The vast majority of testicular tumors are of germ cell origin and, like the totipotent germ cells from which they arise, may differentiate along several pathways. The distinction of seminoma from the nonseminomatous germ cell tumors remains of prime clinical importance. Tumors of nongerminal origin, although relatively uncommon, are frequent diagnostic problems, both from the standpoint of classification and prognosis. These include Leydig cell tumors, Sertoli cell tumors, granulosa cell tumors, and less specific forms of sex cord–stromal tumor. The non–Leydig cell elements of the testicular interstitium may also be sources for neoplasms, as may the ductal system of the testis. The mesothelium that lines the tunica vaginalis testis may give rise to mesotheliomas as well as provide the basis, through müllerian metaplasia, for the formation of epithelial tumors more typically identified in the ovary. Müllerian remnants, especially the appendix testis, may also give rise to ovarian-type neoplasms. A distinctive form of mesothelioma with a benign natural history, the adenomatoid tumor, also derives from paratesticular mesothelium but is categorized separately. Mesothelium may also be the source for the rare cases of desmoplastic round cell tumor that develop in the paratestis (2), although the histogenesis of this neoplasm is not clear. The paratesticular area has a rich component of supporting mesenchymal cells as well as embryonic remnants that allow for a truly diverse number of paratesticular tumors. A general classification of testicular and paratesticular neoplasms is shown in Table 47.1.

GROSS EXAMINATION
A radical orchiectomy consists of the testis and the surrounding tunica vaginalis with a variable length of spermatic cord. The specimen should be measured in three dimensions, and the length of the cord should be specified. Submission of spermatic cord sections, including the margin of the cord, prior to dissection of the testis minimizes artifactual contamination of the cord by tumor (3), but care must be taken not to delay tumor incision and fixation, as poor preservation is a common cause of diagnostic ambiguity. The tunica vaginalis (parietal layer) should be inspected for possible tumor penetration; impairment of free movement of the parietal tunica vaginalis over the testicular surface may be an indication of this. The parietal layer of the tunica vaginalis should subsequently be incised, with submission of any abnormalities. With the opening of the tunica vaginalis, the external aspect of the testis (the tunica albuginea) becomes apparent, and it should be inspected for possible tumor transgression. Next, the testis should be bisected through its long axis in a plane that extends into the testicular hilum (i.e., toward the head of the epididymis). If desired, photographs and samples for electron microscopy and special studies (flow cytometric, molecular biologic, and cytogenetic) may be obtained at this point, although these are primarily for investigative rather than diagnostic purposes. Additional parallel cuts at 2- to 3-mm intervals should be made, followed by thorough fixation in a generous volume of fixative (10% neutral buffered formalin is adequate) if this has not occurred prior to dissection. After fixation, any tumor should be described and measured, and its relationship to the tunica albuginea and the hilar structures should be noted. Blocks of the different-appearing areas should be submitted, with a minimum number of one block per centimeter of maximum tumor dimension. Foci of hemorrhage and necrosis should be noted and included in the blocks for microscopic examination, as should the tumor–parenchyma interface, the testicular hilum, and nonneoplastic parenchyma. We recommend at least 10 blocks of tumors that have the gross appearance of seminoma (or submission of the entire tumor, if it can be accomplished in 10 blocks or less) because nonseminomatous elements may be identified focally in such cases and often determine a different treatment. The epididymis should be incised by serial, parallel cuts from head to tail; any abnormalities noted; and an appropriate block submitted. Care must be taken during the entire dissection because testicular tumors are often cellular and friable, leading to artifactual implantation of tumor on surfaces and tissue spaces, including vascular lumens (3).
STAGING
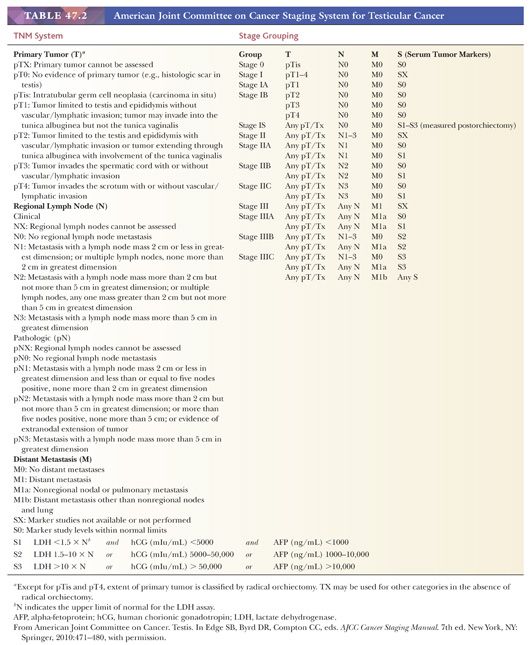
The currently recommended staging system for testicular tumors is the American Joint Committee on Cancer (AJCC) tumor-node-metastasis (TNM) classification, which is summarized in Table 47.2. It should be noted that because tumor markers play such a key role in the management of patients with germ cell tumors and have been shown to have prognostic importance, the degree of elevation of various serum markers is considered in the determination of the stage groups (Table 47.2). A more clinically based system, the International Germ Cell Consensus Classification (4), stratifies the patients into three risk groups on the basis of serum markers and distribution of metastases.
GERM CELL TUMORS
CLASSIFICATION
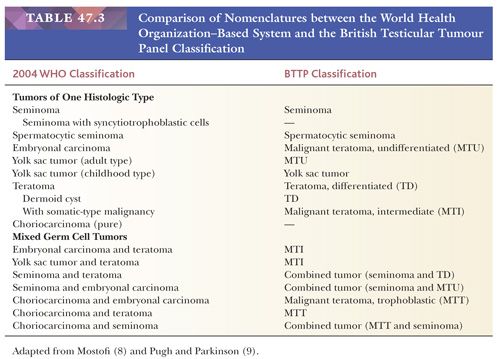
The overwhelming majority of primary testicular tumors are of germ cell origin. The classification in Table 47.1 represents a modification of the most recent classification of the World Health Organization (WHO) (5) and is based on work originally performed by several contributors. Unfortunately, there remains a lack of uniform acceptance of the WHO classification, with a second system in use in Great Britain stemming from the work of Collins and Pugh (6). The classification of the British Testicular Tumour Panel (BTTP) recognizes two major categories of testicular germ cell tumors—seminoma and teratoma—with the further subdivision of the teratoma category into differentiated, intermediate, undifferentiated, and trophoblastic types (7). The use of the modified WHO system is urged. Comparison of the nomenclature of the two systems (8,9) is provided in Table 47.3.
HISTOGENESIS
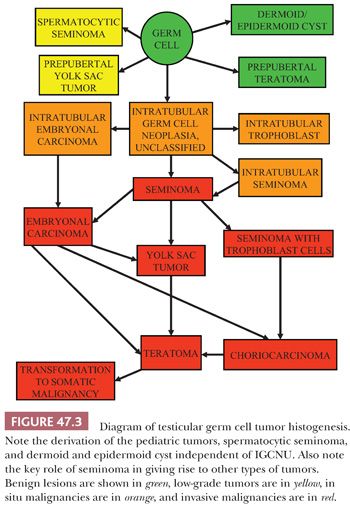
The histogenetic relationship among the various morphologic types of germ cell tumors has been a matter of continued interest and controversy. Recent evidence from several studies supports that the initial event in the origin of germ cell tumors is an in utero mutational event or transcriptional modification in intratubular germ cells that causes them to retain immature characteristics in postnatal life (10). There is support that defective development of Sertoli cells, likely induced in some cases by exposure to estrogenic substances during development, is permissive of delayed germ cell maturation (11). Additional events and exposure to pubertal hormones lead to the development of an intratubular malignant germ cell that shares many of the features of seminoma cells. Continued proliferation of such cells within the seminiferous tubules yields the lesion that is termed “intratubular germ cell neoplasia, unclassified type” (IGCNU) or “carcinoma in situ,” although the latter term is not strictly accurate given that the cells are not epithelial. These cells eventually develop into an invasive tumor, and a critical event in the development of invasion appears to be the acquisition of additional copies of genes on the short arm of chromosome 12, often in the form of an isochromosome, i(12p), that inhibit apoptosis and permit extratubular growth of invasive tumor cells that do not rely on Sertoli cells for their survival.(12). The direct invasive derivative of IGCNU appears to be seminoma, given the remarkable similarity of these two entities, not only morphologically but also by immunohistochemistry (13–18), ultrastructure (19,20), ploidy analysis (21), lectin-binding patterns (22), number of nucleolar-organizer regions (23), and genetic analysis (24). This supports that seminoma is a common precursor to many other forms of invasive germ cell tumor, a concept further supported by reports showing foci of nonseminomatous elements at the periphery of nodules of seminoma (25,26), the frequent presence of nonseminomatous elements in autopsy studies of patients who died of metastatic germ cell tumor subsequent to an orchiectomy showing pure seminoma (27,28), ultrastructural evidence of epithelial differentiation in some seminomas (29), the greater DNA content of seminoma compared to nonseminomatous tumors (supporting evolution from seminoma secondary to gene loss), and studies showing similar patterns of loss of heterozygosity in the seminomatous and nonseminomatous components of mixed germ cell tumors (24).
However, other forms of intratubular germ cell neoplasia are also recognized, and these may also be precursor lesions. Intratubular embryonal carcinoma has been identified by morphology (30) and immunochemistry for CD30 (31) in a significant percentage of nonseminomas and may be more common than previously recognized; however, this is disputed (32). Intratubular embryonal carcinoma is always associated with IGCNU, suggesting that it may be derived from it, and provides an alternative path for the development of nonseminomatous tumors. Intratubular trophoblast has also been identified in association with seminomas that have a component of trophoblastic giant cells (33). A further lesion, intratubular seminoma, where the seminiferous tubules are packed and expanded with seminoma-like cells, probably represents an end stage of IGCNU and may be associated with all germ cell tumor types (34). A current model of germ cell tumor histogenesis, therefore, recognizes the pivotal role of IGCNU in the development of the various germ cell tumor types, which may be by several alternative pathways. However, primacy of the pathway whereby most tumors are derived through seminoma is assumed (Fig. 47.3). It is important to understand, however, that this model does not apply to the pediatric germ cell tumors, spermatocytic seminoma, and two forms of presumed teratoma in adult patients, dermoid and epidermoid cyst.
EPIDEMIOLOGY
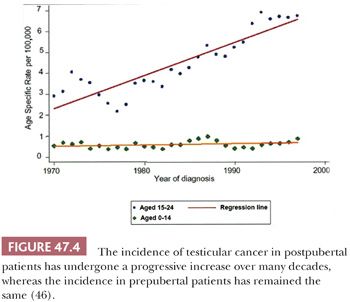
Germ cell tumors of the testis have characteristic associations. They occur predominantly (with the exception of spermatocytic seminoma) in young men, most commonly from 15 to 45 years of age, with a much smaller peak occurs in childhood, and there are rare cases in the elderly. The incidence of testicular germ cell tumors increased progressively in the twentieth century (Fig. 47.4), as verified by independent studies in several different countries (35–43), with the exception of cohorts born during World War II in Nazi-occupied countries (44,45). Some authorities consider this increase to be of “epidemic” proportions. This alarming increase in incidence, however, only applies to the more common postpubertal cases, with the rate in prepubertal patients remaining relatively constant throughout this same interval (Fig. 47.4) (46). The recent annual incidences are on the order of 10 per 100,000 male population in Denmark and Switzerland, the countries with the highest rates of testicular germ cell tumors; the annual incidence in the United States white population and in the United Kingdom is approximately 6 per 100,000 male population. Racial variations in the incidence of testicular cancer are quite apparent; the nonwhite populations in the United States have a substantially lower incidence than does the white population. In fact, the only nonwhite population with a comparably high incidence is the Maoris of New Zealand (47,48). Testicular germ cells tumors furthermore occur more commonly among professional workers and those of higher socioeconomic class than among laborers and those of lower socioeconomic class (49–52). Isolated reports have implicated exposures to certain agents or association with particular occupations as being of potential etiologic importance (40,49,53–56), but no consistent pattern has emerged. Exposure to estrogenic compounds in utero is implicated in some studies (57–59), as is a familial history of breast cancer (60), early birth order (61), certain human leukocyte antigen (HLA) haplotypes (62–65), Marfan syndrome (66), Down syndrome (66–68), Klinefelter syndrome (69), and the dysplastic nevus syndrome (70). It has been suggested that a hyperestrogenic state in utero leads to defective Sertoli cells which, in turn, contribute to the process of malignant transformation of germ cells. Hence, testicular germ cell tumors are associated with testicular maldevelopment, so-called testicular dysgenesis syndrome (71–74). No linkage of testicular cancer with smoking, alcohol consumption, radiation exposure, or prior vasectomy is established (47,75–77).
There are several well-defined positive associations with testicular germ cell tumors: cryptorchidism, a prior testicular germ tumor, a familial history of testicular germ cell tumors, certain disorders of sex development (intersex syndromes), and oligospermic infertility.
Cryptorchidism remains the most common risk factor for testicular germ cell tumors. In many series, approximately 10% of the cases are associated with past (corrected) or present cryptorchidism (78–84). Overall, cryptorchid patients have approximately a fourfold elevated risk of testicular germ cell tumors (85–87). Because of this elevated risk, it has been suggested that cryptorchid patients have diagnostic testicular biopsies to evaluate for the presence of the precursor lesion, IGCNU (see page 2181), which is present in 2% to 4% of the cases (88–90). Follow-up studies of patients with IGCNU on biopsy have verified a high rate of development of invasive testicular germ cell tumors (50% at 5 years [91]) but only very rare cases in patients with negative biopsies (88,92). The normally placed testis opposite a cryptorchid one is also at increased risk but to a lesser degree (50,87,93).
Two percent to 5% of patients with a testicular germ cell tumor develop another germ cell tumor in the residual testis (94–99). There is an especially increased risk if the residual testis is atrophic or cryptorchid (100–104), there is a family history of testicular cancer (105,106), or if the first tumor occurred at a young age (104). Intervals of more than a decade may occur between such metachronous testicular cancers (107), although some may present synchronously, and 50% of second tumors occur within 3 to 5 years (94,101). Again, biopsy of the remaining testis appears to be an effective method of identifying patients at risk for a second primary testicular germ cell tumor (92,103,108). Some advocate biopsy of the opposite testis at the initial orchiectomy (109). In one study, there was a remarkably high (>90%) frequency of C-KIT gene mutations in bilateral tumors compared to their virtual absence (1%) in unilateral cases (110), suggesting their utility in prospectively identifying patients at risk for a contralateral tumor. Unfortunately, other studies have failed to verify this result (111–114).
There is good evidence for a familial basis to some testicular germ cell tumors (115–117), with an approximate 2% frequency of testicular cancer in the first-degree male relatives of patients with testicular germ cell tumors compared to 0.4% in a control population (118). Brothers are at highest risk (10 times), sons at intermediate risk (6 times), and fathers at lowest risk (4 times) (119). Patients with testicular cancer and a familial history are also at increased risk for bilateral tumors, with bilateral involvement occurring in 8% to 14% of such cases (105,115,119). However, specific genes associated with the increased risk of testis cancer are not known (120), although a deletion on the Y chromosome has been shown to be associated with some cases of familial germ cell malignancy (121). Linkage studies implicating genes at Xq27 (122,123) have not been consistently positive (120).
