289e | Tumors and Trauma of the Heart |
TUMORS OF THE HEART
PRIMARY TUMORS
Primary tumors of the heart are rare. Approximately three-quarters are histologically benign, and the majority of these tumors are myxomas. Malignant tumors, almost all of which are sarcomas, account for 25% of primary cardiac tumors. All cardiac tumors, regardless of pathologic type, have the potential to cause life-threatening complications. Many tumors are now surgically curable; thus, early diagnosis is imperative.
Clinical Presentation Cardiac tumors may present with a wide array of cardiac and noncardiac manifestations. These manifestations depend in large part on the location and size of the tumor and are often nonspecific features of more common forms of heart disease, such as chest pain, syncope, heart failure, murmurs, arrhythmias, conduction disturbances, and pericardial effusion with or without tamponade. Additionally, embolic phenomena and constitutional symptoms may occur.
Myxoma Myxomas are the most common type of primary cardiac tumor in adults, accounting for one-third to one-half of all cases at postmortem examination, and about three-quarters of the tumors treated surgically. They occur at all ages, most commonly in the third through sixth decades, with a female predilection. Approximately 90% of myxomas are sporadic; the remainder are familial with autosomal dominant transmission. The familial variety often occurs as part of a syndrome complex (Carney complex) that includes (1) myxomas (cardiac, skin, and/or breast), (2) lentigines and/or pigmented nevi, and (3) endocrine overactivity (primary nodular adrenal cortical disease with or without Cushing’s syndrome, testicular tumors, and/or pituitary adenomas with gigantism or acromegaly). Certain constellations of findings have been referred to as the NAME syndrome (nevi, atrial myxoma, myxoid neurofibroma, and ephelides) or the LAMB syndrome (lentigines, atrial myxoma, and blue nevi), although these syndromes probably represent subsets of the Carney complex. The genetic basis of this complex has not been elucidated completely; however, patients frequently have inactivating mutations in the tumor-suppressor gene PRKAR1A, which encodes the protein kinase A type I-α regulatory subunit.
Pathologically, myxomas are gelatinous structures that consist of myxoma cells embedded in a stroma rich in glycosaminoglycans. Most are solitary, arise from the interatrial septum in the vicinity of the fossa ovalis (particularly the left atrium), and are often pedunculated on a fibrovascular stalk. In contrast to sporadic tumors, familial or syndromic tumors tend to occur in younger individuals, are often multiple, may be ventricular in location, and are more likely to recur after initial resection.
Myxomas commonly present with obstructive signs and symptoms. The most common clinical presentation mimics that of mitral valve disease: either stenosis owing to tumor prolapse into the mitral orifice or regurgitation resulting from tumor-induced valvular trauma. Ventricular myxomas may cause outflow obstruction similar to that caused by subaortic or subpulmonic stenosis. The symptoms and signs of myxoma may be sudden in onset or positional in nature, owing to the effects of gravity on tumor position. A characteristic low-pitched sound, a “tumor plop,” may be appreciated on auscultation during early or mid-diastole and is thought to result from the impact of the tumor against the mitral valve or ventricular wall. Myxomas also may present with peripheral or pulmonary emboli or with constitutional signs and symptoms, including fever, weight loss, cachexia, malaise, arthralgias, rash, digital clubbing, Raynaud’s phenomenon, hypergammaglobulinemia, anemia, polycythemia, leukocytosis, elevated erythrocyte sedimentation rate, thrombocytopenia, and thrombocytosis. These features account for the frequent misdiagnosis of patients with myxomas as having endocarditis, collagen vascular disease, or a paraneoplastic syndrome.
Two-dimensional transthoracic or omniplane transesophageal echocardiography is useful in the diagnosis of cardiac myxoma and allows assessment of tumor size and determination of the site of tumor attachment, both of which are important considerations in the planning of surgical excision (Fig. 289e-1). Computed tomography (CT) and magnetic resonance imaging (MRI) may provide important information regarding size, shape, composition, and surface characteristics of the tumor (Fig. 289e-2).
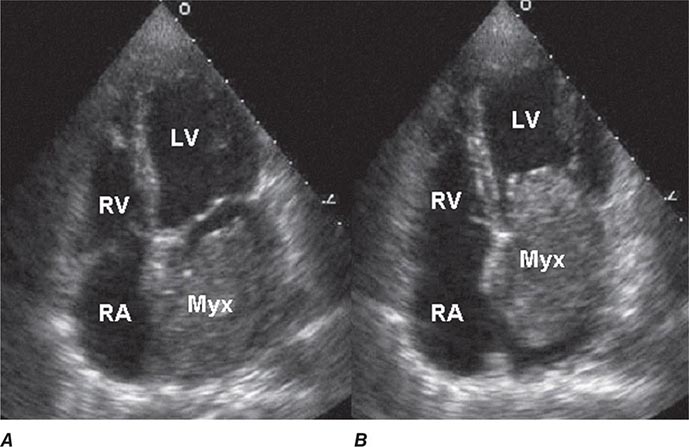
FIGURE 289e-1 Transthoracic echocardiogram demonstrating a large atrial myxoma. The myxoma (Myx) fills the entire left atrium in systole (A) and prolapses across the mitral valve and into the left ventricle (LV) during diastole (B). RA, right atrium; RV, right ventricle. (Courtesy of Dr. Michael Tsang; with permission.)
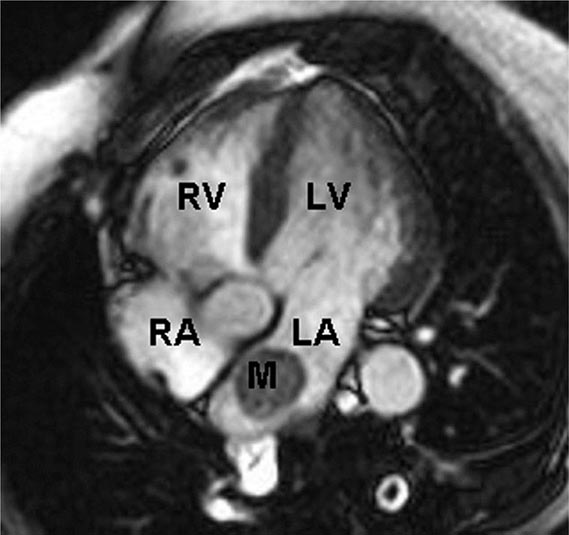
FIGURE 289e-2 Cardiac magnetic resonance imaging demonstrating a rounded mass (M) within the left atrium (LA). Pathologic evaluation at the time of surgery revealed it to be an atrial myxoma. LV, left ventricle; RA, right atrium; RV, right ventricle.
Although cardiac catheterization and angiography were previously performed routinely before tumor resection, they no longer are considered mandatory when adequate noninvasive information is available and other cardiac disorders (e.g., coronary artery disease) are not considered likely. Additionally, catheterization of the chamber from which the tumor arises carries the risk of tumor embolization. Because myxomas may be familial, echocardiographic screening of first-degree relatives is appropriate, particularly if the patient is young and has multiple tumors or evidence of myxoma syndrome.
Other Benign Tumors Cardiac lipomas, although relatively common, are usually incidental findings at postmortem examination; however, they may grow as large as 15 cm and may present with symptoms owing to mechanical interference with cardiac function, arrhythmias, or conduction disturbances or as an abnormality of the cardiac silhouette on chest x-ray. Papillary fibroelastomas are the most common tumors of the cardiac valves. Although usually clinically silent, they can cause valve dysfunction and may embolize distally, resulting in transient ischemic attacks, stroke, or myocardial infarction. In general, these tumors should be resected even when asymptomatic, although a more conservative approach may be considered for small, right-sided lesions. Rhabdomyomas and fibromas are the most common cardiac tumors in infants and children and usually occur in the ventricles, where they may produce mechanical obstruction to blood flow, thereby mimicking valvular stenosis, congestive heart failure (CHF), restrictive or hypertrophic cardiomyopathy, or pericardial constriction. Rhabdomyomas are probably hamartomatous growths, are multiple in 90% of cases, and are strongly associated with tuberous sclerosis. These tumors have a tendency to regress completely or partially; only tumors that cause obstruction require surgical resection. Fibromas are usually single, are often calcified, tend to grow and cause obstructive symptoms, and should be resected. Hemangiomas and mesotheliomas are generally small tumors, most often intramyocardial in location, and may cause atrioventricular (AV) conduction disturbances and even sudden death as a result of their propensity to develop in the region of the AV node. Other benign tumors arising from the heart include teratoma, chemodectoma, neurilemoma, granular cell myoblastoma, and bronchogenic cysts.
Sarcoma Almost all malignant primary cardiac tumors are sarcomas, which may be of several histologic types. In general, these tumors are characterized by rapid progression that culminates in the patient’s death within weeks to months from the time of presentation as a result of hemodynamic compromise, local invasion, or distant metastases. Sarcomas commonly involve the right side of the heart, are characterized by rapid growth, frequently invade the pericardial space, and may obstruct the cardiac chambers or venae cavae. Sarcomas also may occur on the left side of the heart and may be mistaken for myxomas.
TUMORS METASTATIC TO THE HEART
Tumors metastatic to the heart are much more common than primary tumors, and their incidence is likely to increase as the life expectancy of patients with various forms of malignant neoplasms is extended by more effective therapy. Although cardiac metastases may occur with any tumor type, the relative incidence is especially high in malignant melanoma and, to a somewhat lesser extent, leukemia and lymphoma. In absolute terms, the most common primary originating sites of cardiac metastases are carcinoma of the breast and lung, reflecting the high incidence of those cancers. Cardiac metastases almost always occur in the setting of widespread primary disease, and most often there is either primary or metastatic disease elsewhere in the thoracic cavity. Nevertheless, cardiac metastasis occasionally may be the initial presentation of an extrathoracic tumor.
