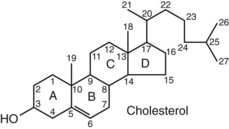
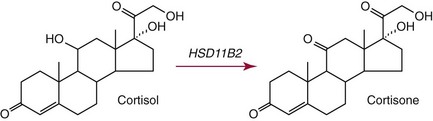
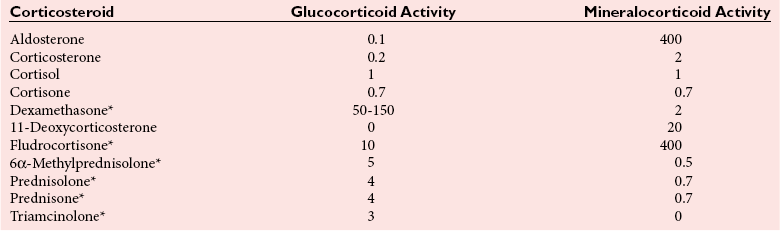
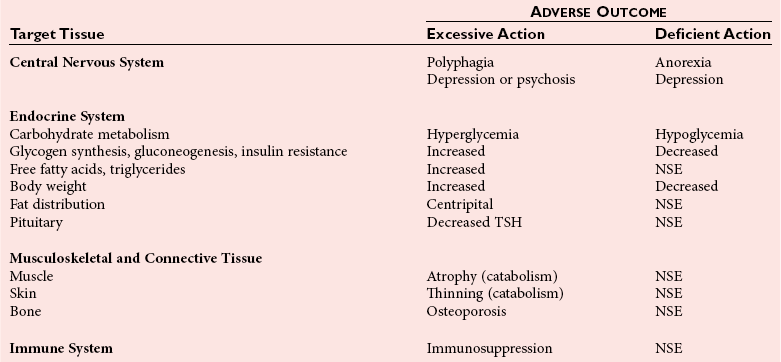
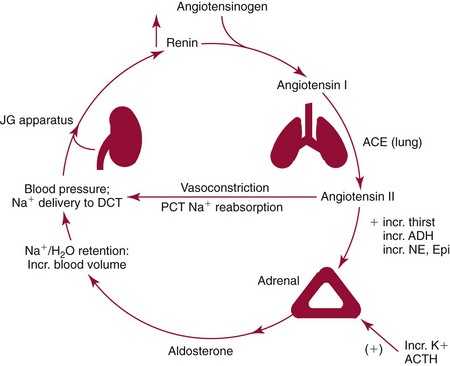
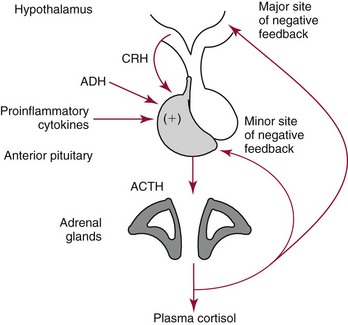
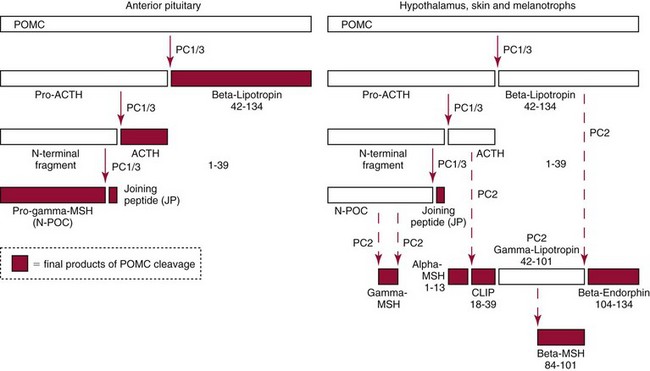
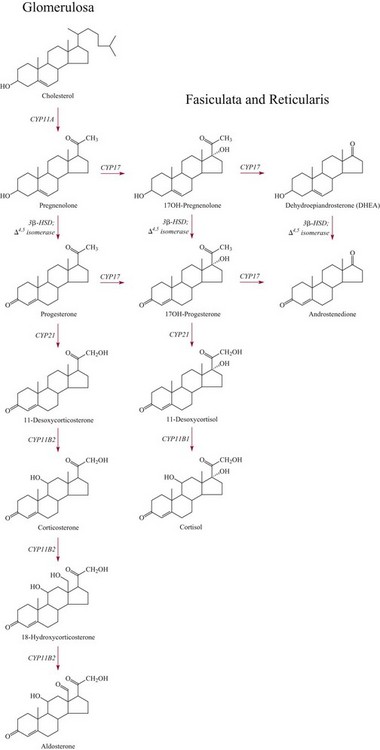
Chapter 54 Roger L. Bertholf, Ph.D., Ishwarlal Jialal, M.D., Ph.D., F.R.C.Path(London), D.A.B.C.C. and William E. Winter, M.D.* The adrenal cortex and gonads produce steroid hormones. Steroids are a class of organic compounds that contain the cyclopentanoperhydrophenanthrene nucleus that is characterized by four aliphatic rings produced by the biosynthetic cyclization of a linear triterpene, squalene, which is produced by the mevalonate pathway (Figure 54-1). Three rings (A, B, and C) are made up of six carbons, and one ring (ring D) is made up of five carbons. Sterols are steroids that have been hydroxylated at position 3 on the A ring; cholesterol is a sterol. Cholesterol has a double bond between carbons 5 and 6 and contains an eight carbon aliphatic side group at position 17. Cholesterol is the precursor for all human steroid hormones. In humans, cholesterol is derived from both dietary sources and de novo synthesis, and is transported principally in low-density lipoproteins (LDLs). As cellular cholesterol concentrations rise, LDL receptor (LDLR) expression declines in an autoregulatory fashion. The synthesis of cholesterol is reviewed in Chapter 27. The adrenal cortex and the gonads share many metabolic pathways in the synthesis of steroid hormones because both are embryologically derived from nearby mesodermal anlagen.148 Two important transcription factors in development of the adrenal cortex are steroidogenic factor-1 (SF-1) and the dosage-sensitive sex reversal adrenal hypoplasia congenita (AHC) on the X-chromosome gene 1 (DAX-1).78 SF-1 regulates DAX-1. Table 54-1 shows the location and action of major products of the adrenal gland. TABLE 54-1 Anatomy and Products of the Adrenal Gland Mineralocorticoids bind to the mineralocorticoid receptor (MR) in the distal convoluted tubule and collecting duct of the nephron, the colon, and the salivary glands to promote sodium reabsorption and potassium and hydrogen ion excretion.352 Similar to receptors for other steroid hormones and thyroid hormone, the MR functions as a transcription factor. When mineralocorticoid binds to the cytoplasmic MRs, the mineralocorticoid-MR complex relocates to the nucleus, where it influences cellular DNA regulating gene transcription. The principal mineralocorticoid is aldosterone, but other compounds with mineralocorticoid activity include 11-desoxycorticosterone (DOC), 18-hydroxycorticosterone, corticosterone, and cortisol. The MR gene encodes a 107 kDa protein and is officially labeled nuclear receptor subfamily 3, group C, member 2 (NR3C2), located on chromosome 4q31.1-31.2. Alternative splicing yields an alpha and beta MR mRNA. Aldosterone stimulates epithelial sodium channel (ENaC) activity via serum and glucocorticoid-induced kinase (SGK) and K-ras, increases expression of mitochondrial ATP-producing genes, and stimulates the basolateral Na+/K+-ATPase pump. ENaC, a highly selective epithelial sodium channel that is amiloride sensitive (amiloride is a K+-sparing diuretic), has three subunits: the alpha subunit (most important of the three subunits: the sodium channel, non–voltage-gated protein 1, alpha coded by SCNN1A, chromosome 12p13), the beta subunit (sodium channel, non–voltage-gated 1, beta SCNN1B, chromosome 16p13-p12), and the gamma subunit (sodium channel, non–voltage-gated 1, gamma SCNN1G, chromosome 16p13-p12).178 All ENaC subunits are similar and contain a large N-terminal extracellular domain, two transmembrane spanning domains (M1 and M2), and a C-terminal, short intracellular domain.140 Although the binding of aldosterone to the MR occurs in the cytoplasm with transit of the complex to the nucleus, some free MR is present in the nucleus. The MR binds cortisol and 11-desoxycorticosterone with affinity equal to that of aldosterone. However, the MR is protected from cortisol and 11-desoxycorticosterone by 11 beta-hydroxysteroid dehydrogenase-2 (HSD11B2), where, for example, HSD11B2 catalyzes the conversion of cortisol to cortisone (Figure 54-2). Cortisone does not bind to the MR. Variations in HSD11B2 may be involved in some cases of otherwise “essential” hypertension.86 Chronic elevations in angiotensin, which controls aldosterone synthesis and release, produces pathologic changes in the heart and kidney by eliciting myocardial fibrosis and inflammatory changes in the renal vasculature.139 The actions of mineralocorticoids are summarized in Table 54-2. TABLE 54-2 Major Actions of Mineralocorticoids Glucocorticoids bind to the glucocorticoid receptor (GR) located in a large number of tissues, including lymphocytes, hepatocytes, and bone.34 The GR gene contains 10 exons (exons 1 through 8 plus 9 alpha and 9 beta). Alternative splicing yields GR alpha and GR beta transcripts. Because of the wide distribution of the GR, glucocorticoid effects are diverse, including changes in intermediary metabolism and immunoregulation. Glucocorticoids raise blood glucose concentrations by enhancing the synthesis of gluconeogenic enzymes (e.g., glucose-6-phosphatase and phosphoenol pyruvate carboxykinase), increase liver glycogen content through activation of glycogen synthase, and inhibit glycogen phosphorylase, producing insulin resistance in both muscle and adipose tissue that further raises blood glucose concentrations. Excessive catabolism of skeletal muscle causes myopathy and consequent weakness. Protein catabolism causes thinning of the skin and loss of strength in connective tissues. With excess glucocorticoids, bone loss may result from collagen catabolism and loss of osteoid, which can lead to fractures and compressed vertebrae. When glucocorticoids bind to the cytoplasmic GR, heat shock proteins (HSPs) are released (HSP70 and HSP90).63 Glucocorticoids are powerful anti-inflammatory hormones that inhibit nuclear factor kappaB (NFkappaB) through the induction of IkappaB synthesis.91 IkappaB binds to NFkappaB in the cytoplasm, impairing the entrance of NFkappaB into the nucleus. Within the nucleus, the GR-cortisol complex binds NFkappaB, preventing its binding to DNA. Finally, within the nucleus, GR-cortisol and NFkappaB compete for cofactors that are available in limited quantities. Although IkappaB synthesis is enhanced, many proinflammatory genes are repressed, such as cyclo-oxygenase 2 (COX-2), inducible nitric oxide synthase (iNOS), various interleukins (IL-1, IL-2, and IL-6), tumor necrosis factor-alpha, interferon-gamma, and E-selectin. Adrenocorticotropic hormone (ACTH) also stimulates the release of IL-1, IL-6, and tumor necrosis factor-alpha.275 Glucocorticoids help maintain vascular tone and cardiac output, and stabilize lysosomal membranes.44 Glucocorticoids suppress hypersensitivity responses by inhibiting the production of histamine by basophils and mast cells. Modest doses of glucocorticoids may improve one’s mood, yet in pharmacologic concentrations, they may produce psychosis. Pathologically elevated concentrations of glucocorticoids are discussed further under the heading of “Cushing syndrome.” The relative potencies of corticosteroids in terms of glucocorticoid and mineralocorticoid activity are given in Table 54-3. The actions of glucocorticoids are summarized in Table 54-4. The adrenal androgens dehydroepiandrosterone (DHEA), dehydroepiandrosterone sulfate (DHEA-S), and