Simple radiographic localization of a mediastinal mass has considerable value in and of itself. Substantial information is available on the relative likelihood of any given lesion in the three anatomically defined regions of the mediastinum. The anterior mediastinal compartment is that which is ventral to the anterior cardiac border and the aortic root; it most commonly harbors thymic epithelial tumors and cysts, germ cell neoplasms, lymphoproliferative lesions, retrosternal thyroid glandular proliferations, parathyroid lesions, aorticopulmonary-type paragangliomas, and nonneurogenic mesenchymal tumors. The “middle” mediastinum is defined on one side by the anterior cardiac silhouette and aortic root and on the other by the posterior aspect of the tracheal carina; lesions in this region are typically benign cysts, although malignant lymphomas also may be encountered therein. Lastly, the posterior mediastinum is dorsal to the large conducting airways; it principally plays host to neurogenous mesenchymal lesions and enteric cysts.
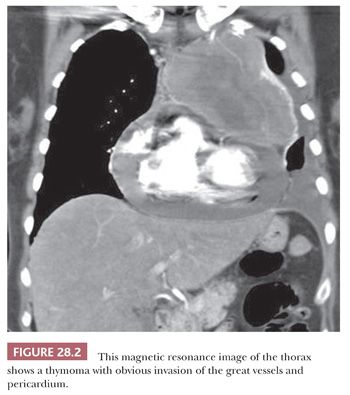
Radiologic findings that tend to correlate with malignancy in mediastinal lesions also have been well documented. These include obvious infiltration of the mediastinal fat, pulmonary hila, or great vessels (Fig. 28.2) (71); “seeding” of the pleural surfaces, the lung fields, or the pericardium (72); and obvious intratumoral foci of spontaneous necrosis or stippled calcification (73). Conversely, peripheral “eggshell” calcification in an anterior mediastinal mass is used as a relatively reliable marker of benign (mature) teratomas, and the circumscription and attenuation of simple mediastinal cysts are easily recognized diagnostically with computed tomography (74).
PERTINENT GROSS FEATURES OF MEDIASTINAL LESIONS
In those cases where the first procedure applied to a mediastinal lesion is an attempt at excision, the pathologist may use selected gross features to begin the process of differential diagnosis (75). Macroscopic invasion into attached fragments of lung, pericardium, or thoracic blood vessels is generally associated with a potential for aggressive behavior. Not all proliferations with such attributes are overtly malignant cytologically, in that desmoid-type fibromatosis, thymoma, and fibrosing mediastinitis may demonstrate invasive growth. Nevertheless, all of the latter lesions do have the capacity to cause significant morbidity and even mortality.
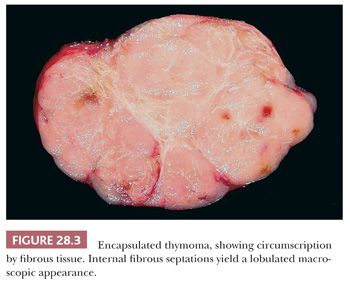
On the other hand, encapsulation is generically a characteristic of biologically indolent processes in the mediastinum; benign cysts of thymic, mesothelial, enteric, and bronchogenic types typically have distinct capsules, as do the majority of thymomas (Fig. 28.3). Conversely, it is uncommon for thymic carcinomas, malignant germ cell tumors, neuroendocrine neoplasms, and sarcomas to be surrounded completely by fibrous tissue at their peripheries, and malignant lymphomas never are encapsulated.
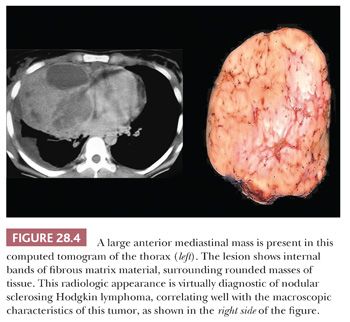
Other macroscopic findings are sometimes useful points of distinction. For example, thymomas often are subdivided internally by broad fibrous bands that intersect one another at acute angles, whereas sclerosing lymphomas, which may sometimes otherwise simulate thymic epithelial neoplasms, exhibit indistinct fibrous trabeculation or stromal bands that connect with one another obliquely (8) (Fig. 28.4). Also, extensive intralesional hemorrhage and necrosis are worthy of note because they are generally uncommon in lymphomas as well as in benign tumors of the mediastinum.
HISTOLOGIC FEATURES OF MEDIASTINAL TUMORS, WITH EMPHASIS ON DIFFERENTIAL DIAGNOSTIC CATEGORIES
The microscopic differential diagnosis of mediastinal lesions is facilitated by three rather simple steps. The pathologist must decide first whether the proliferation being studied is cystic and, second, whether it is composed of cytologically bland or cytologically atypical cells. If the lesion is clearly neoplastic and malignant, then it must be placed into one of five generic categories: small cell, large polygonal cell, mixed large cell and small cell, spindle cell/pleomorphic, and myxoid-adipocytic. Accordingly, the following discussion shall present this broad classification system in a workable context, with ancillary information that is of clinical or conceptual interest.
