44
Tetracyclines, Macrolides, Clindamycin, Chloramphenicol, Streptogramins, & Oxazolidinones
CASE STUDY
A 19-year-old woman with no significant past medical history presents to her college medical clinic complaining of a 2-week history of vaginal discharge. She denies any fever or abdominal pain but does report vaginal bleeding after sexual intercourse. When questioned about her sexual activity, she reports having vaginal intercourse, at times unprotected, with two men in the last 6 months. A pelvic examination is performed and is positive for mucopurulent discharge from the endocervical canal. No cervical motion tenderness is present. A first-catch urine specimen is obtained to be tested for chlamydia and gonorrhea. A pregnancy test is also ordered as the patient reports she “missed her last period.” Pending these results, the decision is made to treat her empirically for gonococcal and chlamydial cervicitis. What are two potential treatment options for her possible chlamydial infection? How does her potential pregnancy affect the treatment decision?
The drugs described in this chapter inhibit bacterial protein synthesis by binding to and interfering with ribosomes. Most are bacteriostatic, but a few are bactericidal against certain organisms. Because of overuse, tetracycline and macrolide resistance is common. Except for tigecycline and the streptogramins, these antibiotics are usually administered orally.
 TETRACYCLINES
TETRACYCLINES
All of the tetracyclines have the basic structure shown at right:
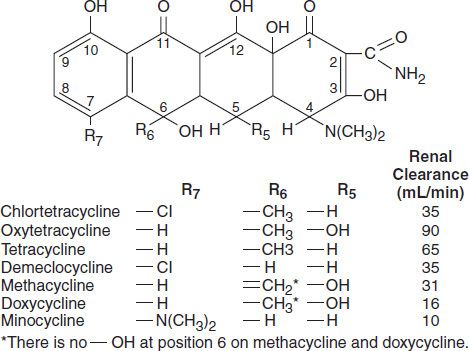
Free tetracyclines are crystalline amphoteric substances of low solubility. They are available as hydrochlorides, which are more soluble. Such solutions are acid and, with the exception of chlortetracycline, fairly stable. Tetracyclines chelate divalent metal ions, which can interfere with their absorption and activity. A newer tetracycline analog, tigecycline, is a glycylcycline and a semisynthetic derivative of minocycline.
Mechanism of Action & Antimicrobial Activity
Tetracyclines are broad-spectrum bacteriostatic antibiotics that inhibit protein synthesis. Tetracyclines enter microorganisms in part by passive diffusion and in part by an energy-dependent process of active transport. Susceptible organisms concentrate the drug intracellularly. Once inside the cell, tetracyclines bind reversibly to the 30S subunit of the bacterial ribosome, blocking the binding of aminoacyl-tRNA to the acceptor site on the mRNA-ribosome complex (Figure 44–1). This prevents addition of amino acids to the growing peptide.
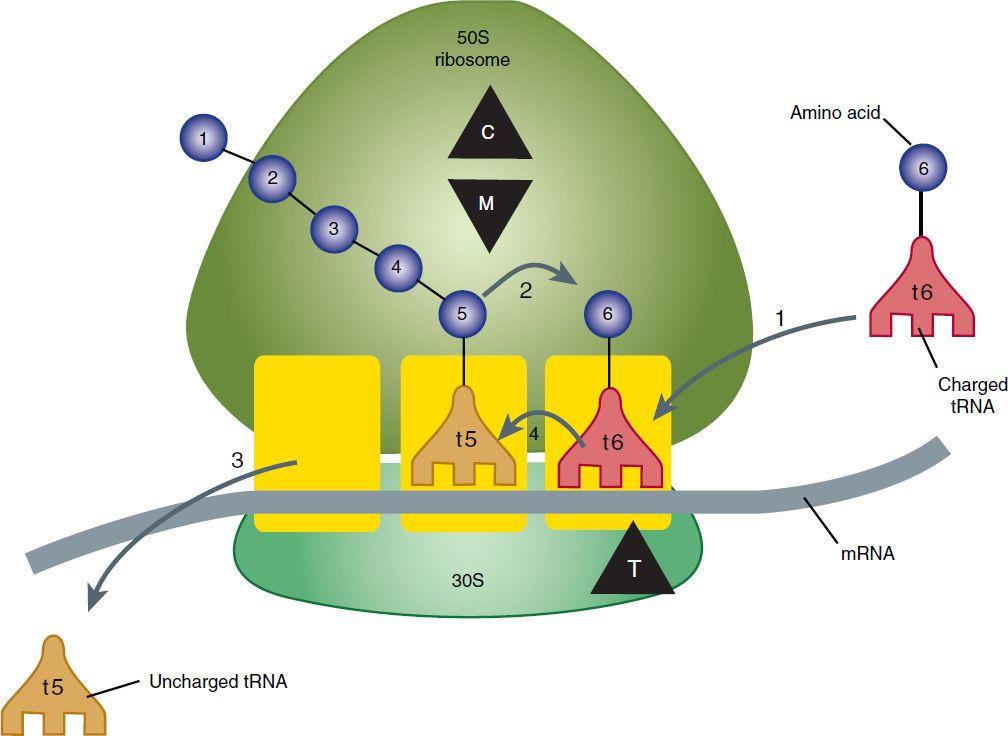
FIGURE 44–1 Steps in bacterial protein synthesis and targets of several antibiotics. Amino acids are shown as numbered circles. The 70S ribosomal mRNA complex is shown with its 50S and 30S subunits. In step 1, the charged tRNA unit carrying amino acid 6 binds to the acceptor site on the 70S ribosome. The peptidyl tRNA at the donor site, with amino acids 1 through 5, then binds the growing amino acid chain to amino acid 6 (peptide bond formation, step 2). The uncharged tRNA left at the donor site is released (step 3), and the new 6-amino acid chain with its tRNA shifts to the peptidyl site (translocation, step 4). The antibiotic binding sites are shown schematically as triangles. Chloramphenicol (C) and macrolides (M) bind to the 50S subunit and block peptide bond formation (step 2). The tetracyclines (T) bind to the 30S subunit and prevent binding of the incoming charged tRNA unit (step 1).