androstenedione provide androgenic effects through their peripheral conversion to testosterone, which in turn binds to the androgen receptor (AR) that is described in Chapter 56.149 Between ages 7 and 8, the urinary excretion of 17-ketosteroids (the breakdown products of adrenal androgens) increases as an early sign that puberty will begin in the coming 3 to 5 years.239 Steroid hormones are not stored in hormone-producing cells and therefore must be produced as needed. As lipophilic molecules, steroids pass through cell membranes to exit the hormone-producing cells, enter the circulation to be distributed throughout the body, and enter target cells passing through the target cell membrane into the cytoplasm, where they bind to receptors. Translocation of the hormone-receptor complex to the nucleus initiates the action of the hormone. Growing evidence suggests that steroid hormones may concurrently act independently of their effect on DNA transcription. In the circulation, steroids exist as free and bound species. This is discussed in greater detail in Chapter 56. Aldosterone production and secretion are controlled through the renin-angiotensin system (Figure 54-3).216,342 The rate-limiting component in this system is renin release, which is regulated by the juxtaglomerular apparatus. Anatomically, the juxtaglomerular apparatus is composed of (1) the juxtaglomerular cells of the afferent arteriole that immediately leads to the glomerulus, (2) lacis cells (extraglomerular mesangial cells located at the vascular pole of the renal corpuscle), and (3) the macula densa. Angiotensinogen (≈60 kDa) is an alpha2-globulin synthesized in hepatocytes.211 Renin acts as an aspartyl proteolytic enzyme cleaving the 10 N-terminal amino acids of angiotensinogen to form the decapeptide angiotensin I (Table 54-5). Angiotensin I has no endocrine, paracrine, or autocrine effects. Angiotensin-converting enzyme (ACE), a zinc metallopeptidase, removes the two C-terminal residues from angiotensin I to generate the octapeptide angiotensin II.315 High concentrations of ACE are expressed in the lung. ACE is pathologically expressed in conditions involving macrophage activation such as sarcoidosis. Further degradation of angiotensin II by aminopeptidase A (a glutamyl aminopeptidase) yields the heptapeptide angiotensin III. The ratio of angiotensin II to angiotensin III is usually 4 to 1. An arginyl aminopeptidase generates angiotensin IV from angiotensin III. TABLE 54-5 Angiotensinogen and Derived Peptides The best characterized of the angiotensin receptors are AT1 and AT2 that involve multiple second messenger systems.134 Most functions of angiotensin II are mediated via the AT1 receptor. Some actions of the stimulated AT2 receptor oppose those of the AT1 receptor (e.g., AT2 receptor engagement causes vasodilatation). Cortisol is controlled through a traditional hypothalamic-pituitary-end organ negative feedback system (Figure 54-4). Corticotropin-releasing hormone (CRH) is released by stress, exercise, and hypoglycemia. Examples of physiologic stress include pain, trauma, surgery, and hemorrhage. Examples of psychologic stress include severe anxiety and major depression. Prolonged administration of supraphysiologic doses of glucocorticoids orally or parenterally will suppress the hypothalamic-pituitary-adrenal axis, leading to adrenal atrophy. As a result, abrupt termination of exogenous steroids may induce an acute and possibly life-threatening glucocorticoid insufficiency. Following hypothalamic secretion, CRH reaches the anterior pituitary gland through the hypothalamic pituitary portal system.244 Corticotrophs represent about 20% of functional anterior pituitary cells and express receptors for CRH that promote synthesis, storage, and release of corticotropin (ACTH; 