring of a woman bearing an mtDNA sequence variant or mutation that has become homoplasmic will also be homoplasmic for that variant and will transmit the sequence variant forward in subsequent generations.
Considerations of reproductive fitness limit the evolutionary or population emergence of homoplasmic mutations that are lethal or cause severe disease in infancy or childhood. Thus, with a number of notable exceptions (e.g., mtDNA mutations causing Leber’s hereditary optic neuropathy; see below), most homoplasmic mutations are considered to be neutral markers of human evolution, which are useful and interesting in the population genetics analysis of shared maternal ancestry but which have little significance in human phenotypic variation or disease predisposition.
More importantly is the understanding that this accumulation of homoplastic mutations occurs at a genetic locus that is transmitted only through the female germline and that lacks recombination. In turn, this enables reconstruction of the sequential topology and radiating phylogeny of mutations accumulated through the course of human evolution since the time of the most recent common mtDNA ancestor of all contemporary mtDNA sequences, some 200,000 years ago. The term haplogroup is usually used to define major branching points in the human mtDNA phylogeny, nested one within the other, which often demonstrate striking continental geographic ancestral partitioning. At the level of the complete mtDNA sequence, the term haplotype is usually used to describe the sum of mutations observed for a given mtDNA sequence and as compared to a reference sequence, such that all haplotypes falling within a given haplogroup share the total sum of mutations that have accumulated since the most recent common ancestor and the bifurcation point they mark. The remaining observed variants are private to each haplotype. Consequentially, human mtDNA sequence is an almost perfect molecular prototype for a nonrecombining locus, and its variation has been extensively used in phylogenetic studies. Moreover, the mtDNA mutation rate is considerably higher than the rate observed for the nuclear genome, especially in the control region, which contains the displacement, or D-loop, in turn comprising two adjacent hypervariable regions (HVR-I and HVR-II). Together with the absence of recombination, this amplifies drift to high frequencies of novel haplotypes. As a result, mtDNA haplotypes are more highly partitioned across geographically defined populations than sequence variants in other parts of the genome. Despite extensive research, it has not been well established that such haplotype-based partitioning has a significant influence on human health conditions. However, mtDNA-based phylogenetic analysis can be used both as a quality assurance tool and as a filter in distinguishing neutral mtDNA variants comprising human mtDNA phylogeny from potentially deleterious mutations.
MITOCHONDRIAL DNA DISEASE
The true prevalence of mtDNA disease is difficult to estimate because of the phenotypic heterogeneity that occurs as a function of heteroplasmy, the challenge of detecting and assessing heteroplasmy in different affected tissues, and the other unique features of mtDNA function and inheritance described above. It is estimated that at least 1 in 200 healthy humans harbors a pathogenic mtDNA mutation with the potential to causes disease, but that heteroplasmic germline pathogenic mtDNA mutations actually affect up to approximately 1 in 8500 individuals.
The true disease burden relating to mtDNA sequence variation will only be known when the following capabilities become available: (1) ability to distinguish a completely neutral sequence variant from a true phenotype-modifying or pathogenic mutation, (2) accurate assessment of heteroplasmy that can be determined with fidelity, and (3) a systems biology approach (Chap. 87e) to determine the network of epistatic interactions of mtDNA sequence variations with mutations in the nuclear genome.
OVERVIEW OF CLINICAL AND PATHOLOGIC FEATURES OF HUMAN MTDNA DISEASE
Given the vital roles of mitochondria in all nucleated cells, it is not surprising that mtDNA mutations can affect numerous tissues with pleiotropic effects. More than 200 different disease-causing, mostly heteroplasmic mtDNA mutations have been described affecting ETC function. Figure 85e-4 provides a partial mtDNA map of some of the better characterized of these disorders. A number of clinical clues can increase the index of suspicion for a heteroplasmic mtDNA mutation as an etiology of a heritable trait or disease, including (1) familial clustering with absence of paternal transmission; (2) adherence to one of the classic syndromes (see below) or paradigmatic combinations of disease phenotypes involving several organ systems that normally do not fit together within a single nuclear genomic mutation category; (3) a complex of laboratory and pathologic abnormalities that reflect disruption in cellular energetics (e.g., lactic acidosis and neurodegenerative and myodegenerative symptoms with the finding of ragged red fibers, reflecting the accumulation of abnormal mitochondria under the muscle sarcolemmal membrane); or (4) a mosaic pattern reflecting a heteroplasmic state.
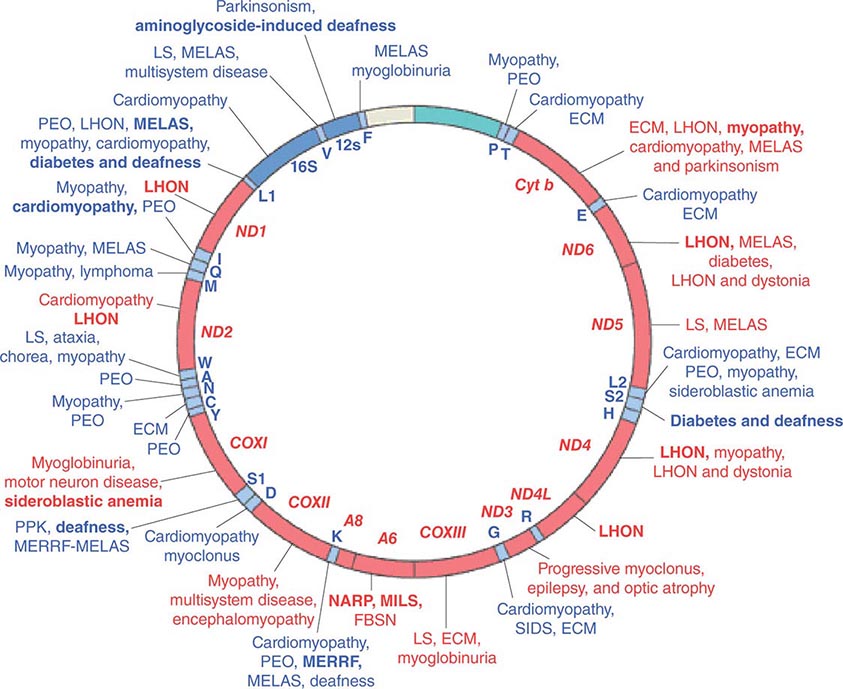
FIGURE 85e-4 Mutations in the human mitochondrial genome known to cause disease. Disorders that are frequently or prominently associated with mutations in a particular gene are shown in boldface. Diseases due to mutations that impair mitochondrial protein synthesis are shown in blue. Diseases due to mutations in protein-coding genes are shown in red. ECM, encephalomyopathy; FBSN, familial bilateral striatal necrosis; LHON, Leber’s hereditary optic neuropathy; LS, Leigh syndrome; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes; MERRF, myoclonic epilepsy with ragged red fibers; MILS, maternally inherited Leigh syndrome; NARP, neuropathy, ataxia, and retinitis pigmentosa; PEO, progressive external ophthalmoplegia; PPK, palmoplantar keratoderma; SIDS, sudden infant death syndrome. (Reproduced with permission from S DiMauro, E Schon: Mitochondrial respiratory-chain diseases. N Engl J Med 348:2656, 2003.)