Cardiac metastases may occur via hematogenous or lymphangitic spread or by direct tumor invasion. They generally manifest as small, firm nodules; diffuse infiltration also may occur, especially with sarcomas or hematologic neoplasms. The pericardium is most often involved, followed by myocardial involvement of any chamber and, rarely, by involvement of the endocardium or cardiac valves.
Cardiac metastases are clinically apparent only ~10% of the time, are usually not the cause of the patient’s presentation, and rarely are the cause of death. The vast majority occur in the setting of a previously recognized malignant neoplasm. As with primary cardiac tumors, the clinical presentation reflects more the location and size of the tumor than its histologic type. When symptomatic, cardiac metastases may result in a variety of clinical features, including dyspnea, acute pericarditis, cardiac tamponade, ectopic tachyarrhythmias, heart block, and CHF. Importantly, many of these signs and symptoms may also result from myocarditis, pericarditis, or cardiomyopathy induced by radiotherapy or chemotherapy.
Electrocardiographic (ECG) findings are nonspecific. On chest x-ray, the cardiac silhouette is most often normal but may be enlarged or exhibit a bizarre contour. Echocardiography is useful for identifying pericardial effusions and visualizing larger metastases, although CT and radionuclide imaging with gallium or thallium may define the tumor burden more clearly. Cardiac MRI offers superb image quality and plays a central role in the diagnostic evaluation of cardiac metastases and cardiac tumors in general. Pericardiocentesis may allow for a specific cytologic diagnosis in patients with malignant pericardial effusions. Angiography is rarely necessary but may delineate discrete lesions.
TRAUMATIC CARDIAC INJURY
Traumatic cardiac injury may be caused by either penetrating or nonpenetrating trauma. Penetrating injuries most often result from gunshot or knife wounds, and the site of entry is usually obvious. Nonpenetrating injuries most often occur during motor vehicle accidents, either from rapid deceleration or from impact of the chest against the steering wheel, and may be associated with significant cardiac injury even in the absence of external signs of thoracic trauma.
NONPENETRATING CARDIAC INJURY
Myocardial contusions are the most common form of nonpenetrating cardiac injury and may initially be overlooked in trauma patients as the clinical focus is directed toward other, more obvious injuries. Myocardial necrosis may occur as a direct result of the blunt injury or as a result of traumatic coronary laceration or thrombosis. The contused myocardium is pathologically similar to infarcted myocardium and may be associated with atrial or ventricular arrhythmias; conduction disturbances, including bundle branch block; or ECG abnormalities resembling those of infarction or pericarditis. Thus, it is important to consider contusion as a cause of otherwise unexplained ECG changes in a trauma patient. Serum creatine kinase, myocardial band (CK-MB) isoenzyme levels are increased in ~20% of patients who experience blunt chest trauma but may be falsely elevated in the presence of massive skeletal muscle injury. Cardiac troponin levels are more specific for identifying cardiac injury in this setting. Echocardiography is useful in detecting structural and functional sequelae of contusion, including wall motion abnormalities (most commonly involving the right ventricle, interventricular septum, or left ventricular apex), pericardial effusion, valvular dysfunction, and ventricular rupture.
Rupture of the cardiac valves or their supporting structures, most commonly of the tricuspid or mitral valve, leads to acute valvular incompetence. This complication is usually heralded by the development of a loud murmur, may be associated with rapidly progressive heart failure, and can be diagnosed by either transthoracic or transesophageal echocardiography.
The most serious consequence of nonpenetrating cardiac injury is myocardial rupture, which may result in hemopericardium and tamponade (free wall rupture) or intracardiac shunting (ventricular septal rupture). Although it generally is fatal, up to 40% of patients with cardiac rupture have been reported to survive long enough to reach a specialized trauma center. Hemopericardium also may result from traumatic rupture of a pericardial vessel or a coronary artery. Additionally, a pericardial effusion may develop weeks or even months after blunt chest trauma as a manifestation of the post–cardiac injury syndrome, which resembles the postpericardiotomy syndrome (Chap. 288).
Blunt, nonpenetrating, often innocent-appearing injuries to the chest may trigger ventricular fibrillation even in absence of overt signs of injury. This syndrome, referred to as commotio cordis, occurs most often in adolescents during sporting events (e.g., baseball, hockey, football, and lacrosse) and probably results from an impact to the chest wall overlying the heart during the susceptible phase of repolarization just before the peak of the T wave. Survival depends on prompt defibrillation. Sudden emotional or physical trauma, even in the absence of direct cardiac trauma, may precipitate a transient catecholamine-mediated cardiomyopathy referred to as tako-tsubo syndrome or the apical ballooning syndrome (Chap. 287).
Rupture or transection of the aorta, usually just above the aortic valve or at the site of the ligamentum arteriosum, is a common consequence of nonpenetrating chest trauma and is the most common vascular deceleration injury. The clinical presentation is similar to that of aortic dissection (Chap. 301); the arterial pressure and pulse amplitude may be increased in the upper extremities and decreased in the lower extremities, and chest x-ray may reveal mediastinal widening. Occasionally, aortic rupture is contained by the aortic adventitia, resulting in a false, or pseudo-, aneurysm that may be discovered months or years after the initial injury.
PENETRATING CARDIAC INJURY
Penetrating injuries of the heart produced by knife or bullet wounds usually result in rapid clinical deterioration and frequently in death as a result of hemopericardium/pericardial tamponade or massive hemorrhage. Nonetheless, up to half of such patients may survive long enough to reach a specialized trauma center if immediate resuscitation is performed. Prognosis in these patients relates to the mechanism of injury, their clinical condition at presentation, and the specific cardiac chamber(s) involved. Iatrogenic cardiac or coronary arterial perforation may complicate placement of central venous or intracardiac catheters, pacemaker leads, or intracoronary stents and is associated with a better prognosis than are other forms of penetrating cardiac trauma.
Traumatic rupture of a great vessel from penetrating injury is usually associated with hemothorax and, less often, hemopericardium. Local hematoma formation may compress major vessels and produce ischemic symptoms, and AV fistulas may develop, occasionally resulting in high-output CHF.
Occasionally, patients who survive penetrating cardiac injuries may subsequently present with a new cardiac murmur or CHF as a result of mitral regurgitation or an intracardiac shunt (i.e., ventricular or atrial septal defect, aortopulmonary fistula, or coronary AV fistula) that was undetected at the time of the initial injury or developed subsequently. Therefore, trauma patients should be examined carefully several weeks after the injury. If a mechanical complication is suspected, it can be confirmed by echocardiography or cardiac catheterization.
290e | Cardiac Manifestations of Systemic Disease |
The common systemic disorders that have associated cardiac manifestations are summarized in Table 290e-1.
COMMON SYSTEMIC DISORDERS AND THEIR ASSOCIATED CARDIAC MANIFESTATIONS |
DIABETES MELLITUS
(See also Chap. 417) Diabetes mellitus, both insulin- and non-insulin-dependent, is an independent risk factor for coronary artery disease (CAD; Chap. 291e) and accounts for 14–50% of new cases of cardiovascular disease. Furthermore, CAD is the most common cause of death in adults with diabetes mellitus. In the diabetic population, the incidence of CAD relates to the duration of diabetes and the level of glycemic control, and its pathogenesis involves endothelial dysfunction, increased lipoprotein peroxidation, increased inflammation, a prothrombotic state, and associated metabolic abnormalities.
Compared to their nondiabetic counterparts, diabetic patients are more likely to have a myocardial infarction, have a greater burden of CAD, have larger infarct size, and have more postinfarct complications, including heart failure, shock, and death. Importantly, diabetic patients are more likely to have atypical ischemic symptoms; nausea, dyspnea, pulmonary edema, arrhythmias, heart block, or syncope may be their anginal equivalent. Additionally, “silent ischemia,” resulting from autonomic nervous system dysfunction, is more common in diabetic patients, accounting for up to 90% of their ischemic episodes. Thus, one must have a low threshold for suspecting CAD in diabetic patients. The treatment of diabetic patients with CAD must include aggressive risk factor management (Chap. 418). Considerations regarding pharmacologic therapy and revascularization strategies are similar in diabetic and nondiabetic patients except that diabetic patients have higher morbidity and mortality rates associated with revascularization, have an increased risk of restenosis after percutaneous coronary intervention (PCI), and have improved survival when treated with surgical bypass compared with PCI for multivessel CAD.
Patients with diabetes mellitus also may have abnormal left ventricular systolic and diastolic function, reflecting concomitant epicardial CAD and/or hypertension, coronary microvascular disease, endothelial dysfunction, ventricular hypertrophy, and autonomic dysfunction. Furthermore, the increase in intramyocardial lipid deposition (predominantly nonesterified fatty acids) that is characteristic of diabetic states may contribute to both systolic and diastolic dysfunction by impairing insulin signaling, reducing trans-sarcolemma calcium flux, and inducing myocyte apoptosis. A restrictive cardiomyopathy may be present with abnormal myocardial relaxation and elevated ventricular filling pressures. Histologically, interstitial fibrosis is seen, and intramural arteries may demonstrate intimal thickening, hyaline deposition, and inflammatory changes. Diabetic patients have an increased risk of developing clinical heart failure, which probably contributes to their excessive cardiovascular morbidity and mortality rates. There is some evidence that insulin therapy may ameliorate diabetes-related myocardial dysfunction.