It is also clear that patients with certain disorders of sex development (formerly known as intersex syndromes) are at increased risk for germ cell tumors. Patients with gonadal dysgenesis who carry a Y chromosome develop germ cell tumors at an increased rate, most often in association with a preexisting gonadoblastoma (124,125). Approximately 30% of such patients develop gonadoblastoma that may then give rise to the various subtypes of invasive germ cell tumor. For certain subsets of this condition, notably Frasier and Denys–Drash syndromes, the risk may be even higher, although the data are limited (126). Interestingly, a common thread for many of these disorders is mutation of the SOX9 gene or genes in the upstream signaling pathway for SOX9, most notably WT1. Patients with the androgen insensitivity (testicular feminization) syndrome are also at increased risk, with 5% to 10% developing germ cell tumors (127–129), which is undoubtedly a low estimate because of prophylactic gonadectomy at an early age in many patients. One study reported a 22% frequency of malignant tumors in androgen insensitivity patients beyond 30 years of age (130), and a consensus statement estimated the risk at 50% for those with partial androgen insensitivity syndrome and a nonscrotal testis (126). The patients with the partial rather than complete form of the androgen insensitivity syndrome are those at risk (131–133). Most germ cell tumors in the androgen insensitivity syndrome occur after the full development of female secondary sexual characteristics (127,134), but occasional cases are reported in younger patients (135), making the timing of prophylactic gonadectomy somewhat controversial. This appears to be a safe strategy for those with the complete form of the syndrome (133).Gonadal biopsy may successfully identify IGCNU in at-risk patients with gonadal dysgenesis and the androgen insensitivity syndrome (136,137), even during childhood (138,139), unlike biopsies in children who have risk factors other than intersex conditions.
The independent linkage of testicular germ cell tumors with oligospermic infertility remains less well established than with the other four factors. There is no question that male infertility patients have an elevated risk of testis cancer, with IGCNU being identified in approximately 1% (19,90,140). The association of cryptorchidism, testicular atrophy, and gonadal dysgenesis with infertility, however, complicates this analysis and casts doubt that infertility is an independent risk factor (141).
INTRATUBULAR GERM CELL NEOPLASIA
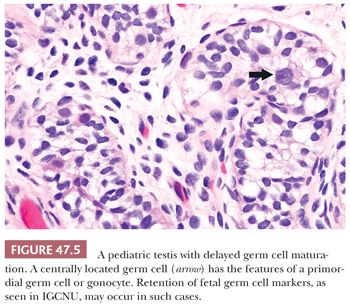
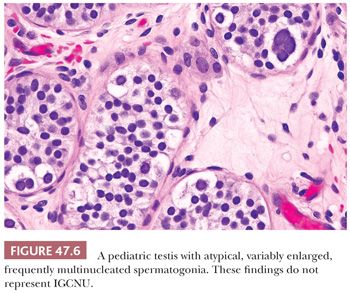
The term intratubular germ cell neoplasia (IGCN) refers both to a lesion that was originally described by Skakkebaek as “carcinoma in situ” of the testis as well as to differentiated forms of intratubular germ cell tumor. The more commonly occurring lesion described by Skakkebaek is now subcategorized as IGCNU because it is associated with the entire spectrum of germ cell tumors, with the exception of spermatocytic seminoma. The term “carcinoma in situ” is not preferred because IGCNU is not epithelial. When patients with IGCNU were prospectively followed, invasive germ cell tumors developed in 50% by 5 years of follow-up (142). In additional support of its precursor role, IGCNU occurs at increased frequency in patients known to be at increased risk for testicular germ cell tumors. Thus, IGCNU is identified in 2% to 4% of cryptorchid patients (88–90), in approximately 5% of the contralateral testes of patients with a prior testicular germ cell tumor (100,103,143–145), in 0.4% to 1% of patients with infertility (19,90,100,140), and at high rates in selected cases of gonadal dysgenesis and the androgen insensitivity syndrome (136,146). IGCNU is also observed in virtually all cases of invasive germ cell tumors of the testis in adults if sufficient residual seminiferous tubules are present (147–149), although it is not seen with spermatocytic seminoma (91). Furthermore, its association with the childhood forms of testicular germ cell tumor (yolk sac tumor and teratoma) is controversial, with some studies claiming an absence of associated IGCNU in these cases (150–152) but others disputing this absence (153,154). At the minimum, there is a much less conspicuous association of IGCNU with the pediatric germ cell tumors than the adult ones. It seems probably to us that many cases of “IGCNU” in pediatric cases represent a reactive phenomenon (155), delayed germ cell maturation as described in other conditions (Fig. 47.5) (132), or malformed spermatogonia of uncertain significance (Fig. 47.6) (156).
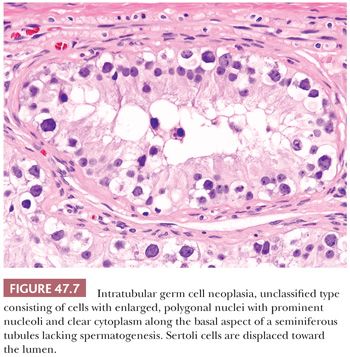
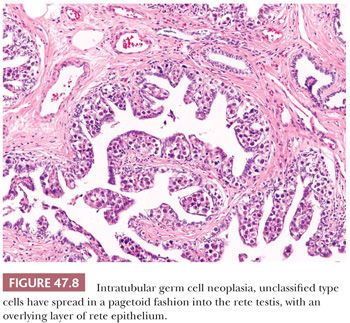
In postpubertal patients, IGCNU most commonly appears as germ cells with enlarged, hyperchromatic nuclei and clear cytoplasm (often having retraction artifact in formalin-fixed material) aligned along the basal portion of the seminiferous tubules (Fig. 47.7). Nucleoli are conspicuous, and mitotic figures are frequent. The Sertoli cells are often displaced toward the lumen, and spermatogenesis in the affected segment of tubule is almost always absent (Fig. 47.7), although it may appear normal in adjacent tubules. The affected tubules usually have thickened peritubular basement membranes. As IGCNU progresses, the Sertoli cells may be replaced, and a pattern resembling so-called “intratubular seminoma” may develop. Pagetoid spread of IGCNU into the rete testis (Fig. 47.8) is common (157). Those rare cases of “IGCNU” described in children reported that the neoplastic cells were not basally located but dispersed at various levels, with the more typical pattern evolving as the patient aged (91,158). More recent studies suggest this reflects germ cells with delayed maturation (Fig. 47.5) that in an unknown proportion of cases may progress to true IGCNU.
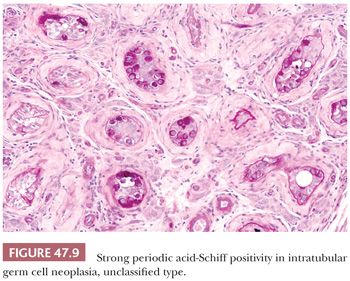
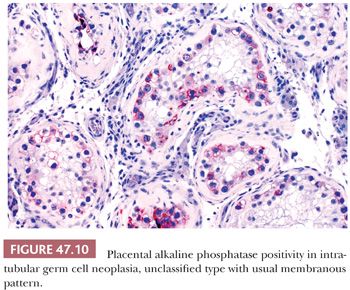
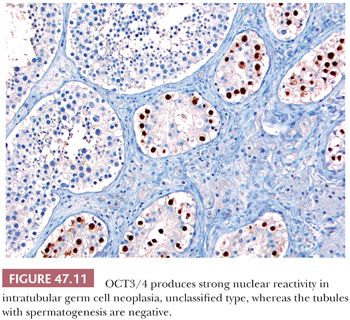
The great majority of cases of IGCNU are periodic acid-Schiff (PAS) positive and diastase sensitive (149) (Fig. 47.9), but similar positivity may also be identified in nonneoplastic spermatogonia and Sertoli cells (159). A more specific method is immunostaining with antibodies directed against placental alkaline phosphatase (PLAP), which highlight IGCNU in more than 95% of the cases (15,159–162). Unlike PAS stains, PLAP positivity does not occur in nonneoplastic spermatogonia, although rare cases show very focal PLAP positivity in spermatocytes (161). PLAP staining is usually membrane accentuated (Fig. 47.10). Antibodies M2A, 43-9F, TRA-1-60, and D2-40 (podoplanin) and those with specificities against glutathione S-transferase π, angiotensin-converting enzyme, OCT3/4, NANOG, and the C-KIT proto-oncogene (CD117) protein have also successfully identified IGCNU with very high sensitivities (13,18,162–169). We currently consider OCT3/4 the marker of choice for the detection of IGCNU because of its remarkably high sensitivity and specificity; it shows easily appreciated nuclear reactivity (Fig. 47.11). Additionally, we have seen a number of cases where nonneoplastic germ cells were CD117 reactive, perhaps because of increasingly sensitive immunostaining methods that, however, have diminished the specificity of this antibody. Several ultrastructural studies of IGCNU have identified features similar to those of seminoma (19,170–173); another study noted a similar distribution of nucleolar organizing regions in IGCNU and seminoma (23).
Testicular biopsies detect IGCNU in at-risk patients with a high sensitivity, and the recommended approach is to take one or two 3-mm biopsies per testis (174). A negative result on adequate biopsies is good evidence of little increased risk of testicular cancer; among almost 2000 patients who had negative biopsies opposite a germ cell tumor, only 0.3% developed a second tumor on follow-up (103). It remains controversial regarding who should be evaluated, although some suggest screening biopsies in patients with a history of cryptorchidism, prior testicular cancer, and somatosexual ambiguity in the presence of a Y chromosome. In patients with these risk factors, there is a much greater probability of a positive biopsy for IGCNU in the presence of testicular atrophy (109).
IGCNU is usually treated by orchiectomy or radiation. The chemotherapy that is given to patients with metastatic germ cell tumors may eradicate IGCNU in the remaining testis, but this is not a consistently effective form of treatment (109,175–178).
Other forms of IGCN, apart from IGCNU, include, most commonly, intratubular embryonal carcinoma and intratubular seminoma. Both are usually encountered in testes with already established invasive tumors. With intratubular embryonal carcinoma, the tubules are often expanded with central necrosis and calcification. The pleomorphic cells are similar to those of embryonal carcinoma and may be highlighted by immunochemistry for CD30, which also may highlight foci not obvious on routine staining. Intratubular seminoma represents filling and distention of seminiferous tubules by cells morphologically identical to IGCNU (Fig. 47.12). Intratubular syncytiotrophoblast cells occur adjacent to seminoma in approximately 15% of cases (33), and intratubular spermatocytic seminoma is virtually universal adjacent to invasive spermatocytic seminomas. Intratubular yolk sac tumor and teratoma are rare.

SEMINOMA
“Pure seminoma” (including cases with scattered trophoblastic elements) represents approximately 50% of all testicular germ cell tumors (179–181) and occurs at an average age of 40 years (179), which is 5 to 10 years older than patients with nonseminomatous germ cell tumors (25,179). Between 80% and 90% of patients with seminoma have initial symptoms of testicular swelling or other palpable abnormalities (7,182), although up to 11% may have normal-sized or atrophic testes (183). Tumors with a predominant or exclusive pattern of intertubular growth often do not present as masses but as metastatic tumor or are discovered incidentally (184). Testicular pain occurs in 10% to 20% of cases (7,182). Presenting symptoms secondary to metastases, most commonly lumbar back pain as a result of retroperitoneal spread, occur in 1% to 3% of cases (7,182). Only rare patients develop gynecomastia as a result of elevations of human chorionic gonadotropin (hCG) secondary to admixed trophoblastic elements. Paraneoplastic exophthalmos (185,186), hypercalcemia (187), limbic encephalopathy (188), polycythemia (189), and hemolytic anemia (190) are rare.
Serum hCG levels are mildly to moderately elevated in approximately 10% of patients with clinical stage I seminoma and in approximately 25% with metastatic involvement (191), although testicular vein hCG is elevated in a much higher proportion of cases (192). Elevated serum hCG correlates with trophoblast cells in the tumor. Alpha-fetoprotein (AFP) is not produced by seminoma cells, and an elevated serum AFP in a patient with an apparently pure seminoma is generally indicative of nonsampled nonseminomatous elements, although mild AFP elevation may reflect liver disease, including metastatic hepatic involvement (193); in one study, minimal AFP elevations in patients with apparently pure seminomas were not associated with a different behavior (194). Serum PLAP levels are also elevated in approximately 50% of patients with seminoma (195).
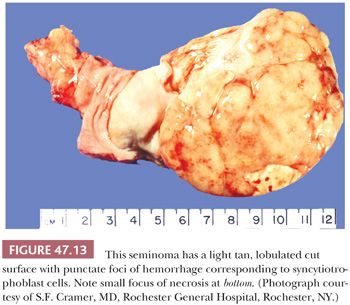
The cut surface of the tumor is cream-colored to tan to pink and lobulated to multinodular (Fig. 47.13), with a fleshy quality and a tendency to bulge above the surrounding parenchyma. Punctate foci of hemorrhage may correspond to foci of syncytiotrophoblast cells (196). Well-defined foci of yellow necrosis may be present, but extensive hemorrhage and necrosis are unusual. Tumors with predominantly interstitial growth may resemble small scars or even be grossly in apparent (184). Extension through the tunica albuginea or into the epididymis occurs in less than 10% of cases (197).
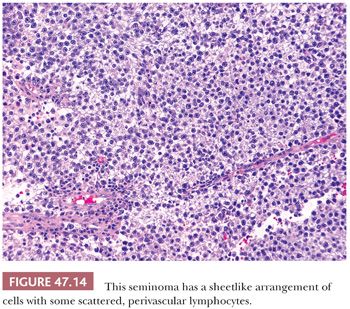
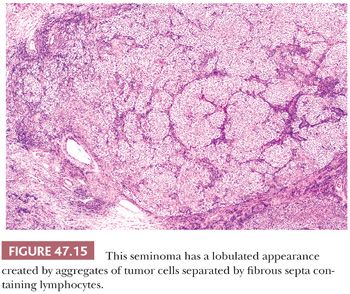
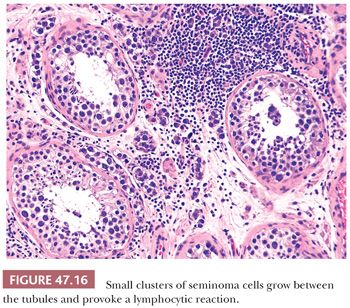
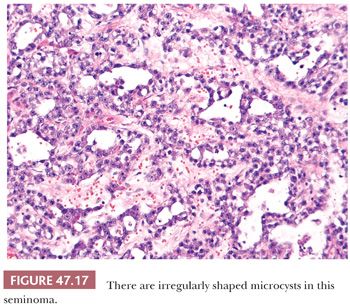
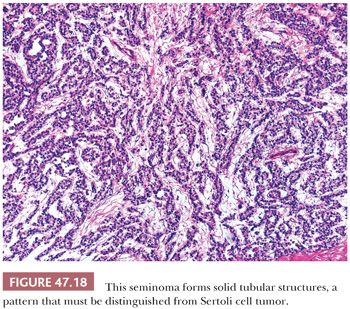
On microscopic examination, seminomas often have a diffuse, sheetlike pattern (Fig. 47.14) or sometimes a lobulated arrangement (Fig. 47.15). There often is an intertubular or cordlike pattern at the periphery of the tumor as neoplastic cells surround but do not destroy seminiferous tubules. Rarely, intertubular growth is predominant (Fig. 47.16), and these tumors may easily be overlooked, although the associated lymphocytes are helpful in their recognition. Branching, fibrous septa often course through seminomas (Fig. 47.15). Edema may cause a microcystic or cribriform arrangement (Fig. 47.17). This pattern may mimic the microcystic pattern of yolk sac tumor, but distinction can usually be made based on the typical seminomatous appearance of other areas; the usually more irregular nature of the cystic spaces, with the frequent presence of intracystic, exfoliated seminoma cells and edema fluid; and the retained cytomorphology of the tumor cells (198). Rarely, a tubular (or pseudotubular) pattern may occur, usually as a focal finding (199,200) (Fig. 47.18). Extensive zones of scarring may occur, with obliteration of major portions of the tumor.