4.5 kDa).110 ACTH is also released by ADH stimulation but to a lesser degree than CRH. The proinflammatory cytokines IL-1, IL-6, and tumor necrosis factor-alpha also release ACTH. There are two CRH receptors (CRH-R1 and CRH-R2) and three splice variants of CRH-2 (CRH-2alpha, CRH-2beta, and CRH-2gamma). CRH mediates its effects on corticotrophs exclusively through CRH-R1. CRH and its receptors are widely distributed throughout the central nervous system (CNS). ACTH is released from its 32 kDa precursor protein, 266 amino acid pro-opiomelanocortin (POMC; gene: 8 kb; chromosome 2p23), by proteolysis (Figure 54-5).133,159 In the corticotrophs, subtilisin-like proprotein convertase (PC1/3) cleaves POMC into two fragments: the 22 kDa pro-ACTH fragment and beta-lipotropin (amino acids 42-134), whose function remains poorly understood. Next, PC1/3 releases ACTH (amino acids 1-39) from pro-ACTH. The resulting N-terminal fragment is further cleaved to pro-gamma-melanocyte-stimulating hormone (MSH) and a joining peptide (JP). ACTH is not further cleaved in corticotrophs. The action of PC2 in the hypothalamus, skin, and melanotrophs of the intermediate lobe is to release gamma-MSH from pro-opiocortin (N-POC); alpha-melanocyte-stimulating hormone (alpha-MSH; amino acids 1-13) and corticotropin-like intermediate lobe peptide (CLIP; amino acids 18-39) from ACTH; and gamma-lipotropin (amino acids 42-101) and beta-endorphin (amino acids 104-134) from beta-lipotropin (see Figure 54-5). Last, beta-MSH (amino acids 84-101) is derived from gamma-lipotropin via PC2. Hyperpigmentation that occurs with ACTH excess appears to be a direct consequence of the MSH-like activity of ACTH and not ACTH cleavage into alpha-MSH.349 Whereas (1) a gamma-MSH sequence is contained within the N-terminal fragment, (2) alpha-MSH is contained within ACTH, and (3) beta-MSH is contained within gamma-lipotropin (a fragment of beta-lipotropin), MSH is not released by the human anterior pituitary gland. ACTH circulates systemically to bind to ACTH receptors located on cells within the adrenal cortex. The ACTH receptor is the G-protein–coupled melanocortin-2 receptor (MC2R; gene location: chromosome 18p11.2).77 The second messenger system involves adenyl cyclase and the generation of cyclic AMP. The actions of ACTH are then triggered via protein kinase A and protein kinase C, leading to steroidogenesis, increased size and number of adrenocortical cells, and increased size and functional complexity of cellular organelles. Cortisol is then synthesized and released. Cortisol feeds back centrally at the hypothalamus and to a lesser degree feeds back negatively at the pituitary to suppress CRH and ACTH secretion.158 Other negative feedback loops include ACTH suppression of hypothalamic CRH and an ultra-short feedback loop whereby ACTH suppresses its own release. The regulation of adrenal androgen synthesis and secretion remains poorly understood. The existence of a pituitary adrenal androgen–stimulating hormone or a cortical androgen–stimulating hormone remains doubtful despite many years of research that sought its existence.116 The best characterized regulator of androstenedione and DHEA secretion is ACTH. This is not surprising, as CYP17 is regulated by ACTH. A diurnal rhythm in adrenal androgen concentrations parallels cortisol variations. Nevertheless, ACTH regulation of adrenal androgens does not explain the normal prepubertal and pubertal increases in adrenal androgen synthesis that occurs in both boys and girls: ACTH does not increase prior to puberty. Evidence indicates that sympathetic innervation of the zona reticularis may exist and may regulate adrenal androgen secretion. Immune regulation of adrenal androgen secretion has also been proposed. An overview of steroid biosynthesis is provided in Figure 54-6, and the steroidogenic enzymes are listed in Table 54-6. TABLE 54-6 Major Steroidogenic Enzymes Expressed in Adrenal Cortex *Usually these terms when italicized refer to the gene name. However, because different sources refer to these enzymes in a variety of ways (gene name vs. enzyme activity), the gene name un-italicized will be used to name the protein. ZF, Zona fasciculata; ZR, zona reticularis. Within the cytoplasm of steroid-producing cells, cholesterol ester is hydrolyzed to free cholesterol and a free fatty acid via the ACTH-responsive steroidogenesis activator protein, which is functionally a cholesterol esterase. Free cholesterol is then transported across the outer mitochondrial membrane by a sterol transfer protein into the mitochondrial intermembranous space. The 30 kDa steroidogenic acute regulatory protein (StAR) next transports cholesterol across the inner membrane into the mitochondria.205,301 StAR-mediated transport of cholesterol into the mitochondria is a rate-limiting step in steroid hormone synthesis. StAR synthesis is enhanced by rising concentrations of cyclic AMP resulting from ACTH binding to its receptor. In the mitochondria, cholesterol is converted to pregnenolone by the action of CYP11A, which is the cytochrome P450 sidechain cleavage enzyme (P450ssc; gene location: chromosome 15q23-24) that is functionally a 20,22-desmolase that releases isocaproaldehyde. CYP11A therefore initiates the conversion of the C27 steroid cholesterol to the other C21 steroids (the subscript indicates the number of carbons in the compound). CYP11B2 (aldosterone synthase) encompasses three enzymatic activities: (1) an 11-hydroxylase (DOC → corticosterone), (2) an 18-hydroxylase [corticosterone methyl oxidase I (CMOI); corticosterone → 18-hydroxycorticosterone], and (3) an 18-hydroxydehydrogenase [corticosterone methyl oxidase II (CMOII); 18-hydroxycorticosterone → aldosterone; see Figure 54-6].234 Aldosterone diffuses out of the mitochondrion into the cytoplasm and across the cell membrane to enter the interstitium and then the circulation. The aldosterone secretion rate per day is 100 to 150 µg, with some estimates varying up to 200 µg. Thus the aldosterone secretion rate is approximately one tenth the secretion rate of cortisol on a weight basis. The half-life of circulating aldosterone is less than 15 minutes. Cortisol production increases within minutes of an increase in circulating ACTH concentrations. In the zona fasciculata, where CYP17 (P450c17; gene location: chromosome 10q24.3) is expressed, CYP17 hydroxylates pregnenolone to 17-hydroxypregnenolone.206 CYP17 also includes P450 17,20-lyase activity, which is important for the formation of adrenal androgens. 3-Beta-HSD and delta(5)-4-isomerase next catalyze the conversion of 17-hydroxypregnenolone to 17-hydroxyprogesterone. The last step outside of the mitochondrion involves CYP21 that 21-hydroxylates 17-hydroxyprogesterone to 11-desoxycortisol. 11-Desoxycortisol travels back to the mitochondrion, where CYP11B1 (P450c11) catalyzes the conversion of 11-desoxycortisol to cortisol via its 11-beta hydroxylase activity. Little CYP11B2 activity is seen in the zona fasciculata. Similar to aldosterone, cortisol diffuses out of the cell to ultimately enter the circulation. The normal cortisol secretion rate is 6 to 14 mg/m2 per 24 hours. In adults, this is approximately 10 to 20 mg/d, with some estimates as high as 25 mg/d. Debate continues as to the anatomic location of adrenal androgen synthesis.258 Traditionally, it has been taught that adrenal androgens are synthesized exclusively in the zona reticularis. However, the enzymatic activity of the 17,20-lyase (17,20-desmolase) that converts 17-hydroxypregnenolone to DHEA [a delta(5) steroid] and 17-hydroxyprogesterone to androstenedione [a delta(4) steroid] is included in the CYP17 protein, whose 17-hydroxylase activity was described previously. Therefore, adrenal androgens theoretically could be synthesized within both the zona fasciculata and the zona reticularis. However, a predominance of adrenal androgen synthesis in the zona reticularis may result from high cytochrome 5b expression, which increases the 17,20-lyase activity of CYP17. The 17,20-lyase activity of CYP17 converts C21 steroids into C19 steroids. Likewise, there is nothing to prevent cortisol synthesis in the zona reticularis. The conversion of DHEA to DHEA-S is catalyzed by DHEA sulfotransferase (SULT2A1; gene location: chromosome 19q13.3). Based on the model presented so far of enzymes partitioned into the mitochondrion or the endoplasmic reticulum, adrenal androgen synthesis is completed within the endoplasmic reticulum because this synthesis does not require CYP21, CYP11B1, or CYP11B2, which are located in the mitochondrion. The fetal adrenal lacks adult concentrations of 3-beta-HSD expression with resulting elevated production of DHEA, DHEA-S, and 16-hydroxy DHEA-S (from hydroxylation of DHEA-S in the fetal liver). In utero, pregnenolone and 17-hydroxypregnenolone from the fetal adrenal enter the fetal circulation to be converted in the placenta, respectively, into progesterone and 17-hydroxyprogesterone via placental 3-beta-HSD. Progesterone and 17-hydroxyprogesterone then return to the adrenal (via the circulation), where they serve as substrates for aldosterone and cortisol, respectively.225 Steroid hormones are 90 to 98% bound to specific carrier proteins or albumin. Steroids that are sulfated or glucuronidated circulate unbound in the plasma. Aldosterone is carried primarily by albumin as cortisol, corticosterone, and 17-hydroxyprogesterone occupy most of the binding sites on corticosteroid-binding globulin (CBG; transcortin), a 58 kDa, 383 amino acid alpha-1 globulin.111,266 Normal concentrations of total cortisol exceed the normal concentrations of aldosterone by many-fold, explaining why little aldosterone is carried on CBG. CBG is a member of the serine protease inhibitor superfamily, specifically, SERPINA6 (clade A—alpha-1 antiprotease, antitrypsin—member 6; gene location: chromosome 14q3).168 Between 80 and 90% of cortisol is carried by CBG, 7% of cortisol is loosely bound to albumin, and 2 to 3% is unbound (free). When total cortisol rises in Cushing syndrome (see later), the increased proportion of plasma free cortisol readily spills into the urine, increasing the urinary free cortisol (UFC) excretion.176 Typically, only 0.25 to 0.5% of total cortisol is excreted in the urine. Because more cortisol than aldosterone is bound to CBG, the half-life of cortisol is longer (60 to 80 minutes) than the half-life of aldosterone (20 to 30 minutes).61 In addition to cortisol, progestins are carried by CBG. Progestin is a generic term for any substance that produces some or all of the biological effects of progesterone. DHEA and its sulfated form, DHEA-S, and estradiol are predominantly carried by albumin. In contrast, testosterone and dihydrotestosterone (DHT) are carried by sex hormone–binding globulin (SHBG), an ≈100 kDa homodimer (each monomer is ≈50 kDa; 373 amino acids; gene location: chromosome 17p1).254 Estrogens and thyroid hormone increase SHBG concentrations, whereas insulin, growth hormone, glucocorticoids, androgens, and progestins lower SHBG concentrations. SHBG concentrations are higher in children than in adults. Aldosterone is reduced to tetrahydroaldosterone. Glucuronidation produces tetrahydroaldosterone 3-glucuronide, which can be excreted by the kidney. Aldosterone is 3 alpha- and 5 alpha-reduced, or 3 alpha- and 5 beta-reduced (Figure 54-7). Only ≈0.5% of aldosterone is excreted