MALNUTRITION AND VITAMIN DEFICIENCY
Malnutrition (See also Chap. 97) In patients whose intake of protein, calories, or both is severely deficient, the heart may become thin, pale, and hypokinetic with myofibrillar atrophy and interstitial edema. The systolic pressure and cardiac output fall, and the pulse pressure narrows. Generalized edema is common and relates to a variety of factors, including reduced serum oncotic pressure and myocardial dysfunction. Such profound states of protein and calorie malnutrition, termed kwashiorkor and marasmus, respectively, are most common in underdeveloped countries. However, significant nutritional heart disease also may occur in developed nations, particularly in patients with chronic diseases such as AIDS, patients with anorexia nervosa, and patients with severe cardiac failure in whom gastrointestinal hypoperfusion and venous congestion may lead to anorexia and malabsorption. Open-heart surgery poses increased risk in malnourished patients; such patients may benefit from preoperative hyperalimentation.
Thiamine Deficiency (Beriberi) (See also Chap. 96e) Generalized malnutrition often is accompanied by thiamine deficiency; however, this hypovitaminosis also may occur in the presence of an adequate protein and caloric intake, particularly in East Asia, where polished rice deficient in thiamine may be a major dietary component. In Western nations where the use of thiamine-enriched flour is widespread, clinical thiamine deficiency is limited primarily to alcoholics, food faddists, and patients receiving chemotherapy. Nonetheless, when thiamine stores are measured using the thiamine-pyrophosphate effect (TPPE), thiamine deficiency has been found in 20–90% of patients with chronic heart failure. This deficiency appears to result from both reduced dietary intake and a diuretic-induced increase in the urinary excretion of thiamine. The acute administration of thiamine to these patients increases the left ventricular ejection fraction and the excretion of salt and water.
Clinically, patients with thiamine deficiency usually have evidence of generalized malnutrition, peripheral neuropathy, glossitis, and anemia. The classic associated cardiovascular syndrome is characterized by high-output heart failure, tachycardia, and often elevated biventricular filling pressures. The major cause of the high-output state is vasomotor depression leading to reduced systemic vascular resistance, the precise mechanism of which is not understood. The cardiac examination may reveal a wide pulse pressure, tachycardia, a third heart sound, and an apical systolic murmur. The electrocardiogram (ECG) may reveal decreased voltage, a prolonged QT interval, and T-wave abnormalities. The chest x-ray generally reveals cardiomegaly and signs of congestive heart failure (CHF). The response to thiamine is often dramatic, with an increase in systemic vascular resistance, a decrease in cardiac output, clearing of pulmonary congestion, and a reduction in heart size often occurring in 12–48 h. Although the response to inotropes and diuretics may be poor before thiamine therapy, these agents may be important after thiamine repletion, since the left ventricle may not be able to handle the increased work load presented by the return of vascular tone.
Vitamin B6, B12, and Folate Deficiency (See also Chap. 96e) Vitamin B6, vitamin B12, and folate are cofactors in the metabolism of homocysteine. Their deficiency probably contributes to the majority of cases of hyperhomocysteinemia, a disorder associated with increased atherosclerotic risk. Supplementation of these vitamins has reduced the incidence of hyperhomocysteinemia in the United States; however, the clinical cardiovascular benefit of normalizing elevated homocysteine levels has not been proved.
OBESITY
(See also Chap. 415e) Obesity is associated with an increased prevalence of hypertension, glucose intolerance, atherosclerotic CAD, atrial fibrillation, obstructive sleep apnea, and pulmonary hypertension, and is associated with increased cardiovascular morbidity and mortality rates. In addition, obese patients have a distinct hemodynamic profile characterized by increased total and central blood volumes, increased cardiac output, and elevated left ventricular filling pressure. The elevated cardiac output appears to be required to support the metabolic demands of the excess adipose tissue. Left ventricular filling pressure is often at the upper limits of normal at rest and rises excessively with exercise, contributing to exertional dyspnea. In part as a result of chronic volume overload, eccentric cardiac hypertrophy with cardiac dilation and ventricular diastolic and/or systolic dysfunction may develop. In addition, altered levels of adipokines secreted by adipose tissue may contribute to adverse myocardial remodeling via direct effects on cardiac myocytes and other cells. Pathologically, there is left and, in some cases, right ventricular hypertrophy and generalized cardiac dilation. Pulmonary congestion, peripheral edema, and exercise intolerance may all ensue; however, the recognition of these findings may be difficult in massively obese patients.
Treatment with angiotensin-converting enzyme inhibitors, sodium restriction, and diuretics may be useful to control heart failure symptoms. Weight reduction, however, is the most effective therapy and results in reduction in blood volume and the return of cardiac output toward normal. However, rapid weight reduction may be dangerous, as cardiac arrhythmias and sudden death owing to electrolyte imbalance have been described.
THYROID DISEASE
(See also Chap. 405) Thyroid hormone exerts a major influence on the cardiovascular system by a number of direct and indirect mechanisms, and not surprisingly, cardiovascular effects are prominent in both hypo- and hyperthyroidism. Thyroid hormone causes increases in total-body metabolism and oxygen consumption that indirectly increase the cardiac workload. In addition, thyroid hormone exerts direct inotropic, chronotropic, and dromotropic effects that are similar to those seen with adrenergic stimulation (e.g., tachycardia, increased cardiac output); they are mediated at least partly by both transcriptional and nontranscriptional effects of thyroid hormone on myosin, calcium-activated ATPase, Na+-K+-ATPase, and myocardial β-adrenergic receptors.
Hyperthyroidism Common cardiovascular manifestations of hyperthyroidism include palpitations, systolic hypertension, and fatigue. Sinus tachycardia is present in ~40% of hyperthyroid patients, and atrial fibrillation is present in ~15%. Physical examination may reveal a hyperdynamic precordium, a widened pulse pressure, increases in the intensity of the first heart sound and the pulmonic component of the second heart sound, and a third heart sound. An increased incidence of mitral valve prolapse has been described in hyperthyroid patients, in which case a midsystolic murmur may be heard at the left sternal border with or without a midsystolic click. A systolic pleuropericardial friction rub (Means-Lerman scratch) may be heard at the left second intercostal space during expiration and is thought to result from the hyperdynamic cardiac motion.
Elderly patients with hyperthyroidism may present with only cardiovascular manifestations of thyrotoxicosis such as sinus tachycardia, atrial fibrillation, and hypertension, all of which may be resistant to therapy until the hyperthyroidism is controlled. Angina pectoris and CHF are unusual with hyperthyroidism unless there is coexistent heart disease; in such cases, symptoms often resolve with treatment of the hyperthyroidism.
Hypothyroidism Cardiac manifestations of hypothyroidism include a reduction in cardiac output, stroke volume, heart rate, systolic blood pressure, and pulse pressure. Pericardial effusions are present in about one-third of patients, rarely progress to tamponade, and probably result from increased capillary permeability. Other clinical signs include cardiomegaly, bradycardia, weak arterial pulses, distant heart sounds, and pleural effusions. Although the signs and symptoms of myxedema may mimic those of CHF, in the absence of other cardiac disease, myocardial failure is uncommon. The ECG generally reveals sinus bradycardia and low voltage and may show prolongation of the QT interval, decreased P-wave voltage, prolonged AV conduction time, intraventricular conduction disturbances, and nonspecific ST-T-wave abnormalities. Chest x-ray may show cardiomegaly, often with a “water bottle” configuration; pleural effusions; and, in some cases, evidence of CHF. Pathologically, the heart is pale and dilated and often demonstrates myofibrillar swelling, loss of striations, and interstitial fibrosis.
Patients with hypothyroidism frequently have elevations of cholesterol and triglycerides, resulting in premature atherosclerotic CAD. Before treatment with thyroid hormone, patients with hypothyroidism frequently do not have angina pectoris, presumably because of the low metabolic demands caused by their condition. However, angina and myocardial infarction may be precipitated during initiation of thyroid hormone replacement, especially in elderly patients with underlying heart disease. Therefore, replacement should be done with care, starting with low doses that are increased gradually.
MALIGNANT CARCINOID
(See also Chap. 113) Carcinoid tumors most often originate in the small bowel and elaborate a variety of vasoactive amines (e.g., serotonin), kinins, indoles, and prostaglandins that are believed to be responsible for the diarrhea, flushing, and labile blood pressure that characterize the carcinoid syndrome. Some 50% of patients with carcinoid syndrome have cardiac involvement, usually manifesting as abnormalities of the tricuspid or pulmonic valves. These patients invariably have hepatic metastases that allow vasoactive substances to circumvent hepatic metabolism. Left-sided cardiac involvement is rare and indicates either pulmonary carcinoid or an intracardiac shunt. Pathologically, carcinoid lesions are fibrous plaques that consist of smooth-muscle cells embedded in a stroma of glycosaminoglycans and collagen. They occur on the cardiac valves, where they cause valvular dysfunction, as well as on the endothelium of the cardiac chambers and great vessels.
Carcinoid heart disease most often presents as tricuspid regurgitation, pulmonic stenosis, or both. In some cases, a high cardiac output state may occur, presumably as a result of a decrease in systemic vascular resistance resulting from vasoactive substances released by the tumor. Treatment with somatostatin analogues (e.g., octreotide) or interferon α improves symptoms and survival in patients with carcinoid heart disease but does not appear to improve valvular abnormalities. Treatment with diuretics usually mitigates the symptoms of right heart failure; in some severely symptomatic patients, valve replacement is indicated. Coronary artery spasm, presumably due to a circulating vasoactive substance, may occur in patients with carcinoid syndrome.
PHEOCHROMOCYTOMA
(See also Chap. 407) In addition to causing labile or sustained hypertension, the high circulating levels of catecholamines resulting from a pheochromocytoma may cause direct myocardial injury. Focal myocardial necrosis and inflammatory cell infiltration are present in ~50% of patients who die with pheochromocytoma and may contribute to clinically significant left ventricular failure and pulmonary edema. In addition, associated hypertension results in left ventricular hypertrophy. Left ventricular dysfunction and CHF may resolve after removal of the tumor.