Heteroplasmy can sometimes be elegantly demonstrated at the tissue level using histochemical staining for enzymes in the oxidative phosphorylation pathway, with a mosaic pattern indicating heterogeneity of the genotype for the coding region for the mtDNA-encoded enzyme. Complex II, CoQ, and cytochrome c are exclusively encoded by nuclear DNA. In contrast, complexes I, III, IV, and V contain at least some subunits encoded by mtDNA. Just 3 of the 13 subunits of the ETC complex IV enzyme, cytochrome c oxidase, are encoded by mtDNA, and, therefore, this enzyme has the lowest threshold for dysfunction when a threshold level of mutated mtDNA is reached. Histochemical staining for cytochrome c oxidase activity in tissues of patients affected with heteroplasmic inherited mtDNA mutations (or with the somatic accumulation of mtDNA mutations, see below) can show a mosaic pattern of reduced histochemical staining in comparison with histochemical staining for the complex II enzyme, succinate dehydrogenase (Fig. 85e-5). Heteroplasmy can also be detected at the genetic level through direct Sanger-type mtDNA genotyping under special conditions, although clinically significant low levels of heteroplasmy can escape detection in genomic samples extracted from whole blood using conventional genotyping and sequencing techniques.
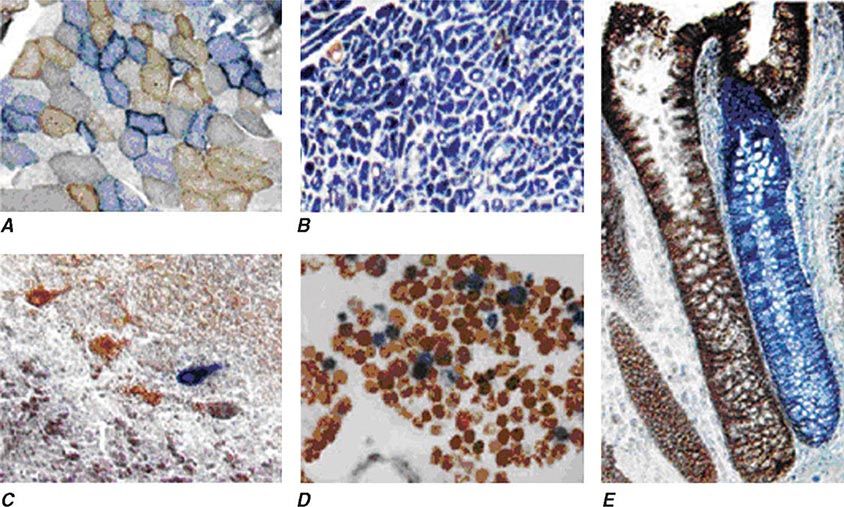
FIGURE 85e-5 Cytochrome c oxidase (COX) deficiency in mitochondrial DNA (mtDNA)–associated disease. Transverse tissue sections that have been stained for COX and succinate dehydrogenase (SDH) activities sequentially, with COX-positive cells shown in brown and COX-deficient cells shown in blue. A. Skeletal muscle from a patient with a heteroplasmic mitochondrial tRNA point mutation. The section shows a typical “mosaic” pattern of COX activity, with many muscle fibers harboring levels of mutated mtDNA that are above the crucial threshold to produce a functional enzyme complex. B. Cardiac tissue (left ventricle) from a patient with a homoplasmic tRNA mutation that causes hypertrophic cardiomyopathy, which demonstrates an absence of COX in most cells. C. A section of cerebellum from a patient with mtDNA rearrangement that highlights the presence of COX-deficient neurons. D, E. Tissues that show COX deficiency due to clonal expansion of somatic mtDNA mutations within single cells—a phenomenon that is seen in both postmitotic cells (D; extraocular muscles) and rapidly dividing cells (E; colonic crypt) in aging humans. (Reproduced with permission from R Taylor, D Turnbull: Mitochondrial DNA mutations in human disease. Nat Rev Genetics 6:389, 2005.)
The emerging next-generation sequencing (NGS) techniques and their rapid penetration and recognition as useful clinical diagnostic tools are expected to also dramatically improve the clinical genetic diagnostic evaluation of mitochondrial diseases at the level of both the nuclear genome and mtDNA. In the context of the larger nuclear genome, the ability of NGS techniques to dramatically increase the speed at which DNA can be sequenced at a fraction of the cost of conventional Sanger-type sequencing technology is particularly beneficial. Low sequencing costs and short turnaround time expedite “first-tier” screening of panels of hundreds of previously known or suspected mitochondrial disease genes or screening for the entire exome or genome in an attempt to identify novel genes and mutations affecting different patients or families. In the context of the mtDNA, NGS approaches hold the particular promise for rapid and reliable detection of heteroplasmy in different affected tissues. Although Sanger sequencing allows for complete coverage of the mtDNA, it is limited by the lack of deep coverage and low sensitivity for heteroplasmy detection when it is much less than 50%. In contrast, NGS technology is an excellent tool for rapidly and accurately obtaining a patient’s predominant mtDNA sequence and also lower frequency heteroplasmic variants. This is enabled by deep coverage of the genome through multiple independent sequence reads. Accordingly, recent studies making use of NGS techniques have demonstrated sequence accuracy equivalent to Sanger-type sequencing, but also have uncovered heretofore unappreciated heteroplasmy rates ranging between 10 and 50% and detection of single-nucleotide heteroplasmy down to levels of <10%.