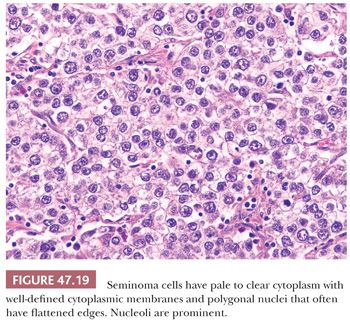
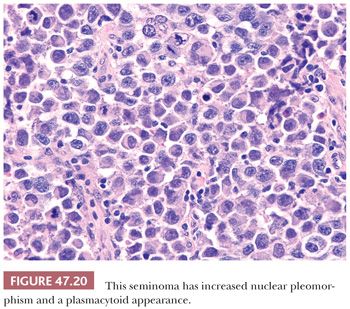
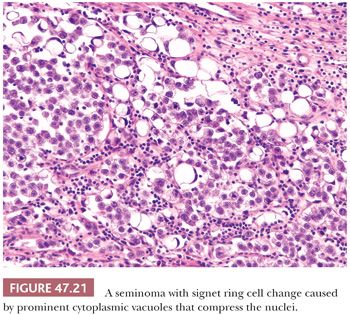
Seminoma cells characteristically have clear to lightly eosinophilic cytoplasm and central nuclei, often with slightly “squared” edges, and one or two large central nucleoli (Fig. 47.19). The cells are closely apposed, with well-defined cytoplasmic borders. An abundant amount of cytoplasm causes the nuclei to be well spaced and nonoverlapping (Fig. 47.19). In poorly fixed specimens, however, cytoplasmic autolysis occurs, obscuring the cell borders and causing apparent nuclear overlapping, thus creating confusion with solid patterns of embryonal carcinoma (see page 2191). Occasionally, the cells have denser, amphophilic to basophilic cytoplasm, imparting a plasmacytoid appearance (Fig. 47.20). These cases often have somewhat more crowded and pleomorphic nuclei and are more apt to be misinterpreted as embryonal carcinoma. We have seen rare cases where empty-appearing cytoplasmic vacuoles imparted a signet ring appearance to some of the tumor cells (Fig. 47.21) (201).
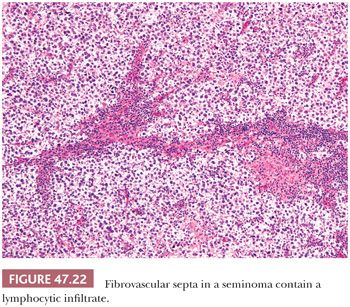
Lymphocytic infiltrates occur in virtually all seminomas. They are often most intense in and around the fibrous trabeculae but also intermingle with the tumor cells (Fig. 47.22). Germinal center formation can develop in some cases, but most of the lymphoid elements are T cells (202–207) that promote granuloma formation. A granulomatous reaction occurs in up to 50% of the cases (197) and varies from scattered clusters of epithelioid histiocytes to well-defined granulomas with characteristic Langhans giant cells. An extensive granulomatous reaction can, in rare cases, obliterate almost all of the underlying seminoma, thus causing a misdiagnosis of granulomatous orchitis (see Chapter 46). It is therefore crucial to examine closely any testis with an extensive granulomatous reaction for residual seminoma cells or IGCNU before accepting such a case as granulomatous orchitis. PLAP or OCT3/4 immunostains can prove useful in identifying residual seminoma cells in this circumstance.
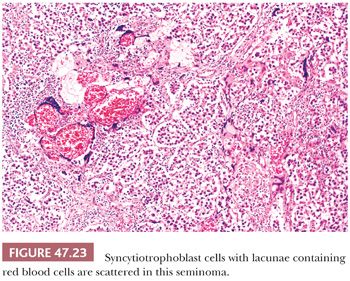
In up to 20% of seminomas, trophoblast cells are identified in a scattered pattern (208,209). Sometimes, these cells have a distinctly syncytiotrophoblastic appearance with multinucleation and/or intracytoplasmic lacunae (Fig. 47.23). In other cases, they appear as large mononucleated cells. They are often located adjacent to capillaries and may be associated with microfoci of hemorrhage. Although small, nodular aggregates of trophoblast cells may be seen, confluent growth is lacking, which, in conjunction with the absence of a mononucleated trophoblast component, permits distinction from choriocarcinoma.
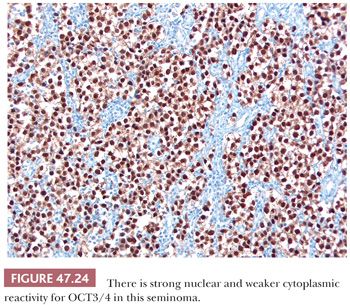
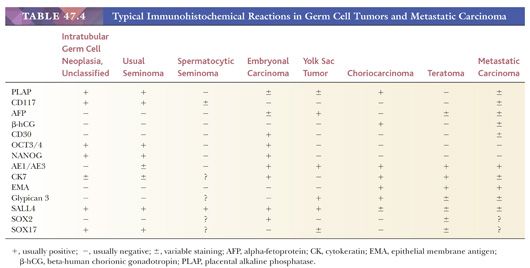
Most seminomas contain glycogen, and PAS stains are usually positive. PLAP immunostains are positive, usually with a membrane-accentuated pattern, in 85% to 98% of cases (17,159,210,211). Reactivity for CD117 and podoplanin (also known as M2A and D2-40) occurs in most (212–217), whereas epithelial membrane antigen (EMA) and CD30 are typically negative (Table 47.4). OCT3/4 is a transcription factor that is positive in seminoma (Fig. 47.24), embryonal carcinoma, and IGCNU (218,219). We consider it the single best marker for seminoma because of its great sensitivity and high specificity. SOX2 negativity and SOX17 positivity in seminomas contrast with the reverse pattern seen in embryonal carcinomas (220–224). Seminomas may also contain cytokeratin (CK) 7, CK8, and CK18 and, less commonly, CK4, CK17, and CK19 (225,226). Intermingled trophoblast cells are positive for CK8, CK18, and CK19 as well as hCG (17,208,209,225,227,228). Positivity of seminomas with antibodies directed against CK8 and CK18 (e.g., CAM 5.2) may, therefore, be seen (usually in isolated cells but rarely more diffusely [17,229,230]) in up to 73% of cases (225). Most routinely processed seminomas, however, are negative or weakly and focally reactive with broad-spectrum CK antibodies lacking these specificities.
On electron microscopic study, seminomas are primitive cells, often having large aggregates of glycogen and simple cellular organelles that are polarized in the cytoplasm (29,231,232). Cell membranes are closely apposed, but junctions are usually primitive and few. The nuclei have intricate nucleoli but an evenly dispersed euchromatin. They may show epithelial differentiation, including well-defined desmosomes and surface microvilli, despite a typical light microscopic morphology (29).
Seminomas have a DNA index of l.66 (233), which is significantly higher than in nonseminomatous tumors (21,233). In addition, isochromosome (12p) or other chromosome 12 anomalies are identified on karyotypic analysis (234–236).
Differential Diagnosis
Solid patterns of embryonal carcinoma may be confused with seminoma. Poorly defined cell borders, nuclear overlapping, and nuclear irregularity and pleomorphism are features of embryonal carcinoma that differ from most well-fixed seminomas. Furthermore, embryonal carcinomas lack the regular fibrous septa of seminoma. CK and CD30 stains (both positive in embryonal carcinoma) may also prove useful in this differential diagnosis (see previous section) (Table 47.4). CD117, podoplanin (D2-40 antibody), and SOX17 may also be useful because they are positive in seminoma but not embryonal carcinoma (237–239). It is clear that some seminomas have more pleomorphic foci than others; we require, however, definite evidence of epithelial differentiation in the form of papillae or glands or supportive immunoreactivity in the pleomorphic zones before recognizing embryonal carcinomatous differentiation.
Solid patterns of yolk sac tumor may also resemble seminoma, but these are usually associated with more characteristic yolk sac tumor patterns (240). If only a small biopsy is available, solid pattern yolk sac tumor often shows suggestions of true microcyst formation, unlike seminoma, and may show intercellular basement membrane deposits or cytoplasmic hyaline globules (241). In difficult cases, strong positivity for AE1/AE3 CK, glypican 3, and AFP and negativity for OCT3/4 support solid yolk sac tumor over seminoma (240). Glypican 3 is more sensitive than AFP in this regard (242,243). Lymphomas involving the testis may be confused with seminoma. Although primary testicular lymphomas do occur, most cases of testicular involvement represent spread from an extratesticular site. Bilateral involvement in an older patient is a clinical feature in favor of lymphoma over seminoma (244–248). Paratesticular involvement on gross examination is more common in lymphoma (248). Lymphomas often have an extensive intertubular pattern with relative tubular preservation (245,246,249) and usually lack the clear cytoplasm and well-defined cell borders of seminoma. The nuclei of lymphomas are frequently irregular and less uniform than those of seminoma. The great majority of seminomas are associated with IGCNU, but lymphomas are not. PLAP and OCT3/4 immunostains are positive in seminomas but almost always negative in lymphomas (218,250), although there are anecdotal reports of rare large B-cell lymphomas that reacted for OCT3/4 (S. Tickoo, personal communication, March 2013). On the other hand, lymphoid markers show the opposite pattern (17). The distinction of seminoma from Sertoli cell tumor is discussed on page 2212.
Seminomas are extremely sensitive to radiation and chemotherapy, and these modalities usually represent the primary forms of treatment following orchiectomy, although clinical stage I patients are increasingly managed by surveillance (251–254). Many patients with clinical stage I seminoma receive radiation directed at the ipsilateral inguinal and iliac lymph nodes as well as to the abdominal para-aortic and paracaval nodes, although some studies suggest that early-stage patients may not need pelvic radiation in the absence of prior inguinal surgery or scrotal involvement (255–257). Cure rates exceeding 95% can be expected for these patients. When recurrence occurs, it is almost always outside of the radiated field. Adjuvant carboplatin is now used in many centers as an alternative to radiotherapy with comparable results, although with less long-term outcome data (258). In patients with metastatic seminoma to the retroperitoneum, those with less bulky disease are treated with radiation with cure rates of 90% to 96% (259–261). With bulky retroperitoneal seminoma or supradiaphragmatic involvement, cisplatin-based chemotherapy is the preferred treatment. Survival for these patients when so treated is approximately 80% (262,263).
The most important prognostic factor in seminoma is tumor stage (which includes tumor bulk). There is controversy concerning the significance of elevated hCG levels in patients with seminoma; there is some evidence that patients with more than a moderate hCG elevation have a poorer overall survival (264–266), although this may be a surrogate for tumor bulk (192,267). The presence of a lymphoid infiltrate and prominent granulomatous reaction may confer a somewhat improved prognosis (268), but this may be of borderline significance (269,270). As seminoma has almost a uniformly favorable prognosis, the possibility of defining pathologic characteristics to define poor risk disease is challenging: Thousands of cases are needed to adequately power any future study.
The recognition of a histologically defined “anaplastic seminoma” with a worse prognosis remains controversial. It is clear that the original criteria (271), based on the mitotic rate of seminomas, are not effective in recognizing an aggressively behaving subset of seminomas (272,273). Often, tumors classified as “anaplastic seminomas” are either poorly fixed seminomas, with obscuring of the diagnostic features, or solid pattern embryonal carcinomas. Tickoo et al. (274) described “seminoma with atypia” based on nuclear pleomorphism and crowding, dense cytoplasm, and few lymphocytes. Such tumors were more likely to present with advanced-stage disease and to express CD30 and lose CD117 reactivity. However, some would regard the CD30 positivity as representing early transformation to nonseminoma.
SPERMATOCYTIC SEMINOMA
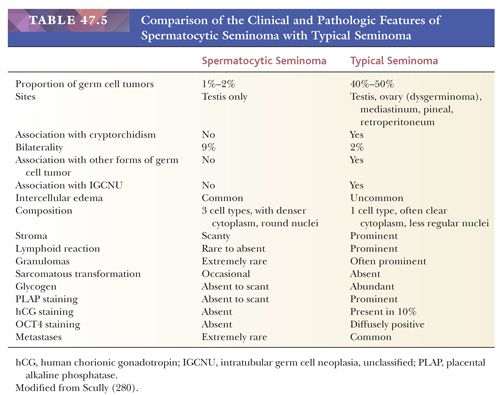
Spermatocytic seminoma is an unusual but generally distinctive germ cell tumor first recognized by Masson (275). It constitutes approximately 1% to 2% of all testicular germ cell tumors (179,276) and therefore is 25 to 40 times less common than usual seminoma. It is particularly important to recognize spermatocytic seminoma and distinguish it from usual seminoma because the implications for treatment differ significantly.
Patients with spermatocytic seminoma are older than most with testicular germ cell tumors, the average being 50 to 60 years (179,276–278), although there is rare occurrence in patients less than 30 years old (277). Most present with painless testicular enlargement (276,277), which can be of several years duration. Bilateral involvement, usually asynchronous, occurs in 9% of cases (276). Unlike usual seminoma, which can occur in extragonadal sites as a primary tumor (where it is designated germinoma) and often occurs with other forms of germ cell tumor, spermatocytic seminoma has never been described as originating in any site other than the testis and is not associated with other germ cell tumor types. It does not appear to be linked to cryptorchidism (276,279), as are other testicular germ cell tumors, and probably does not share other epidemiologic features with the other forms of testicular germ cell tumor. The usual serum markers are negative (276,277).
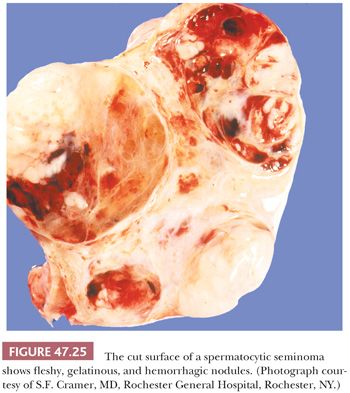
Grossly, spermatocytic seminomas are typically multinodular with a cut surface that varies from gray-white and fleshy to tan and gelatinous (Fig. 47.25). Hemorrhage and necrosis may occur as well as extratesticular extension (277,280).
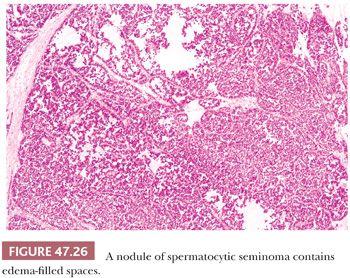
At low power, nodules of diffuse sheets of cells may be interrupted by edema-filled spaces (Fig. 47.26). If edema is extensive, a nested, pseudoglandular, or even trabecular pattern can develop. The stroma is typically not conspicuous, and the regular fibrous septation of classic seminoma is absent. Also, there is usually no, or only a scant, predominantly perivascular lymphoid infiltrate, although a rare case may show a more extensive lymphoid component. Granulomas are virtually always absent, although one of the authors has, courtesy of Dr. R.H. Young, seen a “unique” case with prominent granulomas (Fig. 47.27). Vascular invasion can occasionally be identified (277).
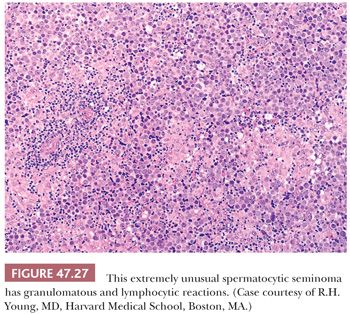
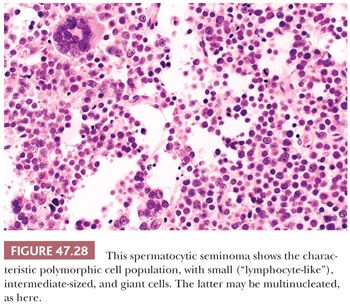
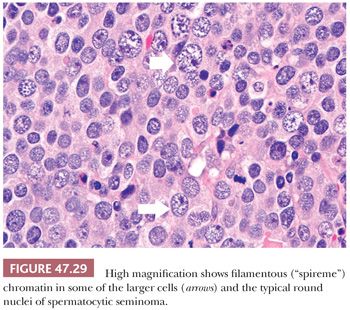
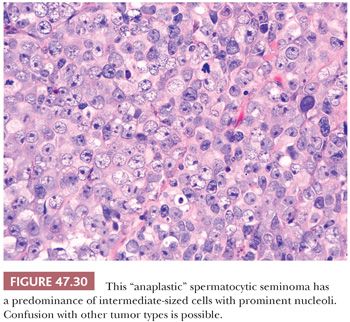
The most distinctive feature of spermatocytic seminoma is its cellular polymorphism. The cells generally have round nuclei, but they vary in diameter from 6 to 100 µm. Three cellular populations occur: small lymphocyte-like cells averaging 6 to 8 µm in diameter, intermediate-sized cells averaging 15 to 20 µm in diameter, and giant cells averaging 50 to 100 µm in diameter (Fig. 47.28). The lymphocyte-like cells are degenerate in nature and have smudged chromatin and more cytoplasm than a true lymphocyte. The intermediate-sized cells are most common and have round nuclei with granular chromatin, variably prominent nucleoli, and pale to eosinophilic cytoplasm. Some have distinctive filamentous chromatin reminiscent of the “spireme” chromatin identified in meiosis of primary spermatocytes (Fig.47.29). The giant cells may be mononucleated or multinucleated with prominent nucleoli; some may also have a “spireme” chromatin. The mitotic rate is often high, and apoptosis is prominent (281). An “anaplastic” variant of spermatocytic seminoma has been described (282); it consists of a relatively monomorphic population of intermediate cells with prominent nucleoli, with foci of typical spermatocytic seminoma elsewhere (Fig. 47.30). The immunohistochemistry, ultrastructure, and clinical course of these cases, however, are typical of spermatocytic seminoma (282).