ACROMEGALY
(See also Chap. 401e) Exposure of the heart to excessive growth hormone may cause CHF as a result of high cardiac output, diastolic dysfunction owing to ventricular hypertrophy (with increased left ventricular chamber size or wall thickness), or global systolic dysfunction. Hypertension occurs in up to one-third of patients with acromegaly and is characterized by suppression of the renin-angiotensin-aldosterone axis and increases in total-body sodium and plasma volume. Some form of cardiac disease occurs in about one-third of patients with acromegaly and is associated with a doubling of the risk of cardiac death.
RHEUMATOID ARTHRITIS AND THE COLLAGEN VASCULAR DISEASES
Rheumatoid Arthritis (See also Chap. 380) Rheumatoid arthritis may be associated with inflammatory changes in any or all cardiac structures, although pericarditis is the most common clinical entity. Pericardial effusions are found on echocardiography in 10–50% of patients with rheumatoid arthritis, particularly those with subcutaneous nodules. Nonetheless, only a small fraction of these patients have symptomatic pericarditis, and when present, it usually follows a benign course, only occasionally progressing to cardiac tamponade or constrictive pericarditis. The pericardial fluid is generally exudative, with decreased concentrations of complement and glucose and elevated cholesterol. Coronary arteritis with intimal inflammation and edema is present in ~20% of cases but only rarely results in angina pectoris or myocardial infarction. Inflammation and granuloma formation may affect the cardiac valves, most often the mitral and aortic valves, and may cause clinically significant regurgitation owing to valve deformity. Myocarditis is uncommon and rarely results in cardiac dysfunction.
Treatment is directed at the underlying rheumatoid arthritis and may include glucocorticoids. Urgent pericardiocentesis should be performed in patients with tamponade, but pericardiectomy usually is required in cases of pericardial constriction.
Seronegative Arthropathies (See also Chap. 384) The seronegative arthropathies, including ankylosing spondylitis, reactive arthritis, psoriatic arthritis, and the arthritides associated with ulcerative colitis and regional enteritis, are all strongly associated with the HLA-B27 histocompatibility antigen and may be accompanied by a pancarditis and proximal aortitis. The aortic inflammation usually is limited to the aortic root but may extend to involve the aortic valve, mitral valve, and ventricular myocardium, resulting in aortic and mitral regurgitation, conduction abnormalities, and ventricular dysfunction. One-tenth of these patients have significant aortic insufficiency, and one-third have conduction disturbances; both are more common in patients with peripheral joint involvement and long-standing disease. Treatment with aortic valve replacement and permanent pacemaker implantation may be required. Occasionally, aortic regurgitation precedes the onset of arthritis, and therefore, the diagnosis of a seronegative arthritis should be considered in young males with isolated aortic regurgitation.
Systemic Lupus Erythematosus (SLE) (See also Chap. 378) A significant percentage of patients with SLE have cardiac involvement. Pericarditis is common, occurring in about two-thirds of patients, and generally follows a benign course, although rarely tamponade or constriction may result. The characteristic endocardial lesions of SLE are verrucous valvular abnormalities known as Libman-Sacks endocarditis. They most often are located on the left-sided cardiac valves, particularly on the ventricular surface of the posterior mitral leaflet, and are made up almost entirely of fibrin. These lesions may embolize or become infected but rarely cause hemodynamically important valvular regurgitation. Myocarditis generally parallels the activity of the disease and, although common histologically, seldom results in clinical heart failure unless associated with hypertension. Although arteritis of epicardial coronary arteries may occur, it rarely results in myocardial ischemia. There is, however, an increased incidence of coronary atherosclerosis that probably is related more to associated risk factors and glucocorticoid use than to SLE itself. Patients with the antiphospholipid antibody syndrome may have a higher incidence of cardiovascular abnormalities, including valvular regurgitation, venous and arterial thrombosis, premature stroke, myocardial infarction, pulmonary hypertension, and cardiomyopathy.
SECTION 5 | CORONARY AND PERIPHERAL VASCULAR DISEASE |
291e | The Pathogenesis, Prevention, and Treatment of Atherosclerosis |
PATHOGENESIS
Atherosclerosis remains the major cause of death and premature disability in developed societies. Moreover, current predictions estimate that by the year 2020 cardiovascular diseases, notably atherosclerosis, will become the leading global cause of total disease burden. Although many generalized or systemic risk factors predispose to its development, atherosclerosis affects various regions of the circulation preferentially and has distinct clinical manifestations that depend on the particular circulatory bed affected. Atherosclerosis of the coronary arteries commonly causes myocardial infarction (MI) (Chap. 295) and angina pectoris (Chap. 293). Atherosclerosis of the arteries supplying the central nervous system frequently provokes strokes and transient cerebral ischemia (Chap. 446). In the peripheral circulation, atherosclerosis causes intermittent claudication and gangrene and can jeopardize limb viability. Involvement of the splanchnic circulation can cause mesenteric ischemia. Atherosclerosis can affect the kidneys either directly (e.g., renal artery stenosis) or as a common site of atheroembolic disease (Chap. 301).
Even within a particular arterial bed, stenoses due to atherosclerosis tend to occur focally, typically in certain predisposed regions. In the coronary circulation, for example, the proximal left anterior descending coronary artery exhibits a particular predilection for developing atherosclerotic disease. Similarly, atherosclerosis preferentially affects the proximal portions of the renal arteries and, in the extracranial circulation to the brain, the carotid bifurcation. Indeed, atherosclerotic lesions often form at branch points of arteries, regions characterized disturbed hydrodynamics. Not all manifestations of atherosclerosis result from stenotic, occlusive disease. Ectasia and the development of aneurysmal disease, for example, frequently occur in the aorta (Chap. 301). In addition to focal, flow-limiting stenoses, nonocclusive intimal atherosclerosis also occurs diffusely in affected arteries, as shown by intravascular imaging and postmortem studies.
Atherogenesis in humans typically occurs over a period of many years, usually many decades. Growth of atherosclerotic plaques probably does not occur in a smooth, linear fashion but discontinuously, with periods of relative quiescence punctuated by periods of rapid evolution. After a generally prolonged “silent” period, atherosclerosis may become clinically manifest. The clinical expressions of atherosclerosis may be chronic, as in the development of stable, effort-induced angina pectoris or predictable and reproducible intermittent claudication. Alternatively, a dramatic acute clinical event such as MI, stroke, or sudden cardiac death may first herald the presence of atherosclerosis. Other individuals may never experience clinical manifestations of arterial disease despite the presence of widespread atherosclerosis demonstrated postmortem.
INITIATION OF ATHEROSCLEROSIS
An integrated view of experimental results in animals and studies of human atherosclerosis suggests that the “fatty streak” represents the initial lesion of atherosclerosis. These early lesions most often seem to arise from focal increases in the content of lipoproteins within regions of the intima. In particular, the fraction of lipoproteins related to low-density lipoprotein (LDL) that bear apolipoprotein B appear causally related to atherosclerosis. This accumulation of lipoprotein particles may not result simply from increased permeability, or “leakiness,” of the overlying endothelium (Fig. 291e-1). Rather, the lipoproteins may collect in the intima of arteries because they bind to constituents of the extracellular matrix, increasing the residence time of the lipid-rich particles within the arterial wall. Lipoproteins that accumulate in the extracellular space of the intima of arteries often associate with proteoglycans of the arterial extracellular matrix, an interaction that may slow the egress of these lipid-rich particles from the intima. Lipoprotein particles in the extracellular space of the intima, particularly those retained by binding to matrix macromolecules, may undergo oxidative modifications. Considerable evidence supports a pathogenic role for products of oxidized lipoproteins in atherogenesis. Lipoproteins sequestered from (plasma) antioxidants in the extracellular space of the intima become particularly susceptible to oxidative modification, giving rise to hydroperoxides, lysophospholipids, oxysterols, and aldehydic breakdown products of fatty acids and phospholipids. Modifications of the apoprotein moieties may include breaks in the peptide backbone as well as derivatization of certain amino acid residues. Local production of hypochlorous acid by myeloperoxidase associated with inflammatory cells within the plaque yields chlorinated species such as chlorotyrosyl moieties. Considerable evidence supports the presence of such oxidation products in atherosclerotic lesions.
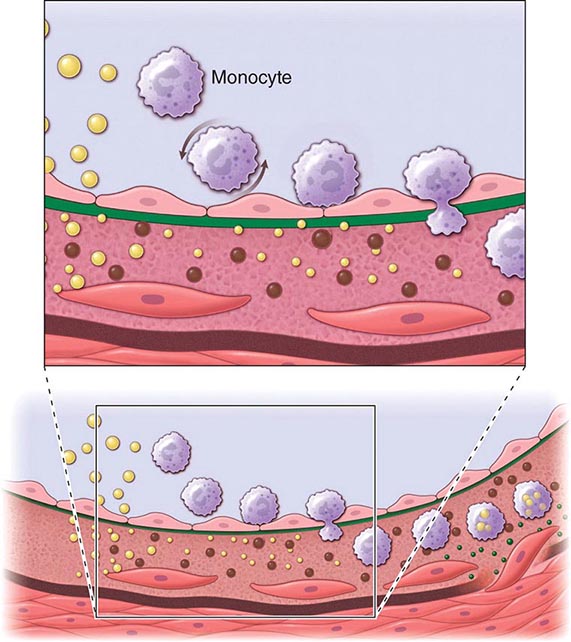
FIGURE 291e-1 Cross-sectional view of an artery depicting steps in development of an atheroma, from left to right. The upper panel shows a detail of the boxed area below. The endothelial monolayer overlying the intima contacts blood. Hypercholesterolemia promotes accumulation of low-density lipoprotein (LDL) particles (yellow spheres) in the intima. The lipoprotein particles often associate with constituents of the extracellular matrix, notably proteoglycans. Sequestration within the intima separates lipoproteins from some plasma antioxidants and favors oxidative modification. Such modified lipoprotein particles (darker spheres) may trigger a local inflammatory response that signals subsequent steps in lesion formation. The augmented expression of various adhesion molecules for leukocytes recruits monocytes to the site of a nascent arterial lesion.
Once adherent, some white blood cells migrate into the intima. The directed migration of leukocytes probably depends on chemoattractant factors, including modified lipoprotein particles themselves and chemoattractant cytokines (depicted by the smaller green spheres), such as the chemokine macrophage chemoattractant protein-1 produced by vascular wall cells in response to modified lipoproteins. Leukocytes in the evolving fatty streak can divide and exhibit augmented expression of receptors for modified lipoproteins (scavenger receptors). These mononuclear phagocytes ingest lipids and become foam cells, represented by a cytoplasm filled with lipid droplets. As the fatty streak evolves into a more complicated atherosclerotic lesion, smooth-muscle cells migrate from the media (bottom of lower panel hairline) through the internal elastic membrane (solid wavy line) and accumulate within the expanding intima, where they lay down extracellular matrix that forms the bulk of the advanced lesion (bottom panel, right side).
Leukocyte Recruitment Accumulation of leukocytes characterizes the formation of early atherosclerotic lesions (Fig. 291e-1). Thus, from its very inception, atherogenesis involves elements of inflammation, a process that now provides a unifying theme in the pathogenesis of this disease. The inflammatory cell types typically found in the evolving atheroma include monocyte-derived macrophages and dendritic cells, T and B lymphocytes, and mast cells. Hypercholesterolemia augments the portion of particularly proinflammatory monocytes in blood that preferentially enter the nascent atheroma in mice. A number of adhesion molecules or receptors for leukocytes expressed on the surface of the arterial endothelial cell probably participate in the recruitment of leukocytes to the nascent atheroma. Proinflammatory cytokines can augment the expression of leukocyte adhesion molecules.
Laminar shear forces such as those encountered in most regions of normal arteries also can suppress the expression of leukocyte adhesion molecules. Sites of predilection for atherosclerotic lesions (e.g., distal to flow dividers) often have low shear stress and/or disturbed flow. Ordered, pulsatile laminar shear of normal blood flow augments the production of nitric oxide by endothelial cells. This molecule, in addition to its vasodilator properties, can act at the low levels constitutively produced by arterial endothelium as a local anti-inflammatory autacoid, e.g., limiting local adhesion molecule expression. Exposure of endothelial cells to laminar shear stress increases the transcription of Krüppel-like factor 2 (KLF2), which augments the activity of numerous salutary endothelial functions including nitric oxide synthase. Laminar shear stress also stimulates endothelial cells to produce superoxide dismutase, an antioxidant enzyme. These examples indicate how hemodynamic forces may influence the cellular events that underlie atherosclerotic lesion initiation and potentially explain the favored localization of atherosclerotic lesions at sites that experience disturbed flow or low shear stress.
Once captured on the surface of the arterial endothelial cell by adhesion receptors, the leukocytes penetrate the endothelial layer and take up residence in the intima. In addition to products of modified lipoproteins, cytokines (protein mediators of inflammation) can regulate the expression of adhesion molecules involved in leukocyte recruitment. For example, interleukin 1 (IL-1) and tumor necrosis factor (TNF) induce or augment the expression of leukocyte adhesion molecules on endothelial cells. Because products of lipoprotein oxidation can induce cytokine release from vascular wall cells, this pathway may provide an additional link between arterial accumulation of lipoproteins and leukocyte recruitment. Chemoattractant cytokines appear to direct the migration of leukocytes into the arterial wall.
Foam-Cell Formation Once resident within the intima, the mononuclear phagocytes mature into macrophages and become lipid-laden foam cells, a conversion that requires the uptake of lipoprotein particles by receptor-mediated endocytosis. One might suppose that the “classic” LDL receptor mediates this lipid uptake; however, humans or animals lacking effective LDL receptors due to genetic alterations (e.g., familial hypercholesterolemia) have abundant arterial lesions and extraarterial xanthomata rich in macrophage-derived foam cells. In addition, the exogenous cholesterol suppresses expression of the LDL receptor; thus, the level of this cell-surface receptor for LDL decreases under conditions of cholesterol excess. Candidates for alternative receptors that can mediate lipid loading of foam cells include a number of macrophage “scavenger” receptors, which preferentially endocytose modified lipoproteins, and other receptors for oxidized LDL or very low-density lipoprotein (VLDL). Monocyte attachment to the endothelium, migration into the intima, and maturation to form lipid-laden macrophages thus represent key steps in the formation of the fatty streak, the precursor of fully formed atherosclerotic plaques.
ATHEROMA EVOLUTION AND COMPLICATIONS
Although the fatty streak commonly precedes the development of a more advanced atherosclerotic plaque, not all fatty streaks progress to form complex atheromata. By ingesting lipids from the extracellular space, the mononuclear phagocytes bearing such scavenger receptors may remove lipoproteins from the developing lesion. Some lipid-laden macrophages may leave the artery wall, exporting lipid in the process. Lipid accumulation, and hence the propensity to form an atheroma, ensues if the amount of lipid entering the artery wall exceeds that removed by mononuclear phagocytes or other pathways. Macrophages also proliferate in plaques in response to hematopoietic growth factors overexpressed in lesions, another aspect of the dynamic regulation and flux of cells during atherogenesis.
Export by phagocytes may constitute one response to local lipid overload in the evolving lesion. Another mechanism, reverse cholesterol transport mediated by high-density lipoproteins (HDLs), probably provides an independent pathway for lipid removal from atheroma. This transfer of cholesterol from the cell to the HDL particle involves specialized cell-surface molecules such as the ATP binding cassette (ABC) transporters. ABCA1, the gene mutated in Tangier disease, a condition characterized by very low HDL levels, transfers cholesterol from cells to nascent HDL particles and ABCG1 to mature HDL particles. “Reverse cholesterol transport” mediated by these ABC transporters allows HDL loaded with cholesterol to deliver it to hepatocytes by binding to scavenger receptor B1 or other receptors. The liver cell can metabolize the sterol to bile acids that can be excreted. Thus, macrophages may play a vital role in the dynamic economy of lipid accumulation in the arterial wall during atherogenesis.
Some lipid-laden foam cells within the expanding intimal lesion perish. Some foam cells may die as a result of programmed cell death, or apoptosis. This death of mononuclear phagocytes results in the formation of the lipid-rich center, often called the necrotic core, in established atherosclerotic plaques. Impaired clearance of dead foam cells (efferocytosis) in plaques may hasten lipid core formation. Macrophages loaded with modified lipoproteins may elaborate microparticles or exosomes (which may contain regulatory microRNAs), cytokines, and growth factors that can further signal some of the cellular events in lesion complication. Whereas accumulation of lipid-laden macrophages characterizes the fatty streak, buildup of fibrous tissue formed by extracellular matrix typifies the more advanced atherosclerotic lesion. The smooth-muscle cell synthesizes the bulk of the extracellular matrix of the complex atherosclerotic lesion. A number of growth factors or cytokines elaborated by mononuclear phagocytes can stimulate smooth-muscle cell proliferation and production of extracellular matrix. Cytokines found in the plaque, including IL-1 and TNF, can induce local production of growth factors, including forms of platelet-derived growth factor (PDGF), fibroblast growth factors, and others, which may contribute to plaque evolution and complication. Other cytokines, notably interferon γ (IFN-γ) derived from activated T cells within lesions, can limit the synthesis of interstitial forms of collagen by smooth-muscle cells. These examples illustrate how atherogenesis involves a complex mix of mediators that in the balance determines the characteristics of particular lesions.
The accumulation of smooth-muscle cells and their elaboration of extracellular matrix probably provide a critical transition, yielding a fibrofatty lesion in place of a simple accumulation of macrophage-derived foam cells. For example, PDGF elaborated by activated platelets, macrophages, and endothelial cells can stimulate the migration of smooth-muscle cells normally resident in the tunica media into the intima. Such growth factors and cytokines produced locally can stimulate the proliferation of resident smooth-muscle cells or resident stem cells in the intima as well as those that may migrate in from the media. Transforming growth factor β (TGF-β), among other mediators, potently stimulates interstitial collagen production by smooth-muscle cells. These mediators may arise not only from neighboring vascular cells or leukocytes (a “paracrine” pathway), but also, in some instances, from the same cell that responds to the factor (an “autocrine” pathway). Together, these alterations in smooth-muscle cells, signaled by these mediators acting at short distances, can hasten transformation of the fatty streak into a more fibrous smooth-muscle cell and extracellular matrix—rich lesion.
In addition to locally produced mediators, products of blood coagulation and thrombosis likely contribute to atheroma evolution and complication. This involvement justifies the use of the term atherothrombosis to convey the inextricable links between atherosclerosis and thrombosis. Fatty streak formation begins beneath a morphologically intact endothelium. In advanced fatty streaks, however, microscopic breaches in endothelial integrity may occur. Microthrombi rich in platelets can form at such sites of limited endothelial denudation, owing to exposure of the thrombogenic extracellular matrix of the underlying basement membrane. Activated platelets release numerous factors that can promote the fibrotic response, including PDGF and TGF-β. Thrombin not only generates fibrin during coagulation, but also stimulates protease-activated receptors that can signal smooth-muscle migration, proliferation, and extracellular matrix production. Many arterial mural microthrombi resolve without clinical manifestation by a process of local fibrinolysis, resorption, and endothelial repair, yet can lead to lesion progression by stimulating these profibrotic functions of smooth-muscle cells (Fig. 291e-2D).