Clinically, the most striking overall characteristic of mitochondrial genetic disease is the phenotypic heterogeneity associated with mtDNA mutations. This extends to intrafamilial phenotypic heterogeneity for the same mtDNA pathogenic mutation and, conversely, to the overlap of phenotypic disease manifestations with distinct mutations. Thus, although fairly consistent and well-defined “classic” syndromes have been attributed to specific mutations, frequently “nonclassic” combinations of disease phenotypes ranging from isolated myopathy to extensive multisystem disease are often encountered, rendering genotype-phenotype correlation challenging. In both classical and nonclassical mtDNA disorders, there is often a clustering of some combination of abnormalities affecting the neurologic system (including optic nerve atrophy, pigment retinopathy, and sensorineural hearing loss), cardiac and skeletal muscle (including extraocular muscles), and endocrine and metabolic systems (including diabetes mellitus). Additional organ systems that may be affected include the hematopoietic, renal, hepatic, and gastrointestinal systems, although these are more frequently involved in infants and children. Disease-causing mtDNA coding region mutations can affect either one of the 13 protein encoding genes or one of the 24 protein synthetic genes. Clinical manifestations do not readily distinguish these two categories, although lactic acidosis and muscle pathologic findings tend to be more prominent in the latter. In all cases, either defective ATP production due to disturbances in the ETC or enhanced generation of ROS has been invoked as the mediating biochemical mechanism between mtDNA mutation and disease manifestation.
MTDNA DISEASE PRESENTATIONS
The clinical presentation of adult patients with mtDNA disease can be divided into three categories: (1) clinical features suggestive of mitochondrial disease (Table 85e-2), but not a well-defined classic syndrome; (2) classic mtDNA syndromes; and (3) clinical presentation confined to one organ system (e.g., isolated sensorineural deafness, cardiomyopathy, or diabetes mellitus).
COMMON FEATURES OF MTDNA-ASSOCIATED DISEASES IN ADULTS |
Table 85e-3 provides a summary of eight illustrative classic mtDNA syndromes or disorders that affect adult patients and highlights some of the most interesting features of mtDNA disease in terms of molecular pathogenesis, inheritance, and clinical presentation. The first five of these syndromes result from heritable point mutations in either protein-encoding or protein synthetic mtDNA genes; the other three result from rearrangements or deletions that usually do not involve the germline.
MITOCHONDRIAL DISEASES DUE TO MTDNA POINT MUTATIONS AND LARGE-SCALE REARRANGEMENTS |
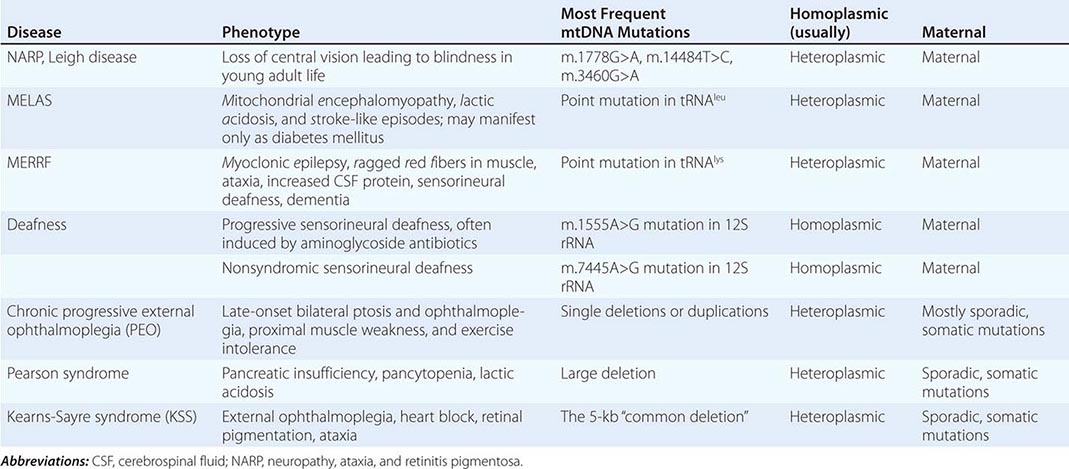
Leber’s hereditary optic neuropathy (LHON) is a common cause of maternally inherited visual failure. LHON typically presents during young adulthood with subacute painless loss of vision in one eye, with symptoms developing in the other eye 6–12 weeks after the initial onset. In some instances, cerebellar ataxia, peripheral neuropathy, and cardiac conduction defects are observed. In >95% of cases, LHON is due to one of three homoplasmic point mutations of mtDNA that affect genes encoding different subunits of complex I of the mitochondrial ETC; however, not all individuals who inherit a primary LHON mtDNA mutation develop optic neuropathy, and males are four to five times more likely than females to be affected, indicating that additional environmental (e.g., tobacco exposure) or genetic factors are important in the etiology of the disorder. Both the nuclear and mitochondrial genomic backgrounds modify disease penetrance. Indeed, a region of the × chromosome containing a high-risk haplotype for LHON was recently identified, supporting the formulation that nuclear genes act as modifiers and affording an explanation for the male prevalence of LHON. This haplotype can be used in predictive genomic testing and prenatal screening for this disease. In contrast to the other classic mtDNA disorders, it is of interest that patients with this syndrome are often homoplasmic for the disease-causing mutation. The somewhat later onset in young adulthood and modifying effect of protective background nuclear genomic haplotypes may have enabled homoplasmic pathogenic mutations to have escaped evolutionary censoring.
Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) is a multisystem disorder with a typical onset between 2 to 10 years of age. Following normal early psychomotor development, the most common initial symptoms are seizures, recurrent headaches, anorexia, and recurrent vomiting. Exercise intolerance or proximal limb weakness can be the initial manifestation, followed by generalized tonic-clonic seizures. Short stature is common. Seizures are often associated with stroke-like episodes of transient hemiparesis or cortical blindness that may produce altered consciousness and may recur. The cumulative residual effects of the stroke-like episodes gradually impair motor abilities, vision, and cognition, often by adolescence or young adulthood. Sensorineural hearing loss adds to the progressive decline of these individuals. A plethora of less common symptoms have been described including myoclonus, ataxia, episodic coma, optic atrophy, cardiomyopathy, pigmentary retinopathy, ophthalmoplegia, diabetes mellitus, hirsutism, gastrointestinal dysmotility, and nephropathy. The typical age of death ranges from 10 to 35 years, but some individuals live into their sixth decade. Intercurrent infections or intestinal obstructions are often the terminal events. Laboratory investigation commonly demonstrates elevated lactate concentrations at rest with excessive increase after moderate exercise. Brain imaging during stroke-like episodes shows areas of increased T2 signal, typically involving the posterior cerebrum and not conforming to the distribution of major arteries. Electrocardiogram (ECG) may show evidence of cardiomyopathy, preexcitation, or incomplete heart block. Electromyography and nerve conduction studies are consistent with a myopathic process, but axonal and sensory neuropathy may coexist. Muscle biopsy typically shows ragged red fibers with the modified Gomori trichrome stain or “ragged blue fibers” resulting from the hyperintense reaction with the histochemical staining for succinate dehydrogenase. The diagnosis of MELAS is based on a combination of clinical findings and molecular genetic testing. Mutations in the mtDNA gene MT-TL1 encoding tRNAleu are causative. The most common mutation, present in approximately 80% of individuals with typical clinical findings, is an A-to-G transition at nucleotide 3243 (m.3243A>G). Mutations can usually be detected in mtDNA from leukocytes in individuals with typical MELAS; however, the occurrence of heteroplasmy can result in varying tissue distribution of mutated mtDNA. In the absence of specific treatment, various manifestations of MELAS are treated according to standard modalities for prevention, surveillance, and treatment.
Myoclonic epilepsy with ragged red fibers (MERRF) is a multisystem disorder characterized by myoclonus, seizures, ataxia, and myopathy with ragged red fibers. Hearing loss, exercise intolerance, neuropathy, and short stature are often present. Almost all MERRF patients have mutation in the mtDNA tRNAlys gene, and the m.8344A>G mutation in the mtDNA gene encoding the lysine amino acid tRNA is responsible for 80–90% of MERRF cases.
Neuropathy, ataxia, and retinitis pigmentosa (NARP) is characterized by moderate diffuse cerebral and cerebellar atrophy and symmetric lesions of the basal ganglia on magnetic resonance imaging (MRI). A heteroplasmic m.8993T>G mutation in the ATPase 6 subunit gene has been identified as causative. Ragged red fibers are not observed in muscle biopsy. When >95% of mtDNA molecules are mutant, a more severe clinical, neuroradiologic. and neuropathologic picture (Leigh syndrome) emerges. Point mutations in the mtDNA gene encoding the 12S rRNA result in heritable nonsyndromic hearing loss. One such mutation causes heritable ototoxic susceptibility to aminoglycoside antibiotics, which opens a pathway for a simple pharmacogenetic test in the appropriate clinical settings.
Kearns-Sayre syndrome (KSS), sporadic progressive external ophthalmoplegia (PEO), and Pearson syndrome are three disease phenotypes caused by large-scale mtDNA rearrangements including partial deletions or partial duplication. The majority of single large-scale rearrangements of mtDNA are thought to result from clonal amplification of a single sporadic mutational event, occurring in the maternal oocyte or during early embryonic development. Because germline involvement is rare, most cases are sporadic rather than inherited. KSS is characterized by the triad of onset before age 20, chronic progressive external ophthalmoplegia, and pigmentary retinopathy. Cerebellar syndrome, heart block, increased cerebrospinal fluid protein content, diabetes mellitus, and short stature are also part of the syndrome. Single deletions/duplication can also result in milder phenotypes such as PEO, characterized by late-onset progressive external ophthalmoplegia, proximal myopathy, and exercise intolerance. In both KSS and PEO, diabetes mellitus and hearing loss are frequent accompaniments. Pearson syndrome is also characterized by diabetes mellitus from pancreatic insufficiency, together with pancytopenia and lactic acidosis, caused by the large-scale sporadic deletion of several mtDNA genes.
Two important dilemmas in classic mtDNA disease have benefited from recent important research insights. The first relates to the greater involvement of neuronal, muscular, renal, hepatic, and pancreatic manifestations in mtDNA disease in these syndromes. This observation has appropriately been mostly attributed to the high energy utilization of the involved tissues and organ systems and, hence, greater dependency on mitochondrial ETC integrity and health. However, because mutations are stochastic events, mitochondrial mutations should occur in any organ during embryogenesis and development. Recently, additional explanations have been suggested based on studies of the common m.3243A>G transition. The proportion of this mutation in peripheral blood cells was shown to decrease exponentially with age. A selective process acting at the stem cell level with a strong bias against the mutated form would have its greatest effect to reduce the mutant mtDNA only in highly proliferating cells, such as those derived from the hematopoietic system. Tissues and organs with lower cell turnover, such as those involved with mtDNA mutations, would not benefit from this effect and, thus, would be the most affected.
The other dilemma arises from the observation that only a subset of mtDNA mutations accounts for the majority of the familial mtDNA diseases. The random occurrence of mutations in the mtDNA sequence should yield a more uniform distribution of disease-causing mutations. However, recent studies using the introduction of one severe and one mild point mutation into the female germline of experimental animals demonstrated selective elimination during oogenesis of the severe mutation and selective retention of the milder mutation, with the emergence of mitochondrial disease in offspring after multiple generations. Thus, oogenesis itself can act as an “evolutionary” filter for mtDNA disease.