Intratubular growth of spermatocytic seminoma is common, and invasion from such foci probably gives rise to separate nodules of tumor. Unlike most other forms of testicular germ cell tumor (with the possible exception of childhood cases [150,151]), spermatocytic seminomas are not associated with IGCNU (283).
Immunostains are negative for vimentin, actin, desmin, AFP, OCT3/4, hCG, carcinoembryonic antigen (CEA), and leukocyte common antigen (LCA) (218,277,284,285). Staining for PLAP is generally reported as negative (277,284,286), although some investigators have reported rare PLAP-positive tumor cells in a minority of cases (277,285). CD117 positivity has been reported (287), and therefore, it is not useful in the differential diagnosis with usual seminoma (Table 47.4). Cytokeratins are usually negative, although a rare case may show cytoplasmic positivity for CK18 (225,284). There is positive reactivity for cancer/testis antigens (222). Studies with maturation-specific markers support differentiation to the stage of spermatogonium-pachytene spermatocyte (288).
On ultrastructural examination, one study described meiotic-like chromosomal structures in spermatocytic seminomas (289), but another failed to find these (290). Primitive junctions can be identified as well as a Golgi apparatus, ribosomes, mitochondria, and a peripheral layer of basement membrane (289,290). Intercytoplasmic bridges may form between adjacent cells (291). Glycogen is usually absent or sparse.
Because of the distinctive meiotic-type chromatin in some cells, it has been proposed that the tumor develops from meiotic cells. However, haploid DNA values have not been identified (283,285,290,292), and lectin-binding studies do not support advanced spermatogenic differentiation (293) However, some preferentially expressed meiotic genes have been shown, supporting origin from the primary spermatocyte (294).
Spermatocytic seminoma is most likely to be confused with usual seminoma. Table 47.5 lists the clinical and pathologic features of these two neoplasms that should permit their distinction. Lack of reactivity of OCT3/4 is helpful in the differential with usual seminoma. The “anaplastic” variant is more prone to confusion with embryonal carcinoma but has more rounded nuclei, has foci of typical appearance, and lacks IGCNU and reactivity for CD30, PLAP, OCT3/4, and cytokeratins. Solid yolk sac tumor is also a possible consideration and, like spermatocytic seminoma, lacks OCT3/4 reactivity. Positivity of solid yolk sac tumor for glypican 3 and cytokeratins is diagnostically useful. Most spermatocytic seminomas also show stronger and more extensive reactivity for two cancer/testis antigens, GAGE7 and NY-ESO-1, than seminomas, embryonal carcinomas, and solid yolk sac tumor, with the exception of the hepatoid variant of the latter (295). Additionally, they express the stage-specific markers of germ cell maturation: synaptonemal complex protein 1, synovial sarcoma on X chromosome, and xeroderma pigmentosum type A (288).
Despite its alarming microscopic appearance, spermatocytic seminoma metastasizes only exceedingly rarely. There are only two well-documented case of metastasis (279,296,297); adequate treatment, therefore, consists of orchiectomy alone.
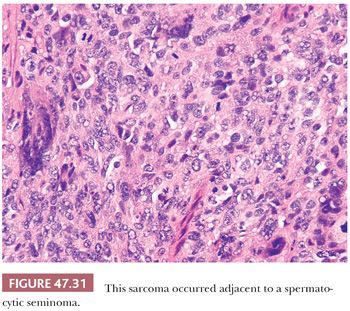
Sarcomatous degeneration is a rare complication of spermatocytic seminoma (277,298,299). These tumors occur in older men, just like uncomplicated spermatocytic seminomas, but there is often a history of recent, accelerated growth. Grossly, they tend to have a more hemorrhagic, necrotic appearance with a whorled aspect on cut surface (298,299). The sarcomatous component is usually intimately intermingled with typical-appearing spermatocytic seminoma and often has an undifferentiated appearance (Fig. 47.31) or shows rhabdomyosarcomatous differentiation (277,298,299). Unlike uncomplicated spermatocytic seminoma, these tumors behave aggressively, with approximately 50% of the patients developing metastatic disease and dying of the tumor (277,298,299). The disseminated tumor consists only of the sarcomatous component and tends to have a hematogenous distribution, involving the lungs most frequently.
EMBRYONAL CARCINOMA
Embryonal carcinoma is a germ cell neoplasm composed of primitive epithelial cells mostly arranged in solid, papillary, and glandular configurations. As a pure neoplasm, it is relatively uncommon, representing approximately 10% of germ cell tumors (179,180); however, an embryonal carcinoma component is found in most (87%) of the nonseminomatous neoplasms (179). Many cases of embryonal carcinoma with yolk sac tumor were previously classified as pure embryonal carcinoma.
Embryonal carcinomas tend to occur in patients approximately 10 years younger (average age, 31 years [179]) than those with seminomas. Most patients present with a testicular mass, which may be associated with pain. Unlike seminoma, which is limited to the testis at presentation in 70% of the cases, 66% of patients with a tumor composed predominantly of embryonal carcinoma have metastases at diagnosis (300), and presenting symptoms may therefore be related to metastatic disease, including back pain, dyspnea, cough, hemoptysis, hematemesis, and neurologic symptoms. Gynecomastia may occur in a minority of patients and generally is a reflection of admixed, hCG-producing trophoblastic elements. Sometimes, widespread metastases are present in the face of a clinically occult primary tumor (301) (see page 2207).
Patients with pure embryonal carcinomas usually do not have serum AFP elevation (302), although rare embryonal carcinoma cells may stain positively for AFP (17,303). The high rate of positivity for AFP reported by some (304) reflects the frequent association of embryonal carcinoma with a yolk sac tumor component. Elevated serum levels of lactate dehydrogenase (LDH) occur in approximately 60% of patients with advanced-stage disease (305), and elevation of PLAP may also be seen (195).
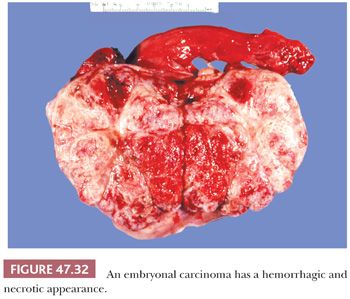
On gross examination, embryonal carcinomas are usually pale-gray, poorly demarcated tumors associated with hemorrhage and necrosis (Fig. 47.32) with an average diameter of 2 to 3 cm (268). Extratesticular spread is present in approximately 20% of cases (197).
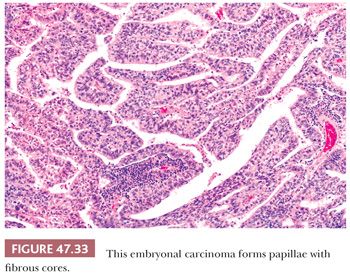
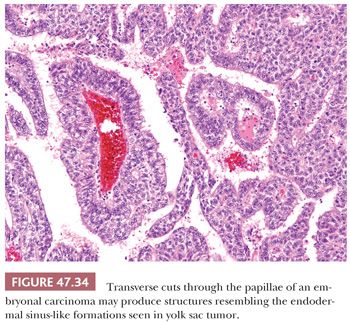
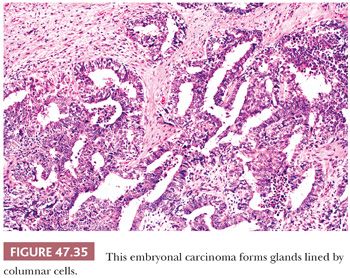
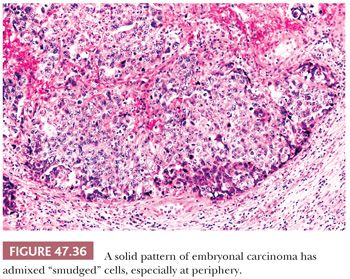
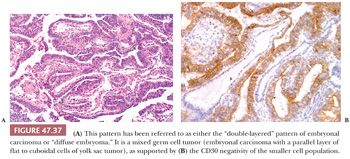
On microscopic examination, there are typically nodules of large cells with prominent zones of eosinophilic, coagulative necrosis. Within these nodules, several patterns may occur. The tumor may be arranged in papillae that may have stromal cores (Fig. 47.33) or consist purely of epithelium, with intervening slitlike spaces. Cross sections of papillae with prominent vessels result in a “pseudoendodermal sinus” pattern (Fig. 47.34) (306). Glandular patterns are common, with cuboidal to columnar tumor cells arranged around luminal spaces (Fig. 47.35). Solid patterns have a sheetlike proliferation of tumor cells (Fig. 47.36). Intermingled cells with a dark, smudged appearance are common and characteristic (Fig. 47.36) but are nonspecific. This feature has been termed an “appliqué” appearance because the degenerate cells appear to be “applied” to the periphery of tumor nodules. Occasionally, such cells can be difficult to distinguish from trophoblastic elements, causing concern for choriocarcinoma. Immunostaining for hCG is useful in resolving this problem. Additional patterns include anastomosing, sievelike, pseudopapillary, and blastocyst-like, but these are uncommon (307).Two patterns with a prominent embryonal carcinoma component include the polyembryoma-like and diffuse embryoma. In the former, embryonal carcinoma is arranged in intimate association with yolk sac tumor to create variably formed simulations of early embryos (see “Mixed Germ Cell Tumors,” page 2206). In the latter, also termed a “double layer pattern” of embryonal carcinoma, ribbons of columnar embryonal carcinoma cells are accompanied by a parallel layer of smaller, flattened cells (306) (Fig. 47.37). The AFP positivity and CD30 negativity of the smaller cell component, as well as its appearance, indicate that this pattern represents a mixed germ cell tumor with a yolk sac tumor component (see page 2207).
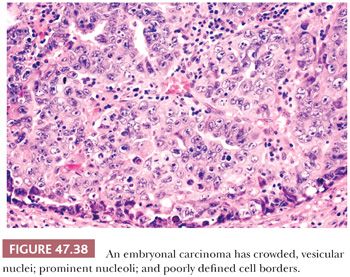
On high-power examination, embryonal carcinoma cells, in routine paraffin sections, are generally polygonal with amphophilic to lightly basophilic cytoplasm and ill-defined cytoplasmic borders (Fig. 47.38). The nuclei are large and often irregularly shaped, with vesicular chromatin interspersed with clumps of heterochromatin, and have one or more centrally located, large nucleoli. Often, the routine histologic appearance is that of nuclear crowding such that nuclei appear to overlap; although in plastic sections, this finding is seen to be an effect of the section thickness. The mitotic rate is generally very brisk.
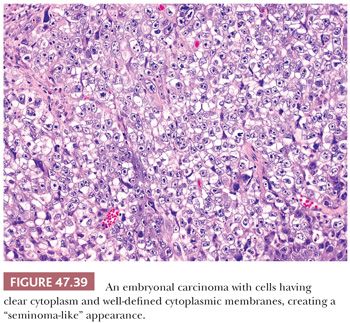
It is not uncommon, however, for some embryonal carcinomas to have foci reminiscent of seminoma where the cells have distinct cytoplasmic membranes and pale or clear cytoplasm (Fig. 47.39). The nuclei, however, have the more irregular and pleomorphic appearance that is expected. When well-formed glands are present, the cells may be cuboidal or columnar, sometimes with subnuclear and or supranuclear clear vacuoles reminiscent of secretory endometrium.
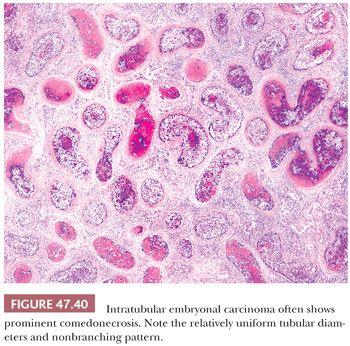
It is common to identify intratubular embryonal carcinoma adjacent to an invasive lesion. Such intratubular foci often show extensive necrosis and may ultimately undergo total necrosis and calcify (Fig. 47.40). If there is similar regression of the invasive neoplasm (see page 2207), the presence of such coarse intratubular calcifications (“hematoxylin-staining bodies”) provides good evidence of a regressed germ cell tumor (308,309).
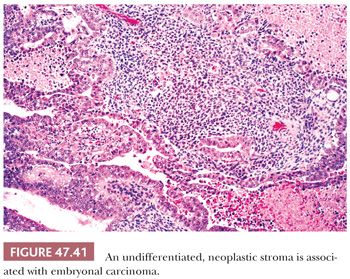
A source of controversy is the extent that an embryonal carcinoma may have a stromal component. There is a tradition of permitting a primitive neoplastic mesenchymal component to be associated with the typical epithelial cells of embryonal carcinoma (Fig. 47.41) and yet retain classification of such tumors as pure embryonal carcinoma (197,310) rather than embryonal carcinoma with a teratomatous component. The rationale for this approach is that such a finding represents a reiteration of primitive embryonic development. There is also some evidence that embryonal carcinomas with and without a primitive mesenchymal component have a similar biology based on the experience of the BTTP (7), although these data antedate current treatments.
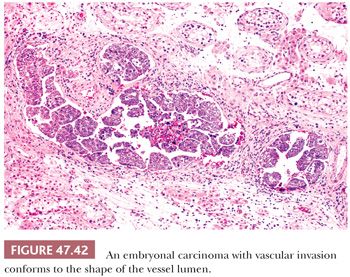
With the advent of “surveillance only” protocols in patients with nonseminomatous tumors (see page 2194), it is important to assess an embryonal carcinoma (and other tumor types) for vascular invasion and extratesticular extension. Artifactual implants of embryonal carcinoma occur easily during tissue cutting because of its extremely cellular and friable nature (3). Intravascular implants consist of loosely cohesive cells that lie randomly in the luminal space. They are often associated with implants that are “buttered” on the surface of the testis and spermatic cord (3). True vascular invasion consists of cohesive groups of cells that conform to the shape of the vessel (Fig. 47.42) or are adherent to its wall by thrombotic material. Vascular invasion is usually easiest to appreciate at the periphery of the main tumor. Intratubular neoplasm may mimic vascular invasion; the identification of residual Sertoli cells in such cases, as well as staining for endothelial markers, can be of assistance. Prominent comedonecrosis supports intratubular rather than intravascular tumor, as does a nonbranching arrangement of rounded tumor nests of relatively uniform diameter (Fig. 47.40).
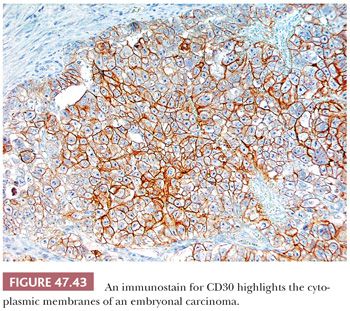
Immunohistochemical stains may assist in the diagnosis of embryonal carcinoma. From 0% to 33% of morphologically typical embryonal carcinomas stain positively for AFP (17,228,303,311,312), with a higher frequency of positivity in the embryonal carcinoma component of a mixed germ cell tumor (303,311), probably indicating its capacity for transformation to yolk sac tumor (Table 47.4). Patchy PLAP reactivity occurs in 86% to 97% of cases (17,159,313), and CK is positive in 95% to 100% (17,314), although EMA positivity occurs in only 2% (17). The presence of CKs other than CK8 and CK18 in embryonal carcinomas contrasts with the limited reactivity (mainly confined to CK8 and CK18 in scattered cells) of most seminomas (225,230,315). OCT3/4 is uniformly positive in tumor nuclei but not helpful in distinction from seminoma (218,219). CD30 positivity (Fig. 47.43) occurs in 84% of cases and is uncommonly seen in other types of germ cell tumor (316–318) (Table 47.4). CD30 positivity is sometimes lost in embryonal carcinomas after chemotherapy (319), in which case, use of SOX2 (positive) and SOX17 (negative) may be helpful (220–224). Although embryonal carcinomas are not difficult to recognize in the testis, an embryonal carcinoma presenting in a metastatic site may be extremely difficult to distinguish from an undifferentiated carcinoma of nongerminal type. The presence of OCT3/4, PLAP, and CD30 positivity and EMA negativity in such cases can be of significant diagnostic assistance. Other stains that may be positive in some embryonal carcinomas include α1-antitrypsin, Leu-7, vimentin, LDH, ferritin, and human placental lactogen (17,303,320,321).
On ultrastructural examination, embryonal carcinomas appear as poorly differentiated adenocarcinomas, forming small glandular spaces with long tight junctional complexes and a peripheral basal lamina (29,322). The Golgi complex is usually prominent, and the nucleus is typically quite irregular in shape with a large, complex nucleolus and cytoplasmic inclusions.
Cytogenetic and molecular analysis of embryonal carcinomas has identified the i(12p) marker chromosome or other 12p amplifications, which can be diagnostically useful when embryonal carcinoma presents in a metastatic site without a known testicular lesion (235,323).
Differential Diagnosis
Embryonal carcinomas must be distinguished from several other types of tumor. It is most important to distinguish embryonal carcinoma from seminoma because seminomas, at least in lower stage disease, are treated with radiation therapy, whereas metastatic embryonal carcinomas are generally treated with polyagent chemotherapy and surgery. This important differential diagnosis has been discussed on page 2186. Solid pattern yolk sac tumors may be confused with embryonal carcinomas; however, other patterns of yolk sac tumor frequently permit this distinction. In small specimens, solid pattern yolk sac tumor can usually be distinguished from embryonal carcinoma by virtue of the smaller size of its cells and nuclei and its less pleomorphic nature. The presence of hyaline globules or basement membrane deposits favors yolk sac tumor (241), as does AFP or glypican 3 positivity and CD30, OCT3/4, or SOX2 negativity (Table 47.4). Often, tumors composed predominantly of embryonal carcinoma have foci of contiguous yolk sac tumor composed of smaller, vacuolated tumor cells, frequently in a myxoid background. Large cell lymphomas may be confused with embryonal carcinomas but usually occur in older patients with extratesticular disease. Lymphomas often have an interstitial pattern; always lack IGCNU; are negative for PLAP, OCT3/4 (with rare exceptions), and CK (250); and are usually positive for LCA and other lymphoid markers (17,248). CD30 reactivity, however, may occur in both types of tumor. The differential with “anaplastic” spermatocytic seminoma has been discussed on page 2189. The most difficult diagnosis is the distinction of embryonal carcinoma in an extragonadal site from a metastatic undifferentiated carcinoma. The helpful immunostaining patterns for this problem have been discussed earlier, with OCT3/4 being particularly valuable. CD30 staining has been shown to be less frequently positive in postchemotherapy embryonal carcinomas (319), an important caveat to keep in mind. Mucin stains may be helpful as well because embryonal carcinomas lack cytoplasmic mucin, which may be identified in poorly differentiated carcinomas of somatic origin. The electron microscopic demonstration of the long tight junctions of embryonal carcinoma may also be helpful in this context (322).
Nonseminomatous germ cell tumors, including embryonal carcinoma, are now effectively managed in most cases by surgery and/or chemotherapy. The form of treatment following radical orchiectomy is stage dependent. For clinical stage I disease, there is controversy regarding the best approach; one school of thought advocates retroperitoneal lymph node dissection (RPLND) of the nerve-sparing type in all such patients (324), whereas others advocate careful follow-up without additional treatment unless relapse develops (“surveillance” management). The overall survival for these two approaches is comparable (approximately 98%), although the identification of certain features in the orchiectomy specimen (notably vascular invasion, large tumor size, and a high proportion of embryonal carcinoma) may be considered relative contraindications to surveillance management because of increased risk of relapse. Therefore, it is important for the examining pathologist to carefully examine the orchiectomy specimen for lymphovascular invasion and to estimate the percentage composition of the various components as well as provide a measurement of the maximum tumor dimension.
For patients with limited retroperitoneal metastases, treatment is RPLND and either close follow-up or postoperative adjuvant chemotherapy. Survival rates exceeding 95% are expected (325). In more advanced-stage disease, the usual treatment is initial cisplatin-based chemotherapy followed by resection of residual masses, if any exist. This group of patients has an overall survival of 70% to 80% (262,326), with a key prognostic finding being the nature of any residual tumor, whether teratomatous, which carries a good prognosis, or nonteratomatous germ cell tumor, which carries a less favorable prognosis. Pathologic findings in the resected residual tissue often determine the need for additional therapy (327). Assessment of completeness of excision and volume of residual malignant elements, in conjunction with accurate classification, are particularly important (328,329).
For patients with disseminated disease, the prognosis appears to depend on factors including tumor bulk (263,326,330–332), levels of serum markers (4,263,330–333), and proliferative index (333). Second- and third-line salvage regimens are used after failure of first-line chemotherapy with some success (334–336).
YOLK SAC TUMOR
Teilum’s (337–339) studies provided the evidence that the neoplasm we now recognize as yolk sac tumor is of germ cell origin with differentiation toward the yolk sac and allantoic membranes. Yolk sac tumor is the most common form of testicular tumor in prepubertal children in whom it occurs as a pure neoplasm at a median age of 16 to 20 months (340,341), with an age range from 3 months to 8 years (340). In postpubertal patients, it is much more commonly a component of a mixed germ cell tumor. In Talerman’s study (342), there was a 44% frequency of a yolk sac tumor component in prospectively examined, nonseminomatous germ cell tumors. Childhood yolk sac tumor is not associated with cryptorchidism and occurs with roughly equivalent frequencies in African American and white populations (343). Both childhood and adult cases most commonly present with a testicular mass, but in a small proportion of adult cases, there are symptoms of metastatic disease or gynecomastia. Eighty percent of children presenting with yolk sac tumor have clinical stage I disease (340,341). Yolk sac tumor remains the most commonly overlooked component of nonseminomatous germ cell tumors (302,344).
From 95% to 100% of patients with tumors containing yolk sac tumor elements have elevated levels of serum AFP (345,346). This provides both an important diagnostic aid, as well as a means of monitoring therapy and detecting recurrences. It is important, however, not to overinterpret high AFP levels in young children because physiologic AFP “elevations” may occur (347).
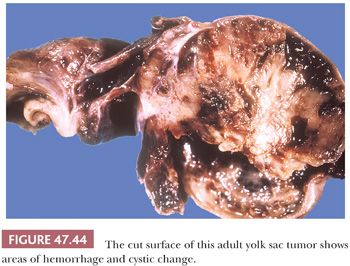
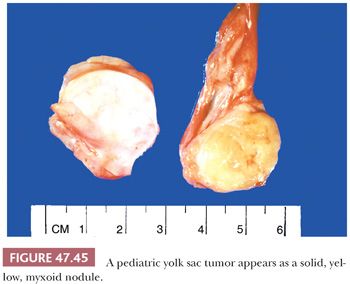
On gross examination, yolk sac tumor may appear as a nonencapsulated, gray to tan to yellow nodule, often with cystic change or a myxoid quality (Fig. 47.44). Hemorrhagic and necrotic areas are common, more so in adult cases. Pediatric cases are usually more homogeneous, often appearing as a solid, yellow/tan myxoid nodule (Fig. 47.45).
Yolk sac tumor has numerous patterns that produce an array of appearances. Fortunately, these patterns are usually admixed, thus facilitating their recognition. The following classification of yolk sac tumor patterns represents a modification of Talerman’s work (348); the 11 patterns are endodermal sinus (also designated perivascular or festoon), reticular (microcystic, honeycomb), macrocystic, papillary, solid, glandular-alveolar, myxomatous, sarcomatoid, polyvesicular vitelline, hepatoid, and parietal.
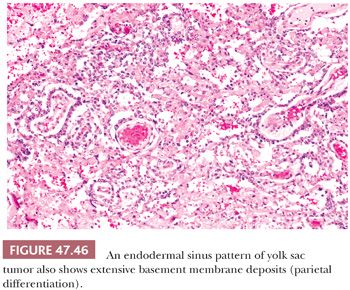
The endodermal sinus pattern has a central vessel in a core of mesenchyme that is mantled by a cuboidal to columnar layer of malignant cells and that is recessed into a cystic space (Fig. 47.46). These structures are referred to as “glomeruloid” or Schiller-Duval bodies. In addition, this pattern has fibrous cores of tissue draped (or “festooned”) by tumor cells that alternate with “labyrinthine” spaces (Fig. 47.47).
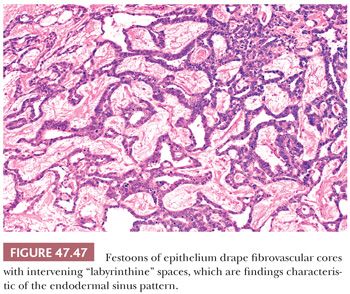
The reticular pattern is most common and consists of a network of tumor cells arranged in cords. Often, the cells have prominent cytoplasmic vacuolation resulting in a microcystic or honeycomb appearance (Fig. 47.48). Sometimes the cells, because of the extensive vacuolation, resemble lipoblasts or signet ring cells. Often, tumor cords are dispersed in a prominent myxoid background that blends gradually into a myxomatous pattern (see page 2196). Larger cysts may develop in some yolk sac tumors, usually in association with a reticular (microcystic) pattern to produce a macrocystic pattern. It is likely that they derive from coalescence of microcysts.
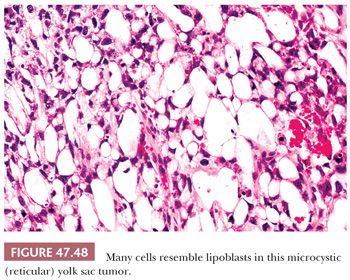
Some yolk sac tumors form cystic spaces into which papillary processes project (Fig. 47.49). These papillae may contain fibrous cores or simply represent piled up epithelium. Typically, the cells have relatively scant cytoplasm, with high nuclear-to-cytoplasmic ratios and some hobnail configurations. Intracystic, detached clusters of cells are frequent.
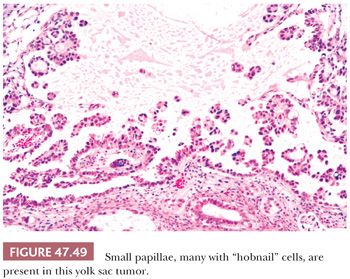
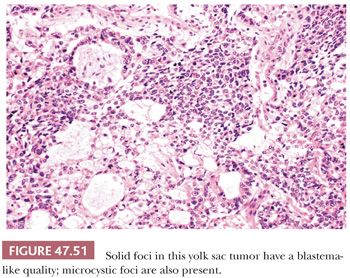
The solid pattern has a sheetlike configuration of cells that may resemble seminoma, although there is usually a focal microcystic tendency, and the fibrous septation and prominent lymphocytes of seminoma are lacking (Fig. 47.50). Often, the cells have clear cytoplasm and well-defined borders, similar to seminoma, but there is association with other yolk sac tumor patterns. They are more pleomorphic than those of seminoma. Occasionally, the cells are small and primitive, resembling blastema (Fig. 47.51). CK, glypican 3, and OCT3/4 stains (see Table 47.4) are useful in separating these two possibilities.
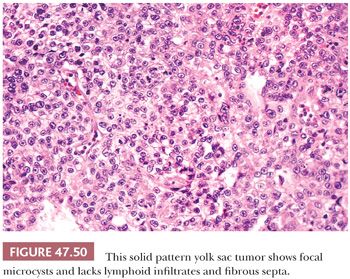
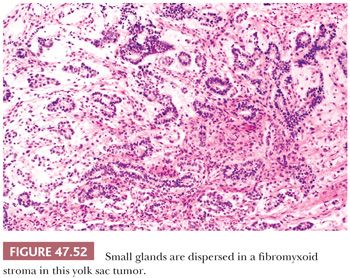
Distinct glands may occur in yolk sac tumor, often developing from cystic, alveolar-like spaces lined by flattened epithelium. Thirty-four percent of yolk sac tumors had such glandular differentiation in one study (241). The glands have a primitive appearance, often with enteric features including an apical brush border (Fig. 47.52). They may have extensive subnuclear vacuolation, similar to early secretory phase endometrial glands. An anastomosing or branching arrangement is common, and rarely, a predominantly or purely glandular pattern occurs, although this is frequent in late recurrences (349). Differentiation of purely glandular yolk sac tumors from teratomas can be problematic; staining for AFP and glypican 3 may assist. The branching, anastomosing pattern is typical of yolk sac tumor, and these glands lack a circumferential smooth muscle component, unlike many teratomatous glands (350).
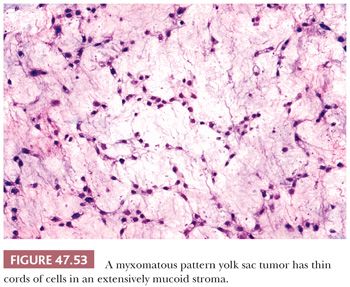
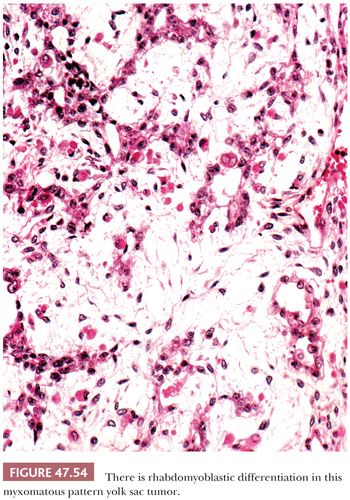
Some yolk sac tumors are extensively myxoid, with trabeculae, thin cords, and individual cells in a mucoid stroma having a high content of hyaluronic acid (Fig. 47.53). Individual stellate to spindle cells trail into the myxoid and vascular stroma in apparent transition from the epithelium but retain CK reactivity (351). Teilum (352) considered this pattern a reiteration of the extraembryonic mesenchyme of the developing embryo. Occasionally, the spindle cells may undergo stromal differentiation, forming skeletal muscle (Fig. 47.54) and cartilage (351), and blurring the distinction between yolk sac tumor and teratoma. We believe that when such elements are intimately associated with yolk sac tumor, they should be classified as yolk sac tumor with, for example, rhabdomyoblastic differentiation. Some of the sarcomatous tumors observed in metastases following chemotherapy may derive from this mesenchymal component of yolk sac tumor (351,353).
It also appears likely that the cellular, sarcomatoid foci that can be identified in occasional yolk sac tumors derive from the proliferation of the spindle and stellate cells originally associated with the myxomatous pattern. Such sarcomatoid areas generally retain CK reactivity. This pattern tends to be especially prominent in postchemotherapy resections where it may be confused with a sarcoma.
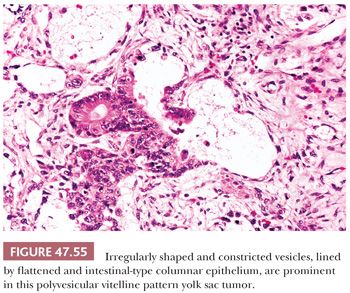
The polyvesicular vitelline pattern is relatively unusual, occurring in 8% of cases in one series (241). It consists of vesicles, often with central constrictions, lined by flattened to cuboidal to columnar cells (Fig. 47.55). A transition in epithelial height may occur at the point of the constriction. The columnar cells resemble primitive enteric epithelium similar to that lining the embryonic allantois. Apical and basal vacuoles may occur. Teilum (354) felt that this pattern was analogous to the embryonic subdivision of the primary yolk sac into the secondary yolk sac. It has always been a minor component in testicular yolk sac tumors, in our experience, unlike in the ovary (355).