Tetracyclines are active against many gram-positive and gram-negative bacteria, including certain anaerobes, rickettsiae, chlamydiae, and mycoplasmas. The antibacterial activities of most tetracyclines are similar except that tetracycline-resistant strains may be susceptible to doxycycline, minocycline, and tigecycline, all of which are poor substrates for the efflux pump, if that is the mechanism of resistance. Differences in clinical efficacy for susceptible organisms are minor and attributable largely to features of absorption, distribution, and excretion of individual drugs.
Resistance
Three mechanisms of resistance to tetracycline analogs have been described: (1) impaired influx or increased efflux by an active transport protein pump; (2) ribosome protection due to production of proteins that interfere with tetracycline binding to the ribosome; and (3) enzymatic inactivation. The most important of these are production of an efflux pump and ribosomal protection. Tet(AE) efflux pump-expressing gram-negative species are resistant to the older tetracyclines, doxycycline, and minocycline. They are susceptible, however, to tigecycline, which is not a substrate of these pumps. Similarly, the Tet(K) efflux pump of staphylococci confers resistance to tetracycline, but not to doxycycline, minocycline, or tigecycline, none of which are pump substrates. The Tet(M) ribosomal protection protein expressed by gram-positives produces resistance to tetracycline, doxycycline, and minocycline, but not to tigecycline, which, because of its bulky t-butylglycylamido substituent, has a steric hindrance effect on Tet(M) binding to the ribosome. Tigecycline is a substrate of the chromosomally encoded multidrug efflux pumps of Proteus sp and Pseudomonas aeruginosa, accounting for their intrinsic resistance to all tetracyclines including tigecycline.
Pharmacokinetics
Tetracyclines differ in their absorption after oral administration and in their elimination. Absorption after oral administration is approximately 30% for chlortetracycline; 60–70% for tetracycline, oxytetracycline, demeclocycline, and methacycline; and 95–100% for doxycycline and minocycline. Tigecycline is poorly absorbed orally and must be administered intravenously. A portion of an orally administered dose of tetracycline remains in the gut lumen, alters intestinal flora, and is excreted in the feces. Absorption occurs mainly in the upper small intestine and is impaired by food (except doxycycline and minocycline); by multivalent cations (Ca2+, Mg2+, Fe2+, Al3+); by dairy products and antacids, which contain multivalent cations; and by alkaline pH. Specially buffered tetracycline solutions are formulated for intravenous administration.