THE INVESTIGATION OF SUSPECTED MTDNA DISEASE
The clinical presentations of classic syndromes, groupings of disease manifestations in multiple organ systems, or unexplained isolated presentations of one of the disease features of a classic mtDNA syndrome should prompt a systematic clinical investigation as outlined in Fig. 85e-6. Indeed, mitochondrial disease should be considered in the differential diagnosis of any progressive multisystem disorder. Despite the centrality of disruptive oxidative phosphorylation, an elevated blood lactate level is neither specific nor sensitive because there are many causes of blood lactic acidosis, and many patients with mtDNA defects presenting in adulthood have normal blood lactate. An elevated cerebrospinal fluid lactate is a more specific test for mitochondrial disease if there is central nervous system involvement. The serum creatine kinase may be elevated but is often normal, even in the presence of a proximal myopathy. Urinary organic and amino acids may also be abnormal, reflecting metabolic and kidney proximal tubule dysfunction. Every patient with seizures or cognitive decline should have an electroencephalogram. A brain computed tomography (CT) scan may show calcified basal ganglia or bilateral hypodense regions with cortical atrophy. MRI is indicated in patients with brainstem signs or stroke-like episodes.
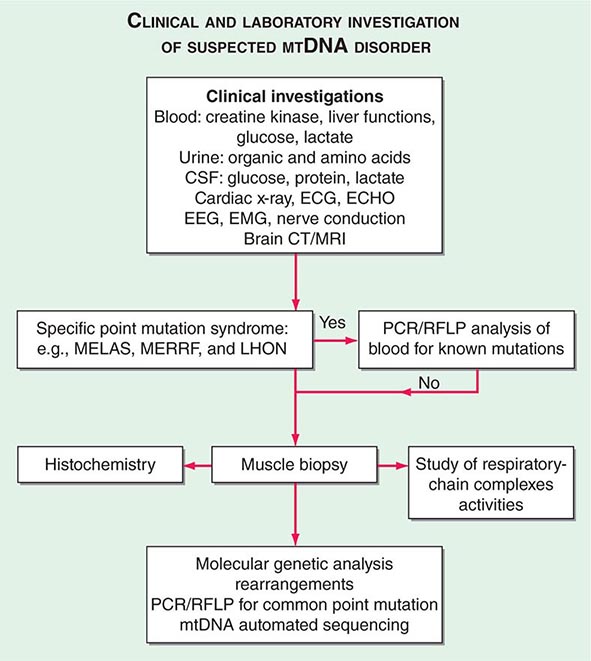
FIGURE 85e-6 Clinical and laboratory investigation of a suspected mitochondrial DNA (mtDNA) disorder. CSF, cerebrospinal fluid; CT, computed tomography; ECG, electrocardiogram; ECHO, echocardiography; EEG, electroencephalogram; EMG, electromyogram; LHON, Leber’s hereditary optic neuropathy; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stoke-like episodes; MERFF, myoclonic epilepsy with ragged red fibers; MRI, magnetic resonance imaging; PCR, polymerase chain reaction; RFLP, restriction fragment length polymorphism.
For some mitochondrial diseases, it is possible to obtain an accurate diagnosis with a simple molecular genetic screen. For examples, 95% of patients with LHON harbor one of three mtDNA point mutations (m.11778A>G, m.A3460A>G, or m.14484T>C). These patients have very high levels of mutated mtDNA in peripheral blood cells, and therefore, it is appropriate to send a blood sample for molecular genetic analysis by polymerase chain reaction (PCR) or restriction fragment length polymorphism. The same is true for most MERRF patients who harbor a point mutation in the lysine tRNA gene at position 8344. In contrast, patients with the m.3243A>G MELAS mutation often have low levels of mutated mtDNA in blood. If clinical suspicion is strong enough to warrant peripheral blood testing, then patients with a negative result should be investigated further by performing a skeletal muscle biopsy.
Muscle biopsy histochemical analysis is the cornerstone for investigation of patients with suspected mitochondrial disease. Histochemical analysis may show subsarcolemmal accumulation of mitochondria with the appearance of ragged red fibers. Electron microscopy might show abnormal mitochondria with paracrystalline inclusions. Muscle histochemistry may show cytochrome c oxidase (COX)–deficient fibers, which indicate mitochondrial dysfunction (Fig. 85e-5). Respiratory chain complex assays may also show reduced enzyme function. Either of these two abnormalities confirms the presence of a mitochondrial disease, to be followed by an in-depth molecular genetic analysis.
Recent evidence has provided important insights into the importance of nuclear-mtDNA genomic cross-talk and has provided a descriptive framework for classifying and understanding disorders that emanate from perturbations in this cross-talk. Although not strictly considered as mtDNA genetic disorders, manifestations do overlap those highlighted above (Fig. 85e-7).
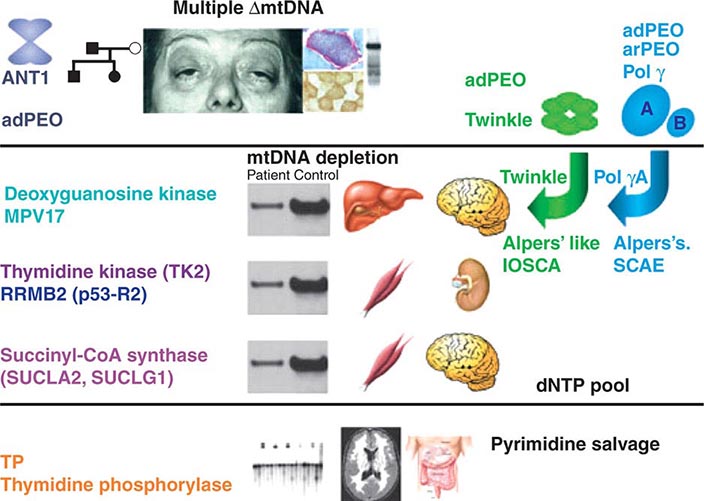
FIGURE 85e-7 Disorders associated with perturbations in nuclear-mitochondrial genomic cross-talk. Clinical features and genes associated with multiple mitochondrial DNA (mtDNA) deletions, mtDNA depletion, and mitochondrial neurogastrointestinal encephalomyopathy syndromes. ANT, adenine nucleotide translocators; adPEO, autosomal dominant progressive external ophthalmoplegia; arPEO, autosomal recessive progressive external ophthalmoplegia; IOSCA, infantile-onset spinocerebellar ataxia; SCAE, spinocerebellar ataxia and epilepsy. (Reproduced with permission from A Spinazzola, M Zeviani: Disorders from perturbations of nuclear-mitochondrial intergenomic cross-talk. J Intern Med 265:174, 2009.)