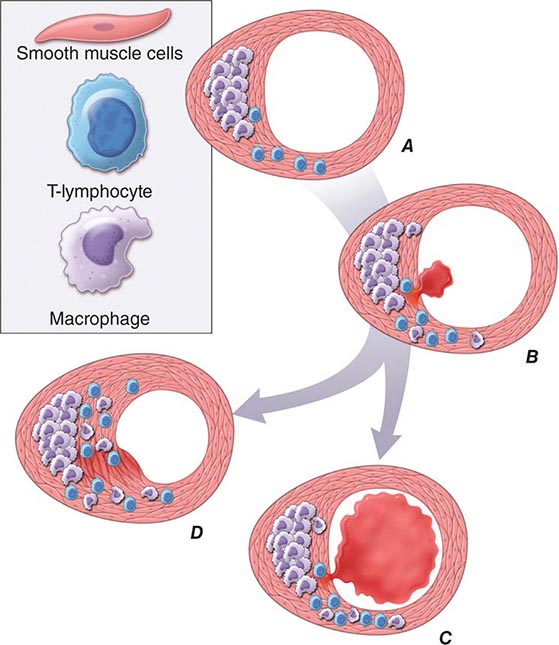
FIGURE 291e-2 Plaque rupture, thrombosis, and healing. A. Arterial remodeling during atherogenesis. During the initial part of the life history of an atheroma, growth is often outward, preserving the caliber of the lumen. This phenomenon of “compensatory enlargement” accounts in part for the tendency of coronary arteriography to underestimate the degree of atherosclerosis. B. Rupture of the plaque’s fibrous cap causes thrombosis. Physical disruption of the atherosclerotic plaque commonly causes arterial thrombosis by allowing blood coagulant factors to contact thrombogenic collagen found in the arterial extracellular matrix and tissue factor produced by macrophage-derived foam cells in the lipid core of lesions. In this manner, sites of plaque rupture form the nidus for thrombi. The normal artery wall has several fibrinolytic or antithrombotic mechanisms that tend to resist thrombosis and lyse clots that begin to form in situ. Such antithrombotic or thrombolytic molecules include thrombomodulin, tissue- and urokinase-type plasminogen activators, heparan sulfate proteoglycans, prostacyclin, and nitric oxide. C. When the clot overwhelms the endogenous fibrinolytic mechanisms, it may propagate and lead to arterial occlusion. The consequences of this occlusion depend on the degree of existing collateral vessels. In a patient with chronic multivessel occlusive coronary artery disease (CAD), collateral channels have often formed. In such circumstances, even a total arterial occlusion may not lead to myocardial infarction (MI), or it may produce an unexpectedly modest or a non-ST-segment elevation infarct because of collateral flow. In a patient with less advanced disease and without substantial stenotic lesions to provide a stimulus for collateral vessel formation, sudden plaque rupture and arterial occlusion commonly produces an ST-segment elevation infarction. These are the types of patients who may present with MI or sudden death as a first manifestation of coronary atherosclerosis. In some cases, the thrombus may lyse or organize into a mural thrombus without occluding the vessel. Such instances may be clinically silent. D. The subsequent thrombin-induced fibrosis and healing causes a fibroproliferative response that can lead to a more fibrous lesion that can produce an eccentric plaque that causes a hemodynamically significant stenosis. In this way, a nonocclusive mural thrombus, even if clinically silent or causing unstable angina rather than infarction, can provoke a healing response that can promote lesion fibrosis and luminal encroachment. Such a sequence of events may convert a “vulnerable” atheroma with a thin fibrous cap that is prone to rupture into a more “stable” fibrous plaque with a reinforced cap. Angioplasty of unstable coronary lesions may “stabilize” the lesions by a similar mechanism, producing a wound followed by healing.
Microvessels As atherosclerotic lesions advance, abundant plexi of microvessels develop in connection with the artery’s vasa vasorum. Newly developing microvascular networks may contribute to lesion complications in several ways. These blood vessels provide an abundant surface area for leukocyte trafficking and may serve as the portal for entry and exit of white blood cells from the established atheroma. Microvessels in the plaques may also furnish foci for intraplaque hemorrhage. Like the neovessels in the diabetic retina, microvessels in the atheroma may be friable and prone to rupture and can produce focal hemorrhage. Such a vascular leak can provoke thrombosis in situ, yielding local thrombin generation, which in turn can activate smooth-muscle and endothelial cells through ligation of protease-activated receptors. Atherosclerotic plaques often contain fibrin and hemosiderin, an indication that episodes of intraplaque hemorrhage contribute to plaque complications.
CALCIFICATION As they advance, atherosclerotic plaques also accumulate calcium. Microvesicles derived from lesional cells can stimulate calcification, and this process co-localizes with regions of heightened inflammation. Mineralization of the atherosclerotic plaque recapitulates many aspects of bone formation, including the regulatory participation of transcription factors such as Runx2.
Plaque Evolution Smooth-muscle cells and macrophages die in the atherosclerotic plaque. Indeed, complex atheromata often have a mostly fibrous character and lack the cellularity of less advanced lesions. This relative paucity of smooth-muscle cells in advanced atheromata may result from the predominance of cytostatic mediators such as TGF-β and IFN-γ (which can inhibit smooth-muscle cell proliferation) and also from smooth-muscle cell apoptosis. Thus, during the evolution of the atherosclerotic plaque, a complex and highly regulated balance between entry and egress of lipoproteins and leukocytes, cell proliferation and cell death, extracellular matrix production, and remodeling, as well as calcification and neovascularization, contribute to lesion formation. Many mediators related to atherogenic risk factors, including those derived from lipoproteins, cigarette smoking, and angiotensin II, provoke the production of proinflammatory cytokines and alter the behavior of the intrinsic vascular wall cells and infiltrating leukocytes that underlie the complex pathogenesis of these lesions. Thus, advances in vascular biology have led to increased understanding of the mechanisms that link risk factors to the pathogenesis of atherosclerosis and its complications.
PATHOPHYSIOLOGIC CONSEQUENCES OF ATHEROSCLEROSIS
Atherosclerotic lesions occur ubiquitously in Western societies, and the prevalence of this disease is on the rise globally. Most atheromata produce no symptoms, and many never cause clinical manifestations. Numerous patients with diffuse atherosclerosis may succumb to unrelated illnesses without ever having experienced a clinically significant manifestation of atherosclerosis. Arterial remodeling during atheroma formation accounts for some of this variability in the clinical expression of atherosclerotic disease (Fig. 291e-2A). During the initial phases of atheroma development, the plaque usually grows outward, in an abluminal direction. Vessels affected by atherogenesis tend to increase in diameter, a phenomenon known as compensatory enlargement, a type of vascular remodeling. The growing atheroma does not encroach on the arterial lumen until the burden of atherosclerotic plaque exceeds ~40% of the area encompassed by the internal elastic lamina. Thus, during much of its life history, an atheroma will not cause stenosis that can limit tissue perfusion.
Flow-limiting stenoses commonly form later in the history of the plaque. Many such plaques cause stable syndromes such as demand-induced angina pectoris or intermittent claudication in the extremities. In the coronary circulation and other circulations, even total vascular occlusion by an atheroma does not invariably lead to infarction. The hypoxic stimulus of repeated bouts of ischemia characteristically induces formation of collateral vessels in the myocardium, mitigating the consequences of an acute occlusion of an epicardial coronary artery. By contrast, many lesions that cause acute or unstable atherosclerotic syndromes, particularly in the coronary circulation, may arise from atherosclerotic plaques that do not produce a flow-limiting stenosis. Such lesions may produce only minimal luminal irregularities on traditional angiograms and often do not meet the traditional criteria for “significance” by arteriography. Thrombi arising from such nonocclusive stenoses may explain the frequency of MI as an initial manifestation of coronary artery disease (CAD) (in at least one-third of cases) in patients who report no prior history of angina pectoris, a syndrome usually caused by flow-limiting stenoses.
Plaque Instability and Rupture Postmortem studies afford considerable insight into the microanatomic substrate underlying the “instability” of plaques that do not cause critical stenoses. A superficial erosion of the endothelium or a frank plaque rupture or fissure usually produces the thrombus that causes episodes of unstable angina pectoris or the occlusive and relatively persistent thrombus that causes acute MI (Fig. 291e-2B). Rupture of the plaque’s fibrous cap (Fig. 291e-2C) permits contact between coagulation factors in the blood and highly thrombogenic tissue factor expressed by macrophage foam cells in the plaque’s lipid-rich core. If the ensuing thrombus is nonocclusive or transient, the episode of plaque disruption may not cause symptoms or may result in episodic ischemic symptoms such as rest angina. Occlusive thrombi that endure often cause acute MI, particularly in the absence of a well-developed collateral circulation that supplies the affected territory. Repetitive episodes of plaque disruption and healing provide one likely mechanism of transition of the fatty streak to a more complex fibrous lesion (Fig. 291e-2D). The healing process in arteries, as in skin wounds, involves the laying down of new extracellular matrix and fibrosis.
Not all atheromata exhibit the same propensity to rupture. Pathologic studies of culprit lesions that have caused acute MI reveal several characteristic features. Plaques that have caused thromboses tend to have thin fibrous caps, relatively large lipid cores, a high content of macrophages, outward remodeling, and spotty (rather than dense) calcification. Morphometric studies of such culprit lesions show that at sites of plaque rupture, macrophages and T lymphocytes predominate and contain relatively few smooth-muscle cells. The cells that concentrate at sites of plaque rupture bear markers of inflammatory activation. In addition, patients with active atherosclerosis and acute coronary syndromes display signs of disseminated inflammation. Inflammatory mediators regulate processes that govern the integrity of the plaque’s fibrous cap and, hence, its propensity to rupture. For example, the T cell–derived cytokine IFN-γ, which is found in atherosclerotic plaques, can inhibit growth and collagen synthesis of smooth-muscle cells, as noted above. Cytokines derived from activated macrophages and lesional T cells can boost production of proteolytic enzymes that can degrade the extracellular matrix of the plaque’s fibrous cap. Thus, inflammatory mediators can impair the collagen synthesis required for maintenance and repair of the fibrous cap and trigger degradation of extracellular matrix macromolecules, processes that weaken the plaque’s fibrous cap and enhance its susceptibility to rupture (so-called vulnerable plaques, Fig. 291e-3). In contrast to plaques with these features of vulnerability, those with a dense extracellular matrix and relatively thick fibrous cap without substantial tissue factor–rich lipid cores seem generally resistant to rupture and unlikely to provoke thrombosis.
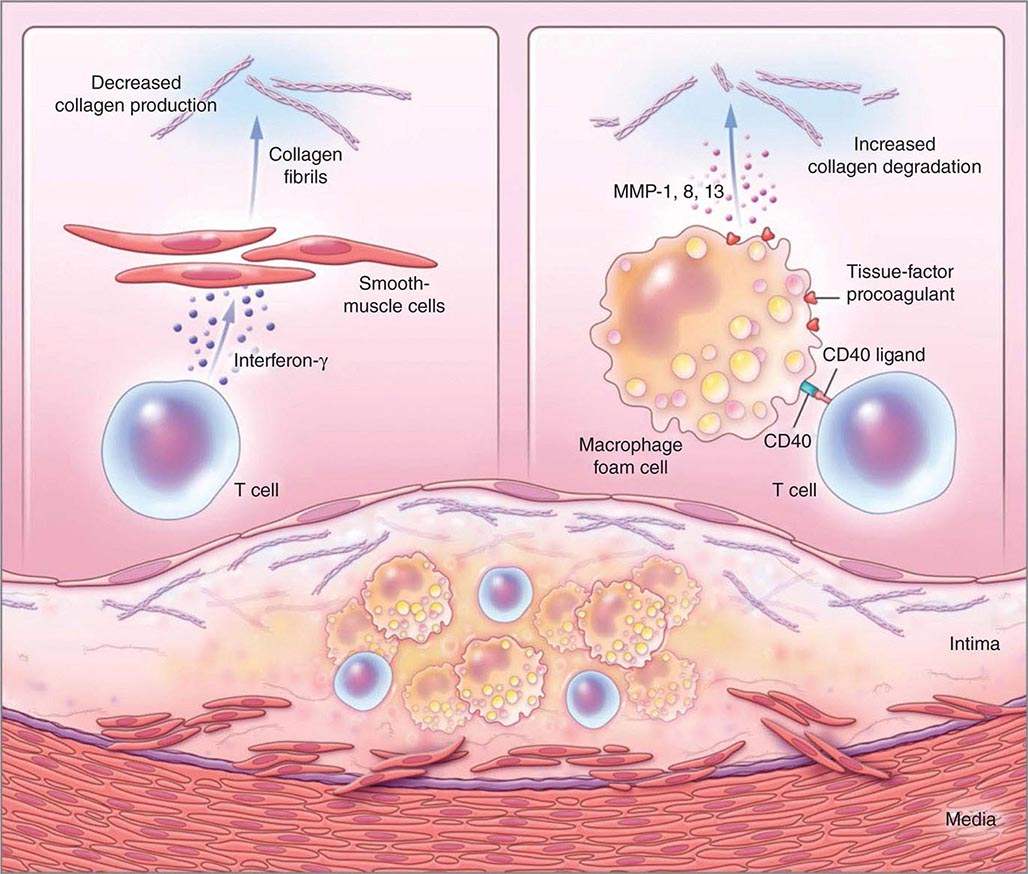
FIGURE 291e-3 Inflammatory pathways that predispose atherosclerotic plaques to rupture and provoke thrombosis. A cross-section of an atheromatous plaque at the bottom of the figure shows the central lipid core that contains macrophage foam cells (yellow) and T cells (blue). The intima and media also contain arterial smooth-muscle cells (red), which are the source of arterial collagen (depicted as triple helical coiled structures). Activated T cells (of the type 1 helper T cell subtype) secrete cytokine interferon γ, which inhibits the production of the new, interstitial collagen that is required to repair and maintain the plaque’s protective fibrous cap (upper left). The T cells can also activate the macrophages in the intimal lesion by expressing the inflammatory mediator CD40 ligand (CD154), which engages its cognate receptor (CD40) on the phagocyte. This inflammatory signalling causes overproduction of interstitial collagenases (matrix metalloproteinases [MMPs] 1, 8, and 13) that catalyze the initial rate-limiting step in collagen breakdown (top right). CD40 ligation also causes macrophages to overproduce tissue-factor procoagulant. Thus, inflammatory signalling puts the collagen in the plaque’s fibrous cap in double jeopardy—decreasing synthesis and increasing breakdown—rendering the cap susceptible to rupture. Inflammatory activation also boosts tissue-factor production, which triggers thrombus formation in the disrupted plaque. These mechanisms link inflammation in the plaque to the thrombotic complications of atherosclerosis, including the acute coronary syndromes. (Adapted from P Libby: N Engl J Med 368:2004, 2013.)
Functional features of the atheromatous plaque, in addition to its degree of luminal encroachment, influence the clinical manifestations of this disease. This enhanced understanding of plaque biology provides insight into the diverse ways in which atherosclerosis can present clinically and the reasons why the disease may remain silent or stable for prolonged periods, punctuated by acute complications at certain times. Increased understanding of atherogenesis provides new insight into the mechanisms linking it to the risk factors discussed below, indicates the ways in which current therapies may improve outcomes, and suggests new targets for future intervention.
PREVENTION AND TREATMENT
THE CONCEPT OF ATHEROSCLEROTIC RISK FACTORS
The systematic study of risk factors for atherosclerosis emerged from a coalescence of experimental results, as well as from cross-sectional and ultimately longitudinal studies in humans. The prospective, community-based Framingham Heart Study provided rigorous support for the concept that hypercholesterolemia, hypertension, and other factors correlate with cardiovascular risk. Similar observational studies performed worldwide bolstered the concept of “risk factors” for cardiovascular disease.
From a practical viewpoint, the cardiovascular risk factors that have emerged from such studies fall into two categories: those modifiable by lifestyle and/or pharmacotherapy, and those that are immutable, such as age and sex. The weight of evidence supporting various risk factors differs. For example, hypercholesterolemia and hypertension certainly predict coronary risk, but the magnitude of the contributions of other so-called nontraditional risk factors, such as levels of homocysteine, levels of lipoprotein (a) [Lp(a)], and infection, remains controversial. Moreover, some biomarkers that predict cardiovascular risk may not participate in the causal pathway for the disease or its complications. Genetic studies using genome-wide association (GWAS) approaches and Mendelian randomization approaches have helped to distinguish between risk markers and factors that contribute causally to the disease. For example, recent genetic studies suggest that C-reactive protein (CRP) does not itself mediate atherogenesis, despite its ability to predict risk, whereas Lp(a) and apolipoprotein C3 have emerged as a causal risk factor. Table 291e-1 lists a number of risk factors implicated in atherosclerosis. The sections below will consider some of these factors and approaches to their modification.
MAJOR RISK FACTORS FOR ATHEROSCLEROSIS |
aHDL cholesterol ≥1.6 mmol/L (≥60 mg/dL) has been viewed as a “negative” risk factor.
Abbreviations: BMI, body mass index; BP, blood pressure; CHD, coronary heart disease; HDL, high-density lipoprotein; LDL, low-density lipoprotein.
Lipid Disorders
Abnormalities in plasma lipoproteins and derangements in lipid metabolism rank among the most firmly established and best understood risk factors for atherosclerosis. Chapter 421 describes the lipoprotein classes and provides a detailed discussion of lipoprotein metabolism.
The American College of Cardiology and American Heart Association (ACC/AHA) promulgated new guidelines on risk assessment, lifestyle measures, and cholesterol management in 2013. The panels that produced these guidelines followed an evidence-based approach. the 2013 cholesterol guideline focused on 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) rather than other classes of lipid-modulating drugs, including fibric acid derivatives, cholesterol absorption inhibitors such as ezetimibe, and niacin products. The guideline cites the lack of contemporary randomized clinical trial evidence that supports the efficacy of these nonstatin lipid-modifying agents in cardiovascular event reduction. The cholesterol guideline defined four statin benefit groups (Table 291e-2): (1) all individuals who have clinical atherosclerotic cardiovascular disease (ASCVD), therefore considered “secondary prevention”; (2) those with LDL cholesterol ≥190 mg/dL without a secondary cause such as a high intake of saturated or trans fats, various drugs, or certain diseases; (3) individuals with diabetes without established cardiovascular disease who are 40–75 years old and have LDL cholesterol of 70–189 mg/dL; and (4) those without established ASCVD without diabetes who are 40–75 years old and who have LDL cholesterol of 70–189 mg/dL and a calculated ASCVD risk ≥7.5%. An online risk calculator based on pooled cohorts was provided to aid clinicians and patients in calculating their risk (http://my.americanheart.org/professional/StatementsGuidelines/PreventionGuidelines/Prevention-Guidelines_UCM_457698_SubHomePage.jsp). Other validated risk calculators that incorporate family history of CAD and a marker of inflammation (high-sensitivity CRP [hsCRP]) that apply to U.S. women and men exist (http://www.reynoldsriskscore.org). Downloadable applications for risk calculation on handheld devices are readily available.
SUMMARY OF THE FOUR STATIN BENEFIT GROUPS DESCRIBED IN THE 2013 ACC/AHA GUIDELINE ON THE TREATMENT OF BLOOD CHOLESTEROL TO REDUCE ATHEROSCLEROTIC CARDIOVASCULAR RISK IN ADULTS |
Abbreviations: ACC/AHA, American College of Cardiology and American Heart Association; ASCVD, atherosclerotic cardiovascular disease; LDL-C, low-density lipoprotein cholesterol.
Source: Adapted from NJ Stone et al: 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults. J Am Coll Cardiol 2013, doi: 10.1016/j.jacc.2013.11.002.