Tetracyclines are 40–80% bound by serum proteins. Oral dosages of 500 mg every 6 hours of tetracycline hydrochloride or oxytetracycline produce peak blood levels of 4–6 mcg/mL. Intravenously injected tetracyclines give somewhat higher levels, but only temporarily. Peak levels of 2–4 mcg/mL are achieved with a 200 mg dose of doxycycline or minocycline. Steady-state peak serum concentrations of tigecycline are 0.6 mcg/mL at the standard dosage. Tetracyclines are distributed widely to tissues and body fluids except for cerebrospinal fluid, where concentrations are 10–25% of those in serum. Minocycline reaches very high concentrations in tears and saliva, which makes it useful for eradication of the meningococcal carrier state. Tetracyclines cross the placenta to reach the fetus and are also excreted in breast milk. As a result of chelation with calcium, tetracyclines are bound to—and damage—growing bones and teeth. Carbamazepine, phenytoin, barbiturates, and chronic alcohol ingestion may shorten the half-life of doxycycline by 50% due to induction of hepatic enzymes that metabolize the drug.
Tetracyclines are excreted mainly in bile and urine. Concentrations in bile exceed those in serum tenfold. Some of the drug excreted in bile is reabsorbed from the intestine (enterohepatic circulation) and may contribute to maintenance of serum levels. Ten to fifty percent of various tetracyclines is excreted into the urine, mainly by glomerular filtration. Ten to forty percent of the drug is excreted in feces. Doxycycline and tigecycline, in contrast to other tetracyclines, are eliminated by nonrenal mechanisms, do not accumulate significantly, and require no dosage adjustment in renal failure.
Tetracyclines are classified as short-acting (chlortetracycline, tetracycline, oxytetracycline), intermediate-acting (demeclocycline and methacycline), or long-acting (doxycycline and minocycline) based on serum half-lives of 6–8 hours, 12 hours, and 16–18 hours, respectively. Tigecycline has a half-life of 36 hours. The almost complete absorption and slow excretion of doxycycline and minocycline allow for once-daily dosing for certain indications, but, by convention, these two drugs are usually dosed twice daily.
Clinical Uses
A tetracycline is the drug of choice in the treatment of infections caused by rickettsiae. Tetracyclines are also excellent drugs for the treatment of Mycoplasma pneumonia, chlamydiae, and some spirochetes. They are used in combination regimens to treat gastric and duodenal ulcer disease caused by Helicobacter pylori. They may be used in various gram-positive and gram-negative bacterial infections, including vibrio infections, provided the organism is not resistant. In cholera, tetracyclines rapidly stop the shedding of vibrios, but tetracycline resistance has appeared during epidemics. Tetracyclines remain effective in most chlamydial infections, including sexually transmitted infections. Doxycycline, in combination with ceftriaxone, is an alternative treatment for gonococcal disease. A tetracycline—in combination with other antibiotics—is indicated for plague, tularemia, and brucellosis. Tetracyclines are sometimes used in the treatment or prophylaxis of protozoal infections, eg, those due to Plasmodium falciparum (see Chapter 52). Other uses include treatment of acne, exacerbations of bronchitis, community-acquired pneumonia, Lyme disease, relapsing fever, leptospirosis, and some nontuberculous mycobacterial infections (eg, Mycobacterium marinum). Tetracyclines formerly were used for a variety of common infections, including bacterial gastroenteritis and urinary tract infections. However, many strains of bacteria causing these infections are now resistant, and other agents have largely supplanted tetracyclines.
Minocycline, 200 mg orally daily for 5 days, can eradicate the meningococcal carrier state, but because of side effects and resistance of many meningococcal strains, ciprofloxacin or rifampin is preferred. Demeclocycline inhibits the action of antidiuretic hormone in the renal tubule and has been used in the treatment of inappropriate secretion of antidiuretic hormone or similar peptides by certain tumors (see Chapter 15).