IMPACT OF HOMOPLASMIC SEQUENCE VARIATION ON HERITABLE TRAITS AND DISEASE
The relationship among the degree of heteroplasmy, tissue distribution of the mutant mtDNA, and disease phenotype simplifies inference of a clear causative relationship between heteroplasmic mutation and disease. With the exception of certain mutations (e.g., those causing most cases of LHON), drift to homoplasmy of such mutations would be precluded normally by the severity of impaired oxidative phosphorylation and the consequent reduction in reproductive fitness. Therefore, sequence variants that have reached homoplasmy should be neutral in terms of human evolution and, hence, useful only for tracing human evolution, demography, and migration, as described above. One important exception is in the case of one or more of the homoplasmic population-level variants, which designate the mtDNA haplogroup J, and the interaction with the mtDNA mutations causing LHON. Reduced disease predilection suggests that one or more of the ancient sequence variants designating mtDNA haplogroup J appears to attenuate predisposition to degenerative disease, in the face of other risk factors. Whether or not additional epistatic interactions between population-level mtDNA haplotypes and common health conditions will be found remains to be determined. If such influences do exist, then they are more likely to be relevant to health conditions in the postreproductive age groups, wherein evolutionary filters would not have had the opportunity to censor deleterious effects and interactions and wherein the effects of oxidative stress may play a role. Although much has been written about the possible associations of population-level common mtDNA variants and human health and disease phenotypes or adaptation to different environmental influences (e.g., climate), a word of caution is in order.
Many studies that purport to show such associations with phenotypes such as longevity, athletic performance, and metabolic and neurodegenerative disease are limited by small sample sizes, possible genotyping inaccuracies, and the possibility of population stratification or ethnic ancestry bias. Because mtDNA haplogroups are so prominently partitioned along phylogeographic lines, it is difficult to rule out the possibility that a haplogroup for which an association has been found is simply a marker for differences in populations with a societal or environmental difference or with different allele frequencies at other genomic loci, which are actually causally related to the heritable trait or disease of interest. The difficulty in generating cellular or animal models to test the functional influence of homoplasmic sequence variants (as a result of mtDNA polyploidy) further compounds the challenge. The most likely formulation is that the risk conferred by different mtDNA haplogroup–defining homoplasmic mutations for common diseases depends on the concomitant nuclear genomic background, together with environmental influences. Progress in minimizing potentially misleading associations in mtDNA heritable trait and disease studies should include ensuring adequate sample size taken from a large sample recruitment base, using carefully matched controls and population structure determination, and performing analysis that takes into account epistatic interactions with other genomic loci and environmental factors.
IMPACT OF ACQUIRED SOMATIC MTDNA MUTATION ON HUMAN HEALTH AND DISEASE
Studies on aging humans and animals have shown a potentially important correlation of age with the accumulation of heterogeneous mtDNA mutations, especially in those organ systems that undergo the most prominent age-related degenerative tissue phenotype. Sequencing of PCR-amplified single mtDNA molecules has demonstrated an average of two to three point mutations per molecule in elderly subjects when compared with younger ones. Point mutations observed include those responsible for known heritable heteroplasmic mtDNA disorders, such as the m.3344A>G and m.3243A>G mutations responsible for the MERRF and MELAS syndromes, respectively. However, the cumulative burden of these acquired somatic point mutations with age was observed to remain well below the threshold expected for phenotypic expression (<2%). Point mutations at other sites not normally involved in inherited mtDNA disorders have also been shown to accumulate to much higher levels in some tissues of elderly individuals, with the description of tissue-specific “hot spots” for mtDNA point mutations. Along the same lines, an age-associated and tissue-specific accumulation of mtDNA deletions has been observed, including deletions involved in known heritable mtDNA disorders, as well as others. The accumulation of functional mtDNA deletions in a given tissue is expected to be associated with mitochondrial dysfunction, as reflected in an age-associated patchy and reduced COX activity on histochemical staining, especially in skeletal and cardiac muscle and brain. A particularly well-studied and potentially important example is the accumulation of mtDNA deletions and COX deficiency observed in neurons of the substantia nigra in Parkinson’s disease patients.
The progressive accumulation of ROS has been proposed as the key factor connecting mtDNA mutations with aging and age-related disease pathogenesis (Fig. 85e-8). As noted above, ROS are a by-product of oxidative phosphorylation and are removed by detoxifying antioxidants into less harmful moieties; however, exaggerated production of ROS or impaired removal results in their accumulation. One of the main targets for ROS-mediated injury is DNA, and mtDNA is particularly vulnerable because of its lack of protective histones and less efficient injury repair systems compared with nuclear DNA. In turn, accumulation of mtDNA mutations results in inefficient oxidative phosphorylation, with the potential for excessive production of ROS, generating a “vicious cycle” of cumulative mtDNA damage. Indeed, measurement of the oxidative stress biomarker 8-hydroxy-2-deoxyguanosine has been used to measure age-dependent increases in mtDNA oxidative damage at a rate exceeding that of nuclear DNA. It should be noted that mtDNA mutation can potentially occur in postmitotic cells as well, because mtDNA replication is not synchronized with the cell cycle. Two other proposed links between mtDNA mutation and aging, besides ROS-mediated tissue injury, are the perturbations in efficiency of oxidative phosphorylation with disturbed cellular aerobic function and perturbations in apoptotic pathways, whose execution steps involve mitochondrial activity.
FIGURE 85e-8 Multiple pathways of mitochondrial DNA (mtDNA) damage and aging. Multiple factors may impinge on the integrity of mitochondria that lead to loss of cell function, apoptosis, and aging. The classic pathway is indicated with blue arrows; the generation of reactive oxygen species (ROS; superoxide anion, hydrogen peroxide, and hydroxyl radicals), as a by-product of mitochondrial oxidative phosphorylation, results in damage to mitochondrial macromolecules, including the mtDNA, with the latter leading to deleterious mutations. When these factors damage the mitochondrial energy-generating apparatus beyond a functional threshold, proteins are released from the mitochondria that activate the caspase pathway, leading to apoptosis, cell death, and aging. (Reproduced with permission from L Loeb et al: The mitochondrial theory of aging and its relationship to reactive oxygen species damage and somatic mtDNA mutations. Proc Natl Acad Sci USA 102:18769, 2005.)