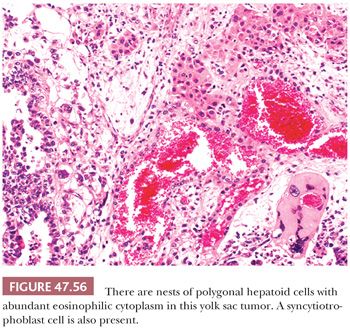
Some yolk sac tumors show hepatic differentiation, with clusters of polygonal cells with eosinophilic cytoplasm; round, vesicular nuclei; and prominent nucleoli (Fig. 47.56). These hepatoid cells may be arranged in sheetlike, trabecular, or nested patterns. Hepatoid differentiation is generally focal in approximately 20% of yolk sac tumors (241,356) but, in rare cases, may be predominant (357), although this would appear to be more common in ovarian tumors (358). Hepatoid foci are intensely positive for AFP and may contain hyaline globules. We have only rarely identified bile.
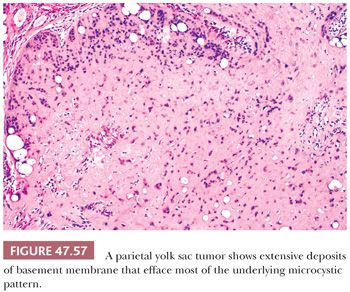
A parietal pattern consists of confluent deposits of extracellular basement membrane between neoplastic cells. The term originates from the parietal layer of the embryonic yolk sac, which synthesizes a thick basement membrane (Reichert membrane). It is common to find scattered foci of parietal differentiation in yolk sac tumors (92% of cases [241]) where it can aid in diagnosis. It is seen in several patterns, including reticular, endodermal sinus, and solid, and consists of focal, wispy to bandlike deposits of eosinophilic matrix (Fig. 47.46). It is rare, however, to have a predominance of extracellular basement membrane in a primary tumor such that the associated pattern is obliterated; when this occurs, it is designated a “parietal pattern” (Fig. 47.57). Parietal patterns occur with greater frequency after chemotherapy and as late recurrences (349,359).
The reticular pattern of yolk sac tumor is the most common one (241,360); solid and macrocystic patterns are also relatively frequent (241,306), whereas hepatoid, polyvesicular vitelline, and endodermal sinus structures are relatively infrequent (241,306).
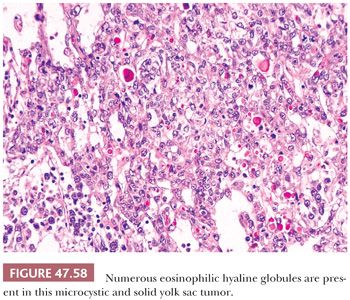
Hyaline globules commonly occur in yolk sac tumor and can aid in its recognition. These are round, eosinophilic, PAS-positive, diastase-resistant globules of variable size from 1 to 50 µm or more in diameter (Fig. 47.58). They also occur in other neoplasms but are uncommon in seminoma and embryonal carcinoma (241). They generally do not stain for AFP. Such globules should not be confused with the more irregularly shaped, bandlike deposits of basement membrane that are the hallmark of parietal differentiation.
Hematopoietic foci, usually consisting of erythroblasts, are rarely identified in yolk sac tumors. IGCNU is commonly present in the seminiferous tubules of postpubertal patients with yolk sac tumor but not in the pediatric cases (16,150).
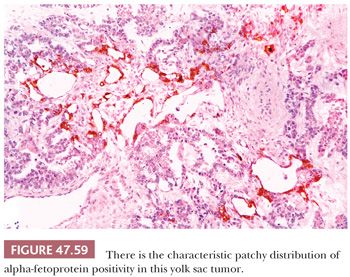
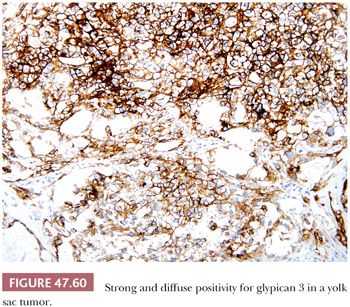
AFP can be identified in the cytoplasm of most yolk sac tumors, with frequencies ranging from 55% to 100% (17,227,229,303); the staining is often patchy (Fig. 47.59 and Table 47.4). Pediatric yolk sac tumors are more likely to be AFP negative (303). Glypican 3 is a newer marker for yolk sac tumor with a greater sensitivity (Fig. 47.60), although it may also be seen in syncytiotrophoblast cells as well as occasional teratomas and embryonal carcinomas (242,243). Hepatoid foci are generally intensely AFP positive and are also usually reactive for HepPar1 and glypican 3. The enteric glandular structures of yolk sac tumor may stain positively for CEA (17,241,321). α1-Antitrypsin can be identified in approximately 50% of cases (17,227). Pankeratins (e.g., AE1/3) are diffusely positive in virtually all yolk sac tumors (17,361), and vimentin can be demonstrated in the stromal component (361). PLAP is not as likely to be reactive in yolk sac tumor as in most other types of germ cell tumor, with 39% to 85% being positive (17,159,313). EMA is usually negative (17), as is CK7, similar to ovarian examples (362) (Table 47.4).
By electron microscopy, yolk sac tumors show epithelial cells with tight junctional complexes and apical microvilli. Extracellular basal laminar deposits may be prominent, with similar appearing material, often with a central lucent zone, within dilated cisternae of endoplasmic reticulum (29). Glycogen is commonly seen, and the nuclei are irregular in shape with a “wandering” nucleolonema.
Differential Diagnosis
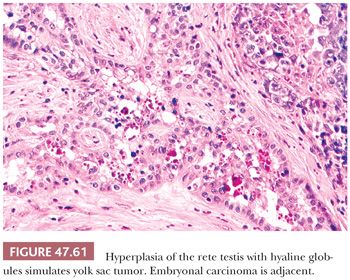
It is important to distinguish solid patterns of yolk sac tumor from seminoma, a differential diagnosis addressed on page 2187. Embryonal carcinomas are distinguished based on the absence of the distinctive patterns of yolk sac tumor and their larger, more pleomorphic nuclei. Hyaline globules and deposits of basement membrane are rarely seen in embryonal carcinoma (241), whereas most express CD30, unlike yolk sac tumor (316,318). There are rare cases that appear to be transitional morphologies between embryonal carcinoma and yolk sac tumor, and examination of other sections usually shows both tumor types. Pediatric yolk sac tumors may be confused with juvenile granulosa cell tumors (363,364) (see page 2215), because both share a solid and cystic pattern, show cytologic atypia, and may have high mitotic rates. Yolk sac tumor typically occurs in older children than the neonates and very young infants who characteristically develop juvenile granulosa cell tumor. Usually, the presence of other yolk sac tumor patterns resolves this differential diagnosis, as does the immunohistochemical demonstration of AFP, which is absent in juvenile granulosa cell tumor. (Serum AFP may, however, be physiologically elevated in neonatal patients [347].) Additionally, juvenile granulosa cell tumors are frequently reactive for inhibin and CD99, whereas those markers are negative in yolk sac tumor (365). Glandular patterns of yolk sac tumor lack the circumferential muscle seen in the glandular components of many teratomas (350). Glandular patterns of yolk sac tumor may be seen as late relapses in the retroperitoneum and mistaken for adenocarcinomatous transformation, with distinction facilitated by AFP and glypican 3 positivity and EMA and CK7 negativity of glandular yolk sac tumor. Invasion of the rete testis by germ cell tumor elements can cause a hyperplastic epithelial reaction with intracytoplasmic hyaline globules that may be misinterpreted as a yolk sac tumor component (366) (Fig. 47.61). This intrarete hyperplasia is differentiated from yolk sac tumor on the basis of the characteristic branching pattern, as seen at low magnification, and the bland cytologic features.
The treatment of adult patients with yolk sac tumor is similar to that of other nonseminomatous germ cell tumors (see page 2194). The presence of a yolk sac tumor component in a testicular primary may be associated with a higher frequency of low-stage disease (367,368). Many children with testicular yolk sac tumors are managed by careful follow-up after radical orchiectomy if there is no clinical evidence of metastases, including postorchiectomy AFP values (369). This approach reflects that 80% to more than 90% of children with yolk sac tumor have pathologic stage I disease (370–372), and there is a lower proportion of retroperitoneal metastases relative to pulmonary (372–375) when compared to adult patients.
A yolk sac tumor component in a metastatic lesion may impart a worse prognosis (376) despite the opposite impact when in the testis (368). This finding probably reflects a greater chemoresistance of yolk sac tumor than other germ cell tumor elements, a conclusion supported by autopsy studies documenting a much greater frequency of residual yolk sac tumor in patients dying in the chemotherapeutic era compared to those dying before effective chemotherapy (377). Chemoresistant spindle cell tumors of variable morphology are often derived from yolk sac tumor elements and may be the source of ultimately fatal sarcomatoid tumors (353).
TERATOMA
Testicular teratomas occur both in pediatric and adult patients, and their biology is significantly different for these two groups. In prepubertal patients, they almost always are pure neoplasms and represent approximately 15% of testicular germ cell tumors (the others being yolk sac tumors) (374,378). The mean age at diagnosis is 20 months, and occurrence beyond 4 years is unusual (374). Most patients present with a testicular mass identified by a parent. In adult patients, on the other hand, teratoma usually occurs as one component of a mixed germ cell tumor, with more than 50% of all mixed germ cell tumors having a teratomatous component (180,379); pure cases in adults are uncommon. The adult patients have the typical features of a testicular germ cell tumor (i.e., they are generally young, present with a testicular mass, but may have symptoms secondary to metastatic involvement and may have serum marker elevations depending on the nature of the associated components).
One puzzling aspect about the biology of testicular teratomas in adults is the occasional development of nonteratomatous metastases in patients with apparently pure teratomas (380). This phenomenon, in the authors’ opinion, is due to the usual development of the adult form of teratoma from a nonteratomatous precursor element (for instance, embryonal carcinoma or yolk sac tumor) that can metastasize as such but also transform to teratoma in the testis (381). Transformation to teratoma in the metastatic site also explains cases reported as “mature teratoma metastasizing as mature teratoma” (382–385). In support of this concept, about 90% of adult patients with pure teratoma also have IGCNU (151), whereas it is absent in pediatric cases (151,386). Karyotypic and molecular studies of pediatric testicular teratomas have yielded normal results (387,388), whereas adult testicular teratomas have had the common abnormalities seen in other malignant germ cell tumors, with the exception of a rare subset of cases (389). Therefore, it is important to recognize that teratoma of the testis in postpubertal patients almost always has malignant potential.
Serum marker studies of patients with pure testicular teratomas are generally negative, although the immunohistochemical detection of AFP in teratomatous glands indicates a potential for abnormal AFP values (209,227,321). One should also be aware that, in very young children, there is a physiologically “high” AFP value (347).
On gross examination, teratomas are often multinodular, and their cut surface varies from multicystic (Fig. 47.62) to solid. Nodules of translucent, white cartilage may be seen. The cysts may be filled with clear or mucoid fluid or keratinous material. Hair, except in the rare dermoid cyst (a specialized form of pure teratoma discussed more fully on page 2203), is not identified. Areas of immature tissue may have a fleshy, encephaloid character.
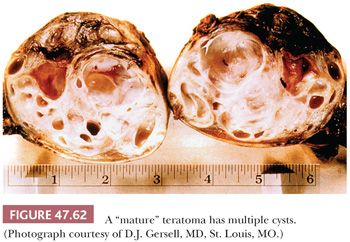
The characterization of testicular teratomas as “mature” or “immature” has been removed from the most recent WHO classification (5). This is because immaturity in the epithelial or stromal components has been shown to have no prognostic significance, although it is likely that overgrowth of highly immature neuroectoderm, thereby constituting a primitive neuroectodermal tumor, does worsen the prognosis. On microscopic examination, pure mature teratomas, by definition, are composed of tissues resembling somatic tissues (Fig. 47.63). Most teratomas in children, somewhat paradoxically, consist solely of mature tissues. In postpubertal patients, there is frequent cytologic atypia of such tissues that reflects their malignant potential and development from IGCNU. This is supported by the demonstration of aneuploidy (390,391). Common findings include nodules of atypical cartilage (Fig. 47.64), glands lined by gastrointestinal or respiratory epithelium with muscular cuffs, squamous islands, transitional epithelium, neuroglia, pigmented retinal epithelium, and fibrous stroma. Bone, liver, pancreas, thyroid, prostate, meninges, kidney, and choroid plexus are uncommon. Sometimes, the glands are architecturally complex (Fig. 47.65). A granulomatous reaction to extravasated keratin is common.
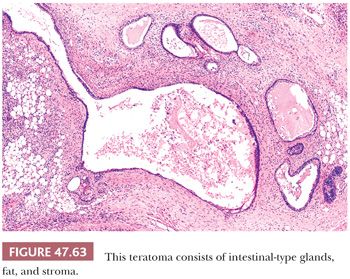
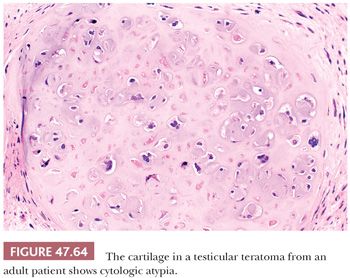
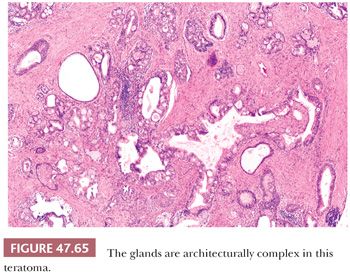
Islands of immature neuroectoderm (Fig. 47.66), as well as primitive tubules, similar to those seen in nephroblastoma and often with accompanying blastema (Fig. 47.67), may also occur, as may embryonic skeletal muscle and nonspecific cellular stroma. There is no point, however, in grading the degree of immaturity in the adult tumors, unlike in ovarian cases, because they derive from invasive malignant germ cell tumors and may therefore have associated metastases even if totally mature. The more important task is to recognize the development of independently evolving neoplasms from overgrowth of neuroectoderm, nephroblastoma-like tissues, rhabdomyoblastic cells, or other types of primitive cells. A guideline for such overgrowth is size in excess of the majority of a low-power field (4× objective); overgrowth of neuroectoderm to this extent results in a primitive neuroectodermal tumor (Fig. 47.68) (392), and similarly for a nephroblastoma-like tumor (Fig. 47.69) (393) and embryonal rhabdomyosarcoma. These lesions represent “teratomas with somatic-type malignancies.” Alternatively, malignant but mature stromal tissues can also overgrow teratomas, giving rise, for example, to “teratoma with leiomyosarcoma.” Secondary carcinomas in teratoma are appreciable when there is evidence of stromal invasion by malignant-appearing epithelium (Fig. 47.70). It should be noted that in some studies, no adverse prognostic significance was associated with secondary teratomatous malignancies when identified in the testis (394), although their presence in metastatic sites and especially in the mediastinum was ominous (392,394–397). In the experience at Indiana University, we found that grading of the sarcomatous tumors into low- and high-grade groups, using the criteria of the French grading system for sarcomas (398), was prognostically useful (399).