The 2013 guideline emphasized a patient-centered approach and recommended that clinicians and patients engage in a risk-benefit conversation before starting statin therapy and not rely solely on calculated risks or arbitrary category assignment. It further emphasizes that medications do not supplant a healthy lifestyle. The guideline also provides some practical suggestions regarding management of muscle symptoms attributed to statins, an issue of considerable concern to many patients and practitioners alike.
In a major departure from prior guidelines, the 2013 guideline eliminates LDL targets as goals of therapy. The panel did so because major clinical trials did not titrate therapy to a goal, but rather used fixed doses of statins. Instead, the new guideline suggests different intensities of statin therapy based on risk category (Fig. 291e-4).
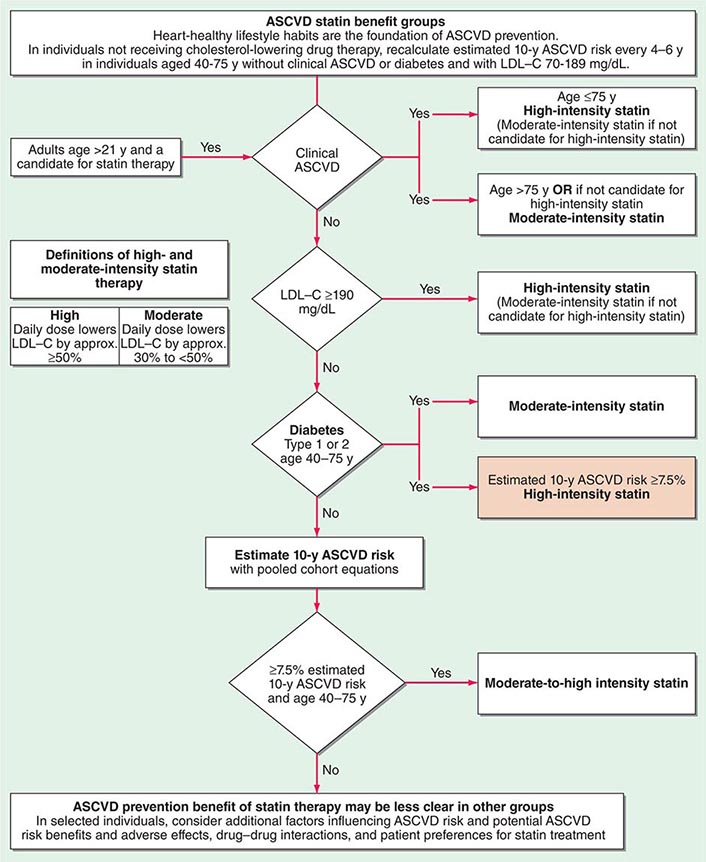
FIGURE 291e-4 Major recommendations for statin therapy for atherosclerotic cardiovascular disease (ASCVD) prevention. LDL-C, low-density lipoprotein cholesterol. (From NJ Stone et al: J Am Coll Cardiol, 2013, doi: 10.1016/j.jacc.2013.11.002.)
The 2013 guideline focus on statins reflects an extensive body of rigorous evidence that supports the effectiveness of this class of drugs in cardiovascular event reduction and an acceptable risk-benefit relationship (Fig. 291e-5). Moreover, because almost all statins are now available as generic statins medications, cost has become much less of an impediment to their use.
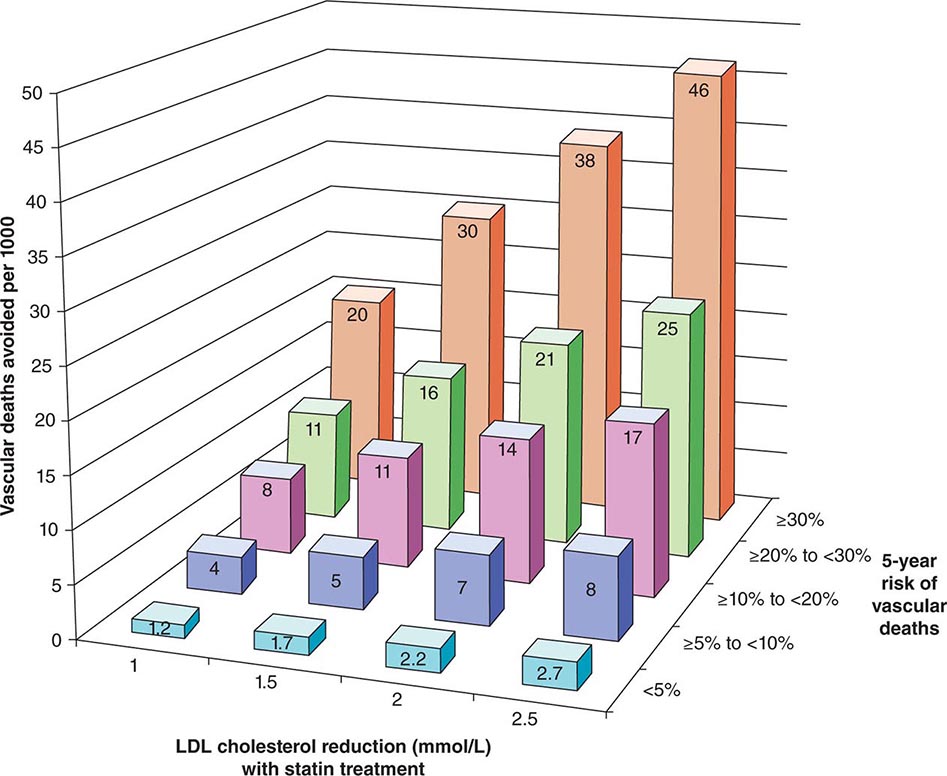
FIGURE 291e-5 The Cholesterol Treatment Trialists Collaboration meta-analyzed 27 randomized clinical trials evaluating statin therapy. They found profound decreases in both major vascular events and vascular death (not shown) proportional to the magnitude of low-density lipoprotein (LDL) cholesterol reduction achieved with statin treatment. This diagram shows the results of this meta-analysis for vascular death. (From Lancet 380:581, 2012.)
The clinical use of effective pharmacologic strategies for lowering LDL has reduced cardiovascular events markedly, but a considerable burden of residual risk remains even in patients treated with high-intensity statins. Hence, current studies are evaluating other avenues to address the residual burden of cardiovascular disease that persists despite statin treatment. Inhibitors of genetic studies identified proprotein convertase subtilisin kexin-like 9 (PCSK9) as a regulator of LDL levels associated with cardiovascular outcomes. Interaction of the LDL receptor with PCSK9 hastens the receptor’s degradation, and hence yields higher circulating LDL concentrations. Genetic variants that lower PCSK9 activity appear to protect against cardiovascular events. Monoclonal antibodies that neutralize PCSK9 lower LDL levels even in statin-treated patients and are currently under investigation as novel therapeutics to lower cardiovascular risk.
LDL-lowering therapies do not appear to exert their beneficial effect on cardiovascular events by causing a marked “regression” of stenoses. Studies of lipid lowering monitored by angiography or by intravascular imaging modalities have shown at best a modest reduction in coronary artery stenoses over the duration of study, despite abundant evidence of event reduction. These results suggest that the beneficial mechanism of lipid lowering by statins does not require a substantial reduction in the fixed stenoses. Rather, the benefit may derive from “stabilization” of atherosclerotic lesions without substantially decreased stenosis. Such stabilization of atherosclerotic lesions and the attendant decrease in coronary events may result from the egress of lipids or from favorably influencing aspects of the biology of atherogenesis discussed above. In addition, as sizable lesions may protrude abluminally rather than into the lumen due to complementary enlargement, shrinkage of such plaques may not be apparent on angiograms. The consistent benefit of statins may depend not only on their salutary effects on the lipid profile, but also on direct modulation of plaque biology independent of lipid lowering.
As the prevalence of metabolic syndrome and diabetes increases, many patients present with low concentrations of HDL (HDL cholesterol <1.0 mmol/L [<40 mg/dL]). A baseline measurement of HDL cholesterol indubitably correlates with future cardiovascular risk. Yet, the utility of therapies that raise HDL cholesterol levels in blood as effective interventions to reduce cardiovascular vascular events has come into question. Blood HDL levels vary inversely with those of triglycerides, and the independent role of HDL versus triglycerides as a cardiovascular risk factor remains unsettled. The 2013 guideline does not advocate any specific therapy for raising HDL. Indeed, multiple recent trials failed to show that raising HDL cholesterol levels improves cardiovascular outcomes, and recent genetic studies cast doubt on low HDL as a causal risk factor for atherosclerotic events. Weight loss and physical activity can raise HDL, and these lifestyle measures merit universal adoption (Table 291e-3). Nicotinic acid, particularly in combination with statins, can robustly raise HDL, but clinical trial data do not support the effectiveness of nicotinic acid in cardiovascular risk reduction. Agonists of nuclear receptors provide another potential avenue for raising HDL levels. Yet patients treated with peroxisome proliferator–activated receptors alpha and gamma (PPAR-α and -γ) agonists have not consistently shown improved cardiovascular outcomes, and at least some PPAR agonists have been associated with worsened cardiovascular outcomes. Other agents in clinical development raise HDL levels by inhibiting cholesteryl ester transfer protein (CETP). Two such agents have undergone large-scale clinical evaluation and have not shown efficacy in improving cardiovascular outcomes. Clinical studies currently under way will assess the effectiveness of two other CETP inhibitors that lack some of the adverse off-target actions encountered with the first agent tested.
HEART HEALTHY NUTRITION AND PHYSICAL ACTIVITY BEHAVIORS RECOMMENDED IN THE 2013 ACC/AHA GUIDELINE ON LIFESTYLE MANAGEMENT TO REDUCE CARDIOVASCULAR RISK |