Tigecycline, the first glycylcycline to reach clinical practice, has several unique features that warrant its consideration apart from the older tetracyclines. Many tetracycline-resistant strains are susceptible to tigecycline because it is not affected by the common resistance determinants. Its spectrum is very broad. Coagulase-negative staphylococci and Staphylococcus aureus, including methicillin-resistant, vancomycin-intermediate, and vancomycin-resistant strains; streptococci, penicillin-susceptible and resistant; enterococci, including vancomycin-resistant strains; gram-positive rods; Enterobacteriaceae; multidrug-resistant strains of Acinetobacter sp; anaerobes, both gram-positive and gramnegative; rickettsiae, Chlamydia sp, and Legionella pneumophila; and rapidly growing mycobacteria all are susceptible. Proteus sp and P aeruginosa, however, are intrinsically resistant.
Tigecycline, formulated for intravenous administration only, is given as a 100 mg loading dose, then 50 mg every 12 hours. As with all tetracyclines, tissue and intracellular penetration is excellent; consequently, the volume of distribution is quite large and peak serum concentrations are low. Elimination is primarily biliary, and no dosage adjustment is needed for patients with renal insufficiency. In addition to the tetracycline class effects, the chief adverse effect of tigecycline is nausea, which occurs in up to one third of patients, and occasionally vomiting. Neither nausea nor vomiting usually requires discontinuation of the drug.
Tigecycline is approved for treatment of skin and skin-structure infection, intra-abdominal infections, and community-acquired pneumonia. However, in a meta-analysis of clinical trials, tigecycline was associated with a small but significant increase in the risk of death compared with other antibiotics used to treat these infections. This has led the FDA to issue a black box warning that tigecycline should be reserved for situations where alternative treatments are not suitable. Because active drug concentrations in the urine are relatively low, tigecycline may not be effective for urinary tract infections and has no indication for this use. Tigecycline has in vitro activity against a wide variety of multidrug-resistant nosocomial pathogens (eg, methicillin-resistant S aureus, extended-spectrum β-lactamase-producing gram-negatives, and Acinetobacter sp); however, its clinical efficacy in infections with multidrug-resistant organisms, compared with other agents, is unproven.
A. Oral Dosage
The oral dosage for rapidly excreted tetracyclines, equivalent to tetracycline hydrochloride, is 0.25–0.5 g four times daily for adults and 20–40 mg/kg/d for children (8 years of age and older). For severe systemic infections, the higher dosage is indicated, at least for the first few days. The daily dose is 600 mg for demeclocycline or methacycline, 100 mg once or twice daily for doxycycline, and 100 mg twice daily for minocycline. Doxycycline is the oral tetracycline of choice because it can be given twice daily, and its absorption is not significantly affected by food. All tetracyclines chelate with metals, and none should be orally administered with milk, antacids, or ferrous sulfate. To avoid deposition in growing bones or teeth, tetracyclines should be avoided in pregnant women and children younger than 8 years.
B. Parenteral Dosage
Several tetracyclines are available for intravenous injection in doses of 0.1–0.5 g every 6–12 hours (similar to oral doses) but doxycycline is the usual preferred agent, at a dosage of 100 mg every 12–24 hours. Intramuscular injection is not recommended because of pain and inflammation at the injection site.
Adverse Reactions
Hypersensitivity reactions (drug fever, skin rashes) to tetracyclines are uncommon. Most adverse effects are due to direct toxicity of the drug or to alteration of microbial flora.
A. Gastrointestinal Adverse Effects
Nausea, vomiting, and diarrhea are the most common reasons for discontinuing tetracyclines. These effects are attributable to direct local irritation of the intestinal tract. Nausea, anorexia, and diarrhea can usually be controlled by administering the drug with food or carboxymethylcellulose, reducing drug dosage, or discontinuing the drug.
Tetracyclines alter the normal gastrointestinal flora, with suppression of susceptible coliform organisms and overgrowth of pseudomonas, proteus, staphylococci, resistant coliforms, clostridia, and candida. This can result in intestinal functional disturbances, anal pruritus, vaginal or oral candidiasis, or Clostridium difficile-associated colitis. However, the risk of C difficile colitis may be lower with tetracyclines than with other antibiotics.
B. Bony Structures and Teeth
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