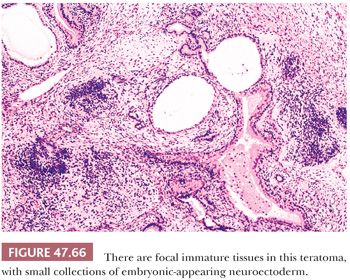
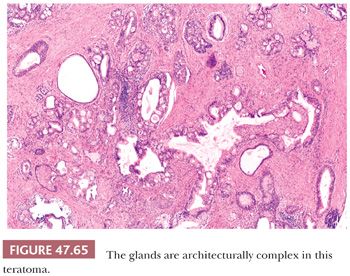
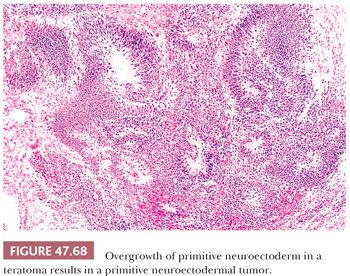
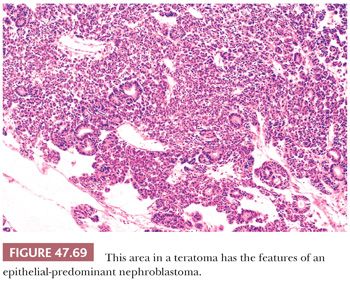
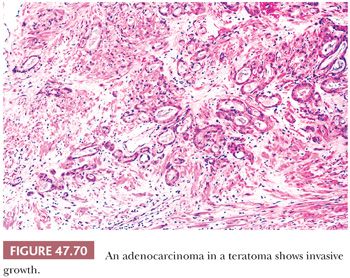
Immunohistochemical staining of teratomas yields the expected results for the specific elements examined. Thus, neural markers are identified in neural tissues (400), chromogranin in neuroendocrine cells (365), cytokeratins in epithelia, vimentin in stromal tissues, and desmin in muscle (361). AFP may be seen in enteric or respiratory-type glandular structures as well as in liver (209,227,321). CEA, α1-antitrypsin, and ferritin occur in teratomatous epithelium in approximately 50% of cases (321), and PLAP is also positive in glandular teratomatous structures in a minority of cases (159,210,313). p53 protein may be identified in teratomatous epithelium (401). SALL4 reactivity occurs in some teratomas, particularly in enteric-type glands and immature neuroectodermal tissue (402).
Differential Diagnosis
It is important to distinguish teratoma in adults from epidermoid and dermoid cysts and the rare benign form of postpubertal teratoma (197,389,403–407). The absence of IGCNU and elements other than a squamous epithelial-lined cyst serve to distinguish epidermoid cyst from mature teratoma. Although dermoid cyst may have other teratomatous elements, the organotypical arrangement of pilosebaceous units to an epidermal surface, the absence of IGCNU, lack of cytologic atypia, and the frequently associated lipogranulomatous reaction allow distinction from teratoma. The benign teratomas of the adult testis also lack IGCNU, have normal associated spermatogenesis, show no cytologic atypia, often display organoid architecture (especially respiratory epithelial-lined glands encircled by cuffs of smooth muscle), and lack i(12p) or other forms of 12p amplification (389).
Prognosis
Pure teratomas in prepubertal children are benign, and orchiectomy is curative (380,408). Pure teratomas in adults are rare, with most examples associated with other germ cell elements, which determine the prognosis. If pure, they may nonetheless have associated metastases of teratomatous or nonteratomatous germ cell tumors for reasons already discussed. At two referral centers for treatment, the proportion of pure mature teratomas with metastases was slightly more than 40% (409,410), a high figure that likely reflects referral bias. In the Armed Forces Institute of Pathology series, 21% metastasized (411).
Epidermoid and Dermoid Cyst and Benign Postpubertal Teratomas
Dermoid cyst should be considered a specialized form of testicular teratoma that merits separate classification, and it is likely that epidermoid cyst is similar. They are both benign lesions.
Epidermoid cyst, by definition, is a squamous epithelial-lined cyst, typically filled with keratin; it represents approximately 1% of testicular masses (403,404). Epidermoid cysts are most common from the second to fourth decades and present as palpable masses (403). They are usually approximately 2 cm in diameter (412) and contain the characteristic yellow-white, “cheesy” debris, often having a laminated appearance typical of keratin (Fig. 47.71). On microscopic examination, a fibrous wall is lined by keratinizing squamous epithelium with a granular cell layer but no skin appendages (Fig. 47.72). Areas of rupture may allow keratin to escape and result in a granulomatous and fibrous reaction. There is no associated IGCNU (151,413), and spermatogenesis, except for the effects of compression atrophy, is generally intact. They are benign lesions (403,404) but must be distinguished from teratoma because of the malignant potential of the latter. The absence of IGCNU and “other” teratomatous elements is key in this regard. Epidermoid cyst may be managed by conservative local excision if the diagnosis can be assured, typically by biopsies of the surrounding testis to exclude IGCNU (413).
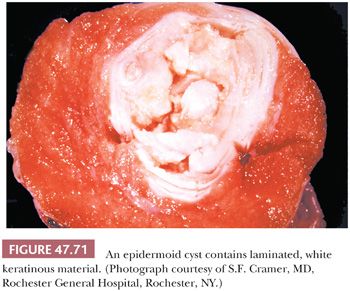
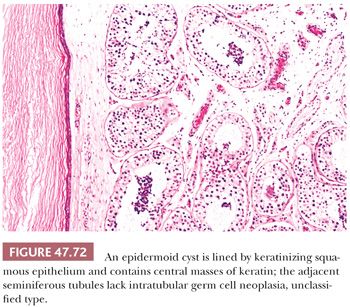
Dermoid cyst is rare and tends to occur in patients in the typical age range for germ cell tumors (407). On gross examination, there is a cyst filled with keratin and, in some cases, hair. On microscopic examination, pilosebaceous units are oriented in a skinlike arrangement to a squamous epithelial surface (Fig. 47.73), and some cases may also contain other elements including cartilage, bone, intestinal mucosa, ciliated glands, and others. It also lacks IGCNU and cytologic atypia and is associated with normal spermatogenesis and absence of chromosome 12p abnormalities (389). Many examples have an associated lipogranulomatous reaction. We now also recognize a “nondermoid” form of benign teratoma of the testis. These cases are analogous to dermoid cysts but lack the cystic, cutaneous-type component of the latter. They frequently, however, have other organoid arrangements, particularly of respiratory type, and otherwise have similar features to the dermoid-type teratomas (389). These cases have had a benign outcome and have been shown to lack the i12p molecular signature characteristic of adult germ cell tumors, including teratomas. It has been suggested that they may represent a prepubertal teratoma that had not been identified in childhood and persisted into adult life (389,414).
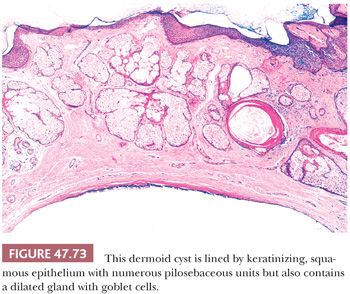
CARCINOID TUMOR
Testicular carcinoid tumors are considered to be monodermal teratomas because, although most are pure neoplasms, approximately 20% are associated with teratomatous elements (415–418). They are rare and occur over an age range from the second to the ninth decades, with a mean age of 46 years (415–417,419,420). Most patients present with a mass, sometimes of several years in duration (417). A clinical carcinoid syndrome is infrequent, occurring in approximately 10% of cases, but serotonin metabolites are detected in a higher proportion (417). On gross examination, they are solid, yellow to tan, well-circumscribed nodules (Fig. 47.74); cysts may be identified if other teratomatous structures are present. Microscopically, they generally appear similar to midgut carcinoid tumors, having islands of cells that form small acini (Fig. 47.75). The nuclei are round with granular chromatin, and there is granular, eosinophilic cytoplasm. Often, discrete pink granules can be seen in the cytoplasm, especially at the periphery of nests and glands. Other teratomatous elements may be identified, most frequently mucinous and glandular epithelium. We have not identified IGCNU in the few pure carcinoids that we have seen, nor have others (421), with one possible exception (422). Serotonin, neuron-specific enolase, chromogranin, synaptophysin, and CK may be identified immunohistochemically (417,423,424). Polymorphous neurosecretory granules are seen by electron microscopy (415,417,423). It is important to distinguish primary testicular carcinoids from metastatic carcinoid tumors. The presence of teratomatous elements supports a primary lesion; metastatic carcinoids are frequently bilateral and multifocal and show lymphatic/vascular invasion, and the patients have evidence of extratesticular tumor. The general prognosis of primary testicular carcinoids is good; 2 of 12 patients in the series of Berdjis and Mostofi (416) developed metastatic disease, which may occur many years following orchiectomy (425).The largest series of Wang et al. (418) showed that 20 typical carcinoids had no recurrences, although recurrence was noted in a case of atypical carcinoid. A clinical carcinoid syndrome correlates with malignant behavior, as does large tumor size (mean size = 7.3 cm for malignant cases) (417). Orchiectomy with follow-up is the usual therapy.
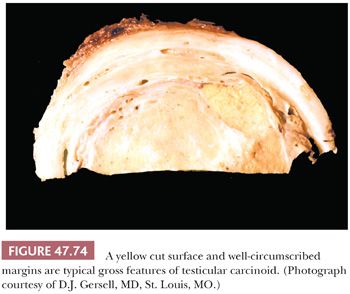
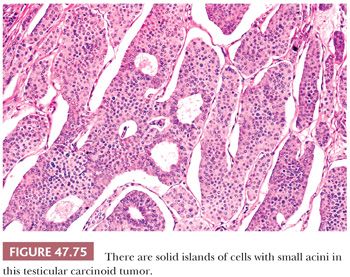
PRIMITIVE NEUROECTODERMAL TUMOR
Primitive neuroectodermal tumors (PNETs) of the testis are also considered monodermal teratomas consisting of small cells arranged in diffuse sheets, tubules, rosettes, and pseudorosettes (Fig. 47.68) (426). They represent overgrowth of primitive neural elements of teratoma and, hence, are usually components of germ cell tumors with teratomatous and other elements (392,427), as discussed on page 2201, in connection with teratomas having a secondary malignant component. Rarely, they represent a pure testicular tumor (426,428,429). On gross examination, the pure cases are fleshy, gray-white, and partially necrotic. Microscopically, small cells with hyperchromatic nuclei are arranged in sheets, neural-type tubules, and rosettes. Neuron-specific enolase, synaptophysin, chromogranin, and CD99 are positive in varying proportions of cases (392). Neurosecretory granules may be present on ultrastructural examination (429). The absence of chromosome 22 rearrangements on fluorescence in situ hybridization (FISH) study supports that they are usually not tumors related to peripheral PNET/Ewing sarcoma (430). Instead, they most resemble central PNETs as seen in the central nervous system of children. Distinction from teratoma with neuroectodermal elements is based on the overgrowth of embryonic-type neuroectoderm (see page 2201). Small cell carcinoma metastatic to the testis typically occurs in older patients, does not form the tubular structures characteristic of PNET, has more impressive CK reactivity, and lacks associated IGCNU. Nephroblastoma-like tumors have better defined tubular structures and usually blastema and stroma (393); the neural markers are negative and WT1 positive. Clinical stage I patients with a component of PNET have a higher rate of relapse than comparable patients lacking such a component (431). Metastatic cases have a very poor outcome (392,431).
TROPHOBLASTIC TUMORS
Choriocarcinoma
Pure testicular choriocarcinomas are rare (0.3% of testicular tumors [197]), but a component of choriocarcinoma is identified in 16% of mixed germ cell tumors on careful examination (179), typically as microfoci. Patients with pure tumors are usually in the second or third decade and often present with metastatic symptoms rather than a testicular mass; some may have no palpable testicular abnormalities on clinical examination. No cases are described in prepubertal children. Frequent presenting symptoms are hemoptysis, lumbar back pain, gastrointestinal bleeding, and neurologic abnormalities. There is often marked elevation of hCG levels and secondary endocrine abnormalities, including gynecomastia (which may also be a presenting complaint) and thyrotoxicosis (because of the thyroid-stimulating hormone–like activity of hCG) (432).
On gross examination, choriocarcinomas are often small, and the testis may appear normal or atrophic from its external aspect. On cut surface, a hemorrhagic and centrally necrotic nodule is generally identified, often with gray tissue at its periphery (Fig. 47.76).
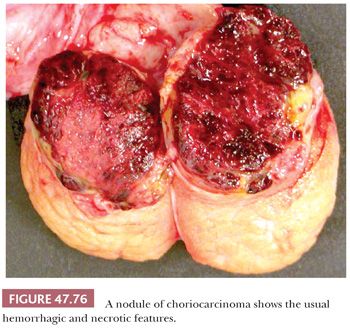
Choriocarcinomas consist of a proliferation of malignant trophoblastic cells. In its classic form, a central area of hemorrhage and necrosis is surrounded by an admixture of two distinct populations of cells—mononuclear cells with generally clear or pale cytoplasm and mild to moderate nuclear pleomorphism (cytotrophoblast and intermediate trophoblast cells) and multinucleated cells, often with intracytoplasmic lacunae containing erythrocytes, that have abundant amphophilic cytoplasm (syncytiotrophoblast cells). Smudged nuclear chromatin is common in the syncytiotrophoblast cells. In the better differentiated cases, the syncytiotrophoblast cells “cap” the mononucleated trophoblast population (Fig. 47.77), similar to the normal arrangement of trophoblast on placental villi. More commonly, the intermingling is random. In some cases, the syncytiotrophoblast cells are scant, occurring as scattered cells among sheets of mononucleated trophoblast cells (Fig. 47.78) (433). This “monophasic” pattern is one that we have more commonly seen following chemotherapy. Not only may the syncytiotrophoblast cells be minor in amount but they may also have a shrunken and degenerate appearance with smudged nuclei. In this circumstance, they are mainly recognizable by their more deeply staining cytoplasm. It can be difficult to distinguish such cases from embryonal carcinoma with cellular degeneration (see page 2190). There are rare cases in which the separation of the trophoblast elements into two separate populations becomes indistinct, but there is a proliferation of a spectrum of malignant trophoblast cells, from small to large; this pattern, in our experience, is also more common in postchemotherapy specimens.
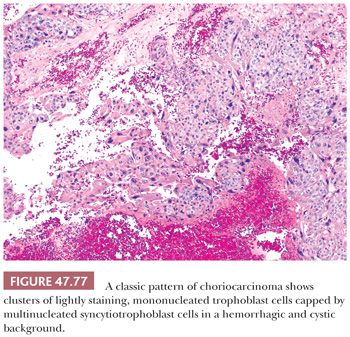
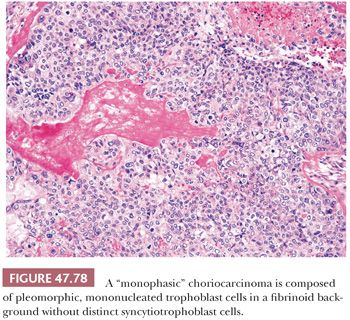
Because of the extensive hemorrhagic necrosis of most choriocarcinomas, diagnostic foci of viable cells may require many sections. Such foci are usually peripheral, and an additional clue to the diagnosis is IGCNU in the adjacent seminiferous tubules. Pyknotic syncytiotrophoblast cells may be identified within the necrotic zone to encourage continued searching for diagnostic foci. Choriocarcinomas, like physiologic trophoblast, have a marked tendency for angioinvasion that correlates with their clinical aggressiveness.
Stains for hCG are valuable in helping to establish a diagnosis of choriocarcinoma and are positive in essentially all cases, mainly in the syncytiotrophoblast cells but also in scattered large mononuclear cells that may be transitional forms between cytotrophoblast and syncytiotrophoblast cells (17,227,434). Syncytiotrophoblast cells are also positive for pregnancy-specific β1-glycoprotein (434) and inhibin-α (365,435,436) Mononucleated trophoblast cells are usually, but not always, hCG negative and SALL4 positive (402). Those having intermediate trophoblast differentiation may stain positively for human placental lactogen and HLA-G (437). PLAP is positive in approximately 50% of choriocarcinomas (17), and CEA is positive in 25% of choriocarcinomas (17,438). Choriocarcinomas contain CK7, CK8, CK18, and CK19 (439). Unlike several forms of germ cell tumor, EMA is positive in a significant number of choriocarcinomas (46%), mainly in the syncytiotrophoblasts (17) (Table 47.4).