unchanged in the urine. Aldosterone is glucuronidated at the carbon 18 position. Cirrhosis, severe congestive heart failure, and ascites impair hepatic aldosterone clearance. Cortisone is formed from cortisol via 11 beta-hydroxysteroid dehydrogenase type 2 (see Figure 54-7). In this reaction, the 11-hydroxyl group is converted to an 11-oxo group by removal of two hydrogens. Cortisone lacks glucocorticoid activity. [Note: When cortisone is used as a drug, it can be activated back to cortisol in the liver via 11-beta hydroxysteroid dehydrogenase type 1, an nicotinamide adenine dinucleotide phosphate (NADPH)-dependent oxo-reductase.] Reduction of the double bonds at carbons 4-5 via delta(4)-5-beta reductase or delta(4)-5-alpha reductase yields dihydrocortisol and dihydrocortisone (DHE). Metabolism with reduction of the ketone groups at carbon 3 results in tetrahydrocortisol (THF) and tetrahydrocortisone (THE), which account for the major portion of cortisol clearance (≈50%). The only difference in the outcome of delta(4)-5-beta reductase versus delta(4)-5-alpha reductase activity is the alpha or beta orientation of the hydrogen (5 beta-THF or 5-alpha-THF). Normally, the beta metabolite predominates (5-beta THF:5-alpha-THF ratio = 2 : 1). Further metabolism of THF and THE via 20-alpha-hydroxysteroid dehydrogenase or 20-beta-hydroxysteroid dehydrogenase produces alpha and beta cortol and cortolone (the cortoic acids), which account for ≈30% of cortisol excretion. Opening the carbon 17-20 bond creates a ketone and gives rise to 11 beta-hydroxyetiocholanolone and 11-ketoetiocholanolone, representing ≈10% of cortisol excretion. Only about 1% of cortisol is normally excreted as free cortisol or cortisone. Minor cortisol metabolites include (1) 20 alpha-hydroxycortisol and 20 beta-hydroxycortisol, which result from reduction of the carbon 20 ketone, and (2) 6 beta-hydroxycortisol, which results from hydroxylation of carbon 6. Oxidation of carbon 17 in THF and THE yields oxo or hydroxy metabolites (Figure 54-8). Minor cortisone metabolites are shown in Figure 54-8. Regarding the metabolism of the adrenal androgens, DHEA is sulfated to DHEA-S (Figure 54-9). These compounds are 7 alpha-hydroxylated or 16-alpha-hydroxylated. Alternatively, 17-ketosteroid reductase reduces the ketone at carbon 17 to a hydroxyl group, yielding androstenediol from DHEA and androstenediol sulfate from DHEA-S. Androstenedione is converted to androsterone via 3-alpha- and 5-alpha-reduction, whereas 3-alpha- and 5-beta-reduction yields etiocholanolone (see Figure 54-9). Similar to metabolites of cortisol, metabolites of the adrenal androgens are glucuronidated or sulfated for urinary excretion. DHEA and androstenedione metabolites are measured in the same way as 17-ketosteroids because both have keto groups in the C-17 position. Because testosterone has a hydroxyl group at the C-17 position, it is not a 17-ketosteroid. DHEA and androstenedione are elevated in untreated and undertreated CYP21 and CYP11B1 deficiencies. Figure 54-10 summarizes these urine steroid measurements. Cortisol clearance (or metabolism) affects cortisol concentrations. If the clearance of cortisol is reduced, plasma cortisol concentrations can increase, whereas enhanced clearance of cortisol decreases its concentration. Rifampin-induced Addisonian crisis from increased cortisol metabolism has been reported. However, in most cases, cortisol, free cortisol, and ACTH are normal in these conditions, presumably because alterations in the free cortisol concentration will be sensed by the hypothalamus, which will respond by secreting CRH to ultimately return the free cortisol concentration to within it’s reference interval. Table 54-7 lists a number of conditions that affect cortisol concentrations. TABLE 54-7 Conditions That Affect Cortisol Clearance Several strategies are used to assess adrenal function. These tests are typically designed to differentiate between primary and secondary causes of disease, or to detect abnormalities that may not be apparent in the results of static, baseline laboratory measurements. For example, provocative stimulation tests are useful in documenting hyposecretion of adrenocortical hormones.226 A specific stimulus is applied, and the release of a given hormone over a specific time frame is measured. Also, suppression tests are used to document hypersecretion of the adrenocortical hormones.76 ACTH stimulation tests, sometimes referred to as the cosyntropin tests, are designed to document the functional capacity of the adrenal glands to synthesize cortisol (Boxes 54-1 and 54-2). Cosyntropin (Cortrosyn) is a synthetic polypeptide that is the N-terminal 24 amino acid sequence of ACTH, which contains the biologically active domain. Another name for cosyntropin is tetracosactrin (Synacthen). Cosyntropin, a potent stimulator of cortisol secretion, has a very brief half-life and minimal antigenicity. The protocols for 1 hour and multiple-day ACTH stimulation tests are shown in Box 54-1. BOX 54-1 Protocol for the 1 Hour (Rapid) Cosyntropin Stimulation Test
The Adrenal Cortex
Anatomy
Adrenal Layer
Major Product(s)
Action
Cortex
Zona glerulosa
Aldosterone
Mineralocorticoid
Zona fasciculata
Cortisol
Glucocorticoid
Zona reticularis
Dehydroepiandrosterone
Androstenedione
Adrenal androgen
Medulla
Epinephrine
Catecholamine
Steroid Biochemistry
Mineralocorticoids (Aldosterone)
Action
Adverse Outcome Excessive Action
Deficient Action
Sodium retention*
Hypertension
Hypotension
Urinary potassium wasting
Hypokalemia
Hyperkalemia
Urinary hydrogen ion wasting
Alkalosis
Glucocorticoids (Cortisol)
Adrenal Androgens (DHEA and Androstenedione)
Physiology and Regulation of Adrenocortical Hormones
Aldosterone
Molecule
Size (Amino Acids; Abbreviation)
Angiotensinogen
485
↓
↓ Renin
↓
Angiotensin I
10 (A1-10)
↓
↓ Angiotensin-converting enzyme (ACE)
↓
Angiotensin II
8 (A1-8)
↓
↓ Amino peptidase A
↓
Angiotensin III
7 (A2-8)
↓
↓ Arginyl aminopeptidase
↓
Angiotensin IV
6 (A3-8)
Cortisol
Adrenal Androgens
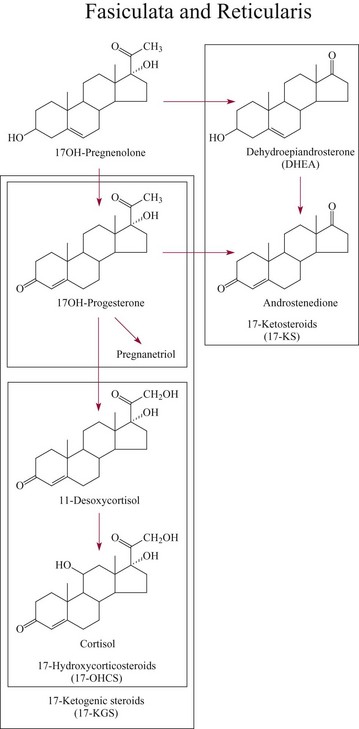
Biosynthesis Of Adrenocortical Hormones100

Aldosterone
Cortisol
Adrenal Androgens
Adrenal Steroid Synthesis in the Fetus
Adrenocortical Hormones In The Circulation
Metabolism Of Adrenal Steroids7
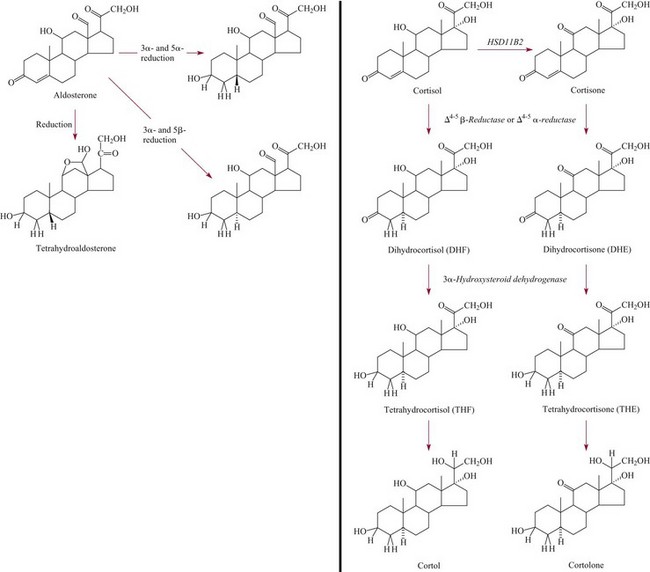
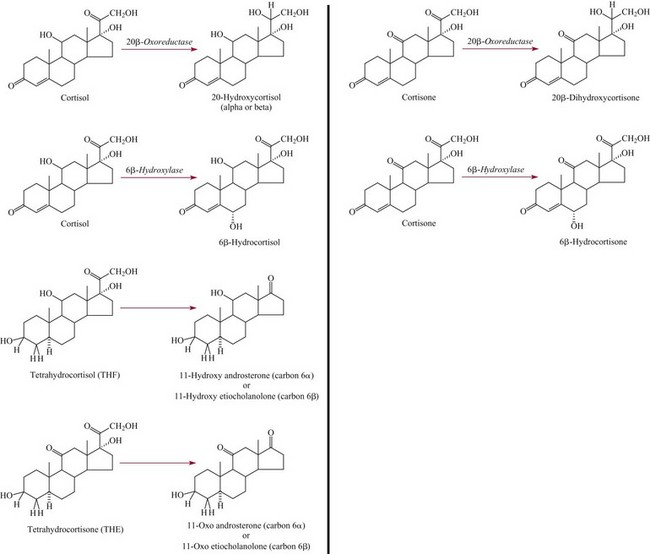
Aldosterone20
Cortisol311
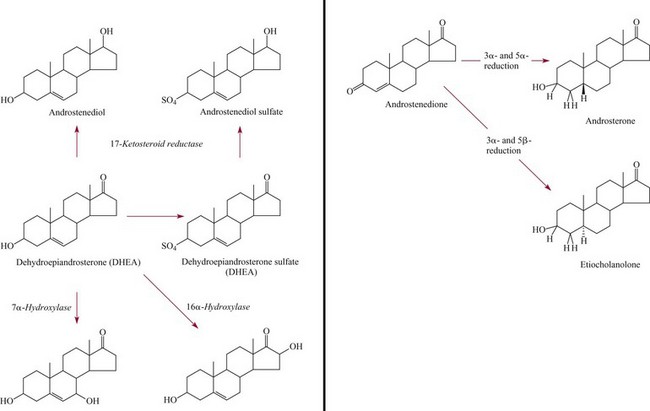
Adrenal Androgens100
Urinary Metabolites
Factors Affecting Adrenal Steroid Metabolism
Endocrinopathy
Cortisol Clearance
Mechanism
Hyperthyroidism
Incr
Inhibition of hepatic 11-beta HSDI and stimulation of 5-alpha reductase and 5-beta reductase
Hypothyroidism
Decr
Stimulation of hepatic 11-beta HSDI and inhibition of 5-alpha reductase and 5-beta reductase
Acromegaly
Incr
Inhibition of hepatic 11-beta HSDI
Cushing syndrome
Incr
Induction of 6 beta-hydroxylase
Solid Organ Dysfunction
Chronic liver disease (disease including alcoholic cirrhosis)
Decr
Decreased cortisol metabolism
Renal disease
Decr
Reduced renal conversion: cortisol → cortisone
Drugs/Toxins
Acute alcohol ingestion
Decr
—
Rifampin
Incr
Induction of 6 beta-hydroxylase
Phenytoin
Incr
Induction of 6 beta-hydroxylase
Phenobarbital
Incr
Induction of hepatic mixed-function oxidases
Other
Aging
Decr
Note: Decreased clearance is balanced by decreased synthesis; therefore cortisol concentrations and responses to ACTH are normal.
Starvation
Decr
Balanced by decreased production
Anorexia nervosa
Decr
—
Dynamic Tests Of Adrenal Function
ACTH Stimulation (Cosyntropin) Test
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


The Adrenal Cortex