Genetic intervention studies in animal models have sought to clarify the potential causative relationship between acquired somatic mtDNA mutation and the aging phenotype, and the role of ROS in particular. Replication of the mitochondrial genome is mediated by the activity of the nuclear-encoded polymerase gamma gene. A transgenic homozygous mouse knock-in mutation of this gene renders the polymerase enzyme deficient in proofreading and results in a three- to fivefold increase in mtDNA mutation rate. Such mice develop a premature aging phenotype, which includes subcutaneous lipoatrophy, alopecia, kyphonia, and weight loss with premature death. Although the finding of increased mtDNA mutation and mitochondrial dysfunction with age has been solidly established, the causative role and specific contribution of mitochondrial ROS to aging and age-related disease in humans has yet to be proved. Similarly, although many tumors display higher levels of heterogeneous mtDNA mutations, a causal relationship to tumorigenesis has not been proved.
Besides the age-dependent acquired accumulation in somatic cells of heterogeneous point mutations and deletions, a quite different effect of nonheritable and acquired mtDNA mutation has been described affecting tissue stem cells. In particular, disease phenotypes attributed to acquired mtDNA mutation have been observed in sporadic and apparently nonfamilial cases involving a single individual or even tissue, usually skeletal muscle. The presentation consists of decreased exercise tolerance and myalgias, sometimes progressing to rhabdomyolysis. As in the case of the sporadic, heteroplasmic, large-scale deletion, classic syndromes of chronic PEO, Pearson syndrome, and KSS, the absence of a maternal inheritance pattern, together with the finding of limited tissue distribution, suggests a molecular pathogenic mechanism emanating from mutations arising de novo in muscle stem cells after germline differentiation (somatic mutations that are not sporadic and occur in tissue-specific stem cells during fetal development or in the postnatal maintenance or postinjury repair stage). Such mutations would be expected to be propagated only within the progeny of that stem cell and affect a particular tissue within a given individual, without evidence of heritability.
PROSPECTS FOR CLINICAL MANAGEMENT OF MTDNA DISEASE
TREATMENT OF MTDNA DISORDERS
No specific curative treatment for mtDNA disorders is currently available; therefore, the management of mitochondrial disease is largely supportive. Management issues may include early diagnosis and treatment of diabetes mellitus, cardiac pacing, ptosis correction, and intraocular lens replacement for cataracts. Less specific interventions in the case of other disorders involve combined treatment strategies including dietary intervention and removal of toxic metabolites. Cofactors and vitamin supplements are widely used in the treatment of diseases of mitochondrial oxidative phosphorylation, although there is little evidence, apart from anecdotal reports, to support their use. This includes administration of artificial electron acceptors, including vitamin K3, vitamin C, and ubiquinone (coenzyme Q10); administration of cofactors (coenzymes) including riboflavin, carnitine, and creatine; and use of oxygen radical scavengers, such as vitamin E, copper, selenium, ubiquinone, and idebenone. Drugs that could interfere with mitochondrial function, such as the anesthetic agent propofol, barbiturates, and high doses of valproate, should be avoided. Supplementation with the nitric oxide synthase substrate, L-arginine, has been advocated as a vasodilator treatment during stroke-like episodes. The physician should also be familiar with environmental interactions, such as the strong and consistent association between visual loss in LHON and smoking. A clinical penetrance of 93% was found in men who smoked. Asymptomatic carriers of an LHON mtDNA mutation should, therefore, be strongly advised not to smoke and to moderate their alcohol intake. Although not a cure, these interventions might stave off the devastating clinical manifestations of the LHON mutation. Another example is strict avodiance of aminoglycosides in the familial syndrome of ototoxic susceptibility to aminoglycosides in the presence of the mtDNA m.1555A>G mutation of the 12SrRNA encoding gene.
GENETIC COUNSELING, PRENATAL DIAGNOSIS, AND PREIMPLANTATION GENETIC DIAGNOSIS IN MTDNA DISORDERS
The provision of accurate genetic counseling and reproductive options to families with mtDNA mutations is challenging due to the unique genetic features of mtDNA inheritance that distinguish it from Mendelian genetics. mtDNA defects are transmitted by maternal inheritance. mtDNA de novo mutations are often large deletions, affect one family member, and usually represent no significant risk to other members of the family. In contrast, mtDNA point mutations or duplications can be transmitted down the maternal line. Accordingly, the father of an affected individual has no risk of harboring the disease-causing mutation, and a male cannot transmit the mtDNA mutation to his offspring. In contrast, the mother of an affected individual usually harbors the same mutation but might be completely asymptomatic. This wide phenotypic variability is primarily related to the phenomena of heteroplasmy and the mutation load carried by different members of the same family. Consequently, a symptomatic or asymptomatic female harboring a disease-causing mutation in a heteroplasmic state will transmit to her offspring variable amounts of the mutant mtDNA molecules. The offspring will be symptomatic or asymptomatic primarily according to the mutant load transmitted via the oocyte and, to some extent, subsequent mitotic segregation during development. Interactions with the mtDNA haplotype background or nuclear human genome (as in the case of LHON) serve as an additional important determinant of disease penetrance. Because the severity of the disease phenotype associated with the heteroplasmic mutation load is a function of the stochastic differential segregation and copy number of mutant mtDNA during the oogenesis bottleneck and, subsequently, following tissue and organ development in the offspring, it is rarely predictable with any degree of accuracy. For this reason, prenatal diagnosis (PND) and PGD techniques that have evolved into integral and well-accepted standards of practice are severely hampered in the case of mtDNA-related diseases.
The value of PND and PGD is limited, partly due to the absence of data on the rules that govern the segregation of wild-type and mutant mtDNA species (heteroplasmy) among tissue in the developing embryo. Three factors are required to ensure the reliability of PND and PGD: (1) a close correlation between the mutant load and the disease severity, (2) a uniform distribution of mutant load among tissues, and (3) no major change in mutant load with time. These criteria are suggested to be fulfilled for the NARP m.8993T>G mutation but do not seem to apply to other mtDNA disorders. In fact, the level of mutant mtDNA in a chorionic villous or amniotic fluid sample may be very different from the level in the fetus, and it would be difficult to deduce whether the mutational load in the prenatal samples provides clinically useful information regarding the postnatal and adult state.
PREVENTION OF MITOCHONDRIAL DISEASE INHERITANCE BY ASSISTED REPRODUCTIVE TECHNOLOGIES
Because the treatment options for patients with mitochondrial disease are rather limited, preventive interventions that eliminate the likelihood of transmission of affected mtDNA into offspring are desirable. The lack of utility of PND and PGD techniques to reliably diagnose and predict mitochondrial disorders at preimplantation-stage products of conception has resulted in the search for alternative preventive approaches for the same problem. One possible approach to “diluting” or even entirely eliminating the mutant mtDNA is applicable only in the earliest embryonic state and in effect represents a form of germline preventive therapy (Fig. 85e-9). This possibility has been explored by using alternative assisted reproduction techniques such as ooplasmic transfer (OT), metaphase chromosome transfer (CT), pronuclear transfer (PNT), and germinal vesicle transfer (GVT) in animal models and, to an extent, in humans. OT is a technique wherein a certain volume (5–15%) of healthy donor oocyte cytoplasm with normal mitochondria is injected into the patient oocyte containing mutated mitochondria. The reasoning behind OT is to supplement the patient’s oocyte with uncompromised cytoplasmic factors such as mtDNA, mRNA, proteins, and other molecules by injecting cytoplasm from healthy oocytes. In PNT, following fertilization, pronuclei of a patient’s zygote are removed with a cytoplasm (“karyoplast”). The karyoplast is transferred to the perivitelline space of a donated zygote, which has been already enucleated. The karyoplast is then fused with enucleated zygote by electric pulses or inactivated Sendai viruses (HVJ). The reconstructed zygote contains a nucleus from the patient (patient nuclear DNA) and cytoplasm from the donor. Thus, the majority of the patient mtDNA is replaced with mtDNA from the donor oocyte. In CT, meiosis II stage of oocyte maturation provides an opportunity for the reconstruction of oocytes with different nuclear and cytoplasmic components before fertilization takes place. Reconstructed oocytes by metaphase chromosome transfer are then fertilized to produce embryos with desired mtDNA haplotypes. In GVT, replacement of compromised cytoplasm with healthy cytoplasm through germinal vesicle transfer before the start of chromosome segregation is carried out.
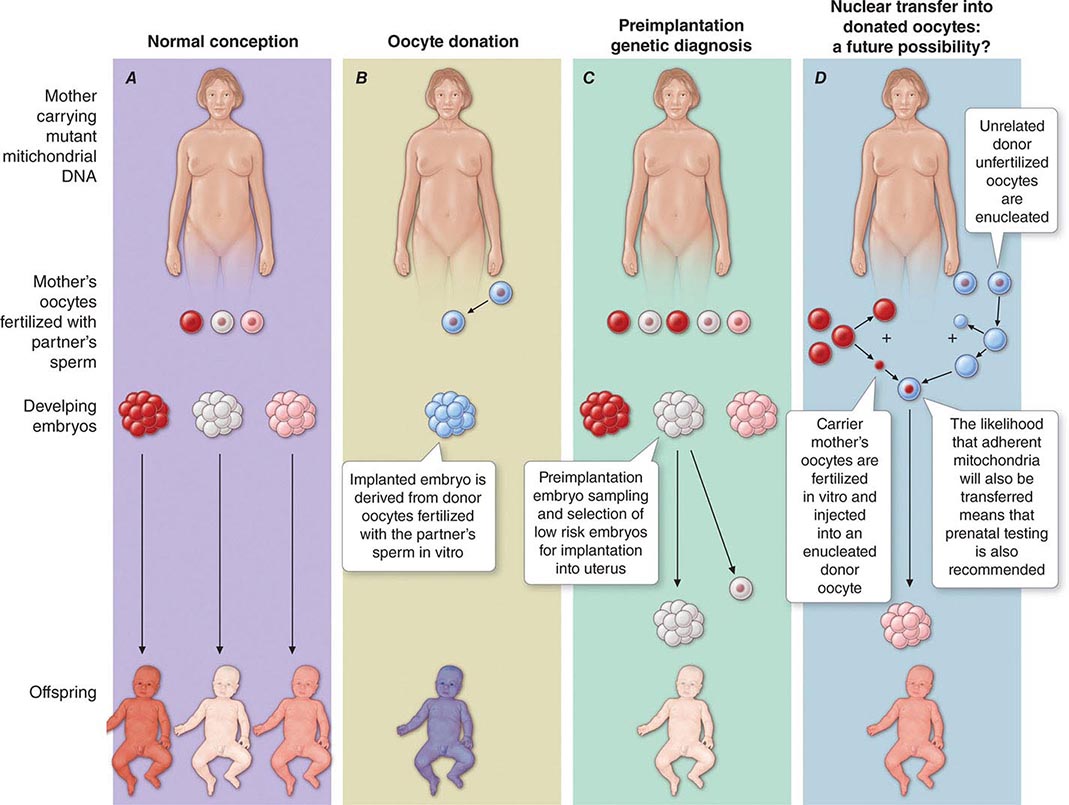
FIGURE 85e-9 Possible approaches for prevention of mitochondrial DNA (mtDNA) disease. A. No intervention: offspring’s mutant mtDNA load will vary greatly. B. Oocyte donation: currently permitted in some constituencies but limited by the availability of oocyte donors. C. Preimplantation genetic diagnosis: available for some mtDNA diseases (reliable in determining background nuclear genomic haplotype risk). D. Nuclear transfer: research stage, including initial studies in nonhuman primates. Red represents mutant mtDNA, pink and white represent successively higher proportions of normal mtDNA. Blue represents genetic material from an unrelated donor. (Adapted with permission from J Poulton et al: Preventing transmission of maternally inherited mitochondrial DNA diseases. Br Med J 338:b94, 2009.)
These approaches have not yet met with widely reported clinical success, yet there is room for optimism. As noted above, analysis of heteroplasmy and inheritance patterns indicates that even a small increase in copies of nonmutant mtDNA can exceed the threshold required to ameliorate serious clinical disease. All of the approaches described above show promise in achieving this goal and thus reducing the burden of clinical mtDNA disease in the future.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