Differential Diagnosis. It is important to distinguish choriocarcinoma from hemorrhagic testicular necrosis as a result of torsion, trauma, vascular lesions, or clotting abnormalities. Clinically, testicular infarction usually produces painful testicular enlargement, whereas patients with choriocarcinomas often have small, nonpainful testes. On cross section, infarction usually has a diffuse hemorrhagic picture, whereas a nodular area of hemorrhagic necrosis is typical of choriocarcinoma. Coagulative necrosis characterizes infarction, but residual tissue structures cannot be identified in the central hemorrhagic and necrotic areas of choriocarcinomas. The presence of IGCNU and hCG immunostains may be of additional help. It is also important to distinguish choriocarcinomas from other types of germ cell tumors that contain trophoblast cells. This distinction is based on the scattered nature and usually minor amounts of the trophoblastic elements, lack of a mononucleated trophoblast component, and absence of hemorrhagic necrosis in the latter. Embryonal carcinoma with cellular degeneration may be distinguished from choriocarcinoma by hCG stains. Monophasic choriocarcinoma should be distinguished from solid pattern yolk sac tumor and seminoma with syncytiotrophoblast cells. The expression of trophoblastic hormones in the mononucleated cells is helpful in this regard, as are the AFP reactivity of yolk sac tumor and the OCT3/4 positivity of seminoma. The placental site trophoblastic tumor (see following section) is distinguished from choriocarcinoma based on the predominant to exclusive presence of intermediate trophoblast cells, as supported by diffuse reactivity for human placental lactogen and absence of or minor reactivity for hCG.
Choriocarcinomas disseminate rapidly, often via hematogenous routes. Brain involvement occurs with disproportionate frequency (27). It is treated similarly to other nonseminomatous tumors, but a significant choriocarcinomatous component in a testicular germ cell tumor probably worsens the prognosis based on several studies in which multivariate analysis correlated a poor outcome with high hCG titers (440–442). In some studies, direct identification of choriocarcinoma or “trophoblastic” elements correlated with a poorer prognosis (440,441,443,444). A “choriocarcinoma” syndrome has been identified in which patients have hemorrhagic visceral metastases and a high mortality despite aggressive therapy (440). Regardless, cures of metastatic choriocarcinoma do occur (440,445). Choriocarcinomas as a component of mixed germ cell tumors may behave less aggressively than do pure choriocarcinomas (268,380,446,447).
Placental Site Trophoblastic Tumor
One of the authors has seen a single case of a trophoblastic tumor of the testis that resembled the placental site trophoblastic tumor of the uterus. This tumor occurred in a 16-month-old infant and consisted of an interstitial proliferation of intermediate trophoblast cells (Fig. 47.79). The patient was well on follow-up, 8 years after orchiectomy (433). Small foci of similar appearance may be seen in some typical choriocarcinomas (434).
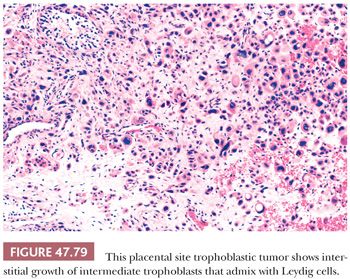
Cystic Trophoblastic Tumor
This lesion is characteristically seen after chemotherapy and therefore usually in a metastatic site (448), but it also occurs de novo in the testis. It consists of a cyst lined by mostly mononucleated trophoblast cells that usually have irregular, pleomorphic nuclei but inconspicuous mitotic activity (Fig. 47.80). Hemorrhage and tumor necrosis are absent. Stains for hCG are usually only focally positive. In metastases, it appears to behave similarly to teratoma (448).
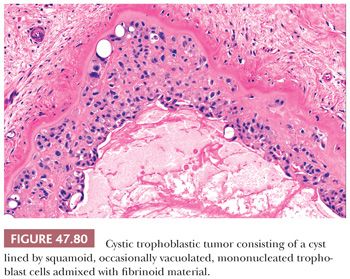
MIXED GERM CELL TUMORS
Although the various types of germ cell tumors discussed in the previous sections may occur as pure tumors, it is much more common, except for seminoma, to see combinations of different neoplastic types. From 69% to 91% of the nonseminomatous testicular germ cell tumors are of mixed types (179,379). In diagnosing such cases, the recommended term is mixed germ cell tumor, followed by a parenthetical listing of the components with an estimate of their relative proportions in percentages. On gross examination, mixed germ cell tumors are characteristically variegated, reflecting their different components, with foci of hemorrhage and necrosis (Fig. 47.81). Microscopically, the appearance is identical to the various pure neoplasms. There is a tendency for many pathologists to overlook foci of yolk sac tumor (302) that characteristically develop in close association with an embryonal carcinoma component. The “double-layered” pattern of embryonal carcinoma (306) (Fig. 47.37) should be regarded as an example of a mixed germ cell tumor composed of embryonal carcinoma and yolk sac tumor. There is evidence that tumors composed of embryonal carcinoma and teratoma behave in a less aggressive fashion than those consisting of pure embryonal carcinoma (449), and yolk sac tumor elements may also favorably alter the behavior of mixed germ cell tumors (368).
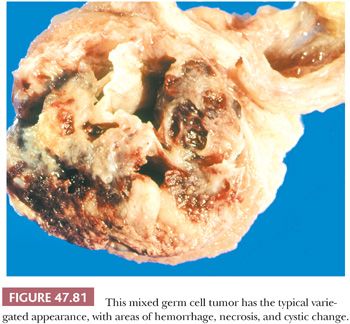
POLYEMBRYOMA AND DIFFUSE EMBRYOMA
Both polyembryoma and diffuse embryoma are distinctive forms of mixed germ cell tumors. Polyembryoma consists of small, scattered, embryo-like bodies having a central core of embryonal carcinoma cells (resembling the embryonic plate), an associated amniotic-like cavity, and a yolk sac tumor component resembling the embryonic yolk sac (450,451) (Fig. 47.82). Syncytiotrophoblasts are often associated with the amniotic-like and yolk sac–like components, and intestinal, hepatic, and squamous differentiation may occur (450). Thus, these tumors are considered to have components of embryonal carcinoma and yolk sac tumor and, sometimes, teratomatous and syncytiotrophoblastic components. Their behavior is typical of nonseminomatous germ cell tumors. In diffuse embryoma, there is a diffuse admixture of approximately equal amounts of embryonal carcinoma and yolk sac tumor elements (Fig. 47.83) (452,453). These components retain their expected immunoreactivities.
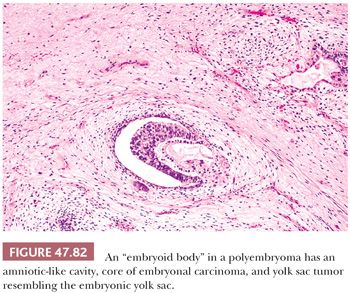
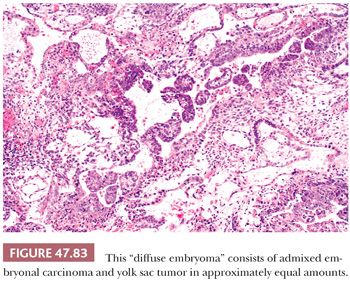
REGRESSION OF GERM CELL TUMORS
Some patients with metastatic germ cell tumors are noted, either at autopsy or orchiectomy, to have testicular scars that may be associated with IGCNU, intratubular coarse calcifications, or teratomatous elements. These patients are felt to have had regression of most or all of their primary neoplasm (308). Approximately 10% of male patients dying of metastatic germ cell tumors have such “burnt-out” primary tumors (454), and it is especially likely to occur in patients with metastatic choriocarcinoma (455) but may be associated with other germ cell tumor types as well. It is likely that most patients with isolated retroperitoneal germ cell tumors have metastatic retroperitoneal disease with tumor regression in the testis (456). On microscopic examination of the testis, there are fibrotic scars with prominent numbers of small blood vessels that may also contain lymphoplasmacytic infiltrates, hemosiderin-laden macrophages, “ghost” tubules, and intratubular coarse calcifications (Fig. 47.84) (309). The latter probably correspond to complete comedo-type necrosis of intratubular embryonal carcinoma with dystrophic calcification. IGCNU may be identified in residual seminiferous tubules in approximately half of the cases (309,456). The testis peripheral to the scar invariably shows atrophy with impaired spermatogenesis (309), reflecting the usual background on which testicular germ cell tumors develop. Many cases of “pure teratoma” were likely originally mixed germ cell tumors with regression of the nonteratomatous elements (457).
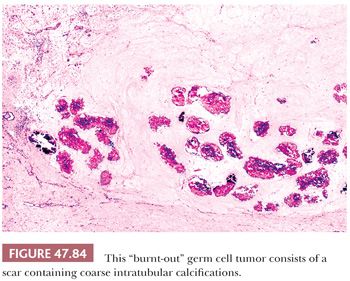
Immunochemistry may be helpful in defining whether a necrotic or mostly necrotic tumor is a seminoma or nonseminoma. A CK, OCT3/4, CD117, and CD30 panel can show differential patterns of expression even in severely necrotic tumors, although only distinct reactivity in the appropriate cellular location ought to be considered as positive.
POSTCHEMOTHERAPY RESECTIONS
Patients with metastatic testicular germ cell tumors commonly undergo postchemotherapy resection of residual masses, and the interpretation of these specimens can pose problems, even to experienced pathologists (327). The diagnosis of residual, nonteratomatous germ cell tumor in these cases is often considered an indication for additional chemotherapy, whereas teratoma, necrosis, and fibrotic or reparative reactions are not (458,459). Some studies have suggested that patients with very small volumes of nonteratoma on RPLND may escape further salvage therapy; thus, quantification of the residual tumor may be important (328,329).
Necrotic foci are often surrounded by a prominent infiltrate of foamy macrophages, sometimes containing hemosiderin, and an active fibroblastic proliferation (Fig. 47.85). The cytoplasmic clarity of the macrophages may cause a misinterpretation as seminoma, but the nuclei, although sometimes “active appearing” with small to moderate-sized nucleoli, lack malignant features. In the central portion of the necrosis, pyknotic nuclei in the “ghostlike” outlines of tumor cells should not be considered evidence of persistent germ cell tumor.
Fibrotic foci often contain widely scattered, spindle-shaped to epithelioid-appearing cells with nuclear atypia. Cytokeratin stains may highlight these cells, confirming they have an epithelial phenotype and supporting the belief they often derive from the “mesenchymal” component of yolk sac tumor (457,737) and, indeed, such stroma has the same genetic profile of the associated germ cell tumor elements (460). Others may represent persistent trophoblastic cells. As long as the atypical cells are widely scattered as individual or small clusters and show no or only rare mitotic figures, we do not qualify our diagnosis of “fibrosis.” A greater degree of cellularity or proliferative activity may merit comment, but it is unlikely to alter the future therapy (i.e., close follow-up rather than additional chemotherapy). Occasionally, repeated recurrences show progressively increased cellularity and atypia of “fibrotic” foci until a clear-cut “sarcoma” is recognizable (353). Many of these sarcomas appear, however, to represent sarcomatoid differentiation of yolk sac tumor.
Persistent teratoma often shows significant cytologic atypia in both mesenchymal and epithelial components. In the absence of stromal invasion or overgrowth, this atypia has no significant impact on prognosis and is not considered an indication for additional chemotherapy (461). The development of carcinoma or sarcoma, manifest by invasion or overgrowth, is associated with an aggressive course, but the treatment primarily remains surgical excision (395,461). Most of the examples of PNET and nephroblastoma-like tumor in patients with testicular germ cell tumors are seen in postchemotherapy resections of residual masses (392,393).
Although most forms of germ cell tumor in postchemotherapy resections have a similar morphology as in untreated cases, immunochemistry for CD30 is often negative in embryonal carcinomas after chemotherapy (31). Also, we have noted a tendency for some choriocarcinomas to lack a well-defined biphasic pattern and mainly consist of mononucleated trophoblast cells, as was noted in treated gestational choriocarcinoma (462). Marked cystic transformation of choriocarcinoma, with a lining epithelium resembling atypical squamoid cells, may also occur and should not be considered a reason for additional chemotherapy (448). Persistent yolk sac tumor may have more prominent parietal, hepatoid, and glandular features than in untreated patients and may be mistaken for adenocarcinoma in the latter circumstance, although AFP positivity and negativity for EMA and CK7 are helpful in recognizing glandular yolk sac tumor. OCT3/4 is useful in identification of seminoma or embryonal carcinoma, but only nuclear positivity should be regarded as cytoplasmic positivity may represent other nonneoplastic tissue such as paraganglia (463).
SEX CORD–STROMAL TUMORS
The sex cord–stromal tumors of the testis represent a distinct minority of testicular neoplasms but a disproportionate number of diagnostic difficulties. Only approximately 4% of testicular tumors fall into this category (271,412), which consists of those neoplasms whose cells resemble Leydig cells, Sertoli cells, theca cells, granulosa cells, and/or fibroblasts. There are six major categories of neoplasms in this group: Leydig cell tumors, Sertoli cell tumors, Sertoli-Leydig cell tumors, granulosa cell tumors, fibromas of gonadal stromal origin, and sex cord–stromal tumors of mixed or indeterminate type.
LEYDIG CELL TUMOR
Leydig cell tumors constitute the single most common type of testicular sex cord–stromal tumor and represent approximately 2% to 3% of all testicular neoplasms (464). They occur over a wide age range in patients from 2 to 90 years old (465) but are most common in the third to sixth decades (464), with a smaller peak in children 5 to 10 years old (466). The presentation depends on the age of the patient, with children, who represent approximately 20% of cases, commonly presenting with isosexual pseudoprecocity as a result of androgenic hormone production by the neoplasm (467) and gynecomastia because of aromatase-mediated conversion of androgens to estrogens in peripheral tissues. These patients may have small, nonpalpable tumors requiring specialized methods for clinical detection. Adults, in whom additional androgenic production is more difficult to detect, more commonly present with a testicular mass (464). Approximately 30% of adults develop gynecomastia (464), which also occurs in approximately 10% of children (466) in whom it is invariably superimposed on virilization. Bilateral Leydig cell tumors occur in approximately 3% of the cases (464). The familial occurrence of Leydig cell tumors in a father and his adult son has been reported (468). Occurrence in nonwhite patients is rare (469). Germline mutations of the fumarate hydratase gene have been reported in some cases (470), and some of the childhood cases are associated with somatic, activating mutations in the gene for the luteinizing hormone (LH) receptor (471–473).
On gross examination, Leydig cell tumors are characteristically solid, well-circumscribed, round to lobulated nodules that vary from yellow to tan to brown to gray, with some cases having coarse fibrous bands (Fig. 47.86). They range from 0.5 to 10 cm in diameter but are most commonly 2 to 5 cm (464), with those in children tending to be smaller. Foci of hemorrhage and necrosis may be identified in a minority of the cases. From 10% to l5% of the cases have extratesticular extension (464). On microscopic examination, a diffuse, sheetlike pattern of cells is most common (Fig. 47.87), although small nests, ribbons, and cords (Fig. 47.88) may also be present in fibrous stroma. We have seen several cases with a prominent microcystic arrangement in areas. Rarely, the tumor cells are spindled and arranged in vague fascicles (474) (Fig. 47.89), and there are uncommon cases of overtly sarcomatoid Leydig cell tumors (474–476). The individual cells are polygonal and usually have eosinophilic cytoplasm, although abundant lipids may cause the cells to have clear, finely vacuolated cytoplasm in some cases (Fig. 47.90). The nuclei are round, often with a moderate-sized central nucleolus, and can show moderate variation in diameter (Fig. 47.91) and a “ground-glass” quality (Fig. 47.92). Mitotic figures are generally infrequent. Lipofuscin can be identified in the cytoplasm in a minority of the cases (Fig. 47.91) (generally those with a tan to brown gross appearance), and rod-shaped intracytoplasmic crystals of Reinke may be identified in up to 40% of the cases (412) (Fig. 47.93); more frequent are globular eosinophilic inclusions that likely represent the precursors to crystals (Fig. 47.92). Occasional tumors have prominent foci of adipose metaplasia, and some may rarely have calcifications (Fig. 47.94) (474). On ultrastructural examination, the distinctive features are those of a steroid hormone–producing cell (i.e., abundant smooth endoplasmic reticulum, mitochondria with tubular cristae, and intracytoplasmic lipid droplets [477,478]); in some cases, the distinctive crystals of Reinke are identified as well as membranous whorls (477). Immunostaining for inhibin-α (365,479,480), melan-A (clone A103) (481), and calretinin is positive in more than 90% of cases. Vimentin is also positive (482), and some cases show patchy low–molecular-weight CK reactivity. Androgenic hormones may be demonstrated by immunohistochemistry. Enkephalin-like reactivity was described in one Leydig cell tumor (483).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