CYSTIC LESIONS OF THE MEDIASTINUM
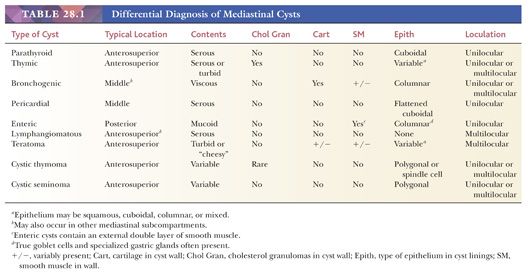
Even when one includes neoplasms that may undergo cystic degeneration, mediastinal cysts are relatively uncommon and account for only 10% to 15% of radiologically detected masses at this site (76). However, several tissue types can be seen in such lesions, including thymic, pericardial, bronchogenic, enteric, and parathyroid cysts (77,78). Indeed, selected mediastinal cysts may contain more than one of these constituents, relating to the fact that many intrathoracic cysts are developmental (congenital) rather than acquired and also to the close proximity in which the embryologic foregut anlage, pleuropericardial membranes, and branchial pouches are found during early morphogenesis (79).
Parathyroid and Thymic Cysts
Because the parathyroid glands and the thymus are all derived from the third and fourth pharyngeal pouches, cysts or solid proliferations containing tissue from either source may both be located in low cervical or anterosuperior mediastinal positions (80–82). “Pure” parathyroid cysts may be seen at any age; they range from 1 to 10 cm in size, are thin-walled, and are filled with clear fluid that contains a high level of PTH (81,83). The nonsecretory nature of such lesions is supported by the observation that hypercalcemia is rare in these cases; accordingly, parathyroid cysts typically present themselves as asymptomatic masses that are found radiologically and have an internal density approximating that of serum (81,83). The lining of these lesions is composed of attenuated parathyroid epithelium, which may be chief cell, oxyphilic, clear cell, or mixed in character. It is uniform in thickness and nonnodular in contrast to the epithelium of parathyroid adenomas, which undergoes secondary cystic change (79).
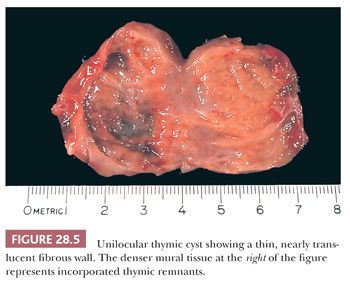
Thymic cysts manifest themselves predominantly between the ages of 20 and 50 years, typically in asymptomatic patients; this observation is germane to the contention offered by some authors that these are all postinflammatory lesions, formed by cystic transformation of Hassall corpuscles or medullary epithelial structures (84,85). However, it is now known that both congenital (unilocular) and acquired (multilocular) thymic cysts exist (85,86), as will be discussed later. Radiologically, these cysts assume the form of rounded, circumscribed masses in the anterior mediastinal subcompartment. Unilocular lesions have an internal water-like density on computed tomography, whereas multilocular cysts are filled with material showing a higher attenuation. Occasionally, plain film x-rays may demonstrate peripheral “rim” calcification. The greatest dimension of thymic cysts may be considerable, extending up to 18 cm (79,87).
As stated previously, macroscopic examination shows two main groups of thymic cysts: unilocular with a thin wall (Fig. 28.5) and multilocular with pericystic fibrous adhesions and a thick wall (85). These may be obviously centered on the thymus or, in some instances, connected to it by a narrow pedicle. Cyst contents have a variable consistency, correlating with other gross findings. Unilocular cysts typically contain only serous fluid, whereas multilocular lesions are filled with turbid, “cheesy,” or hemorrhagic material (85,88).
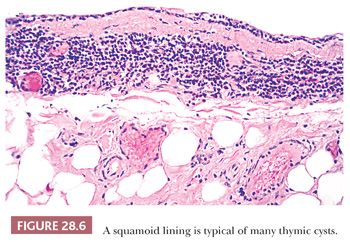
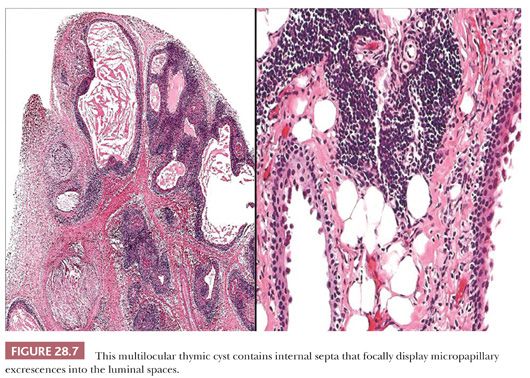
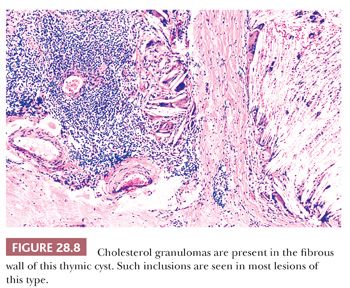
These appearances also correspond to the integrity of the lining as seen microscopically. In unilocular thymic cysts, the epithelial surface of the cyst cavity is only a few layers in thickness—consisting of bland squamoid cells (Fig. 28.6)—and the fibrous wall lacks inflammation, hemorrhage, and cholesterol granulomas. On the other hand, the epithelial component of multilocular thymic cysts is often proliferative in nature; multiple layers of squamous cells may be observed alone or in admixture with simple cuboidal, simple columnar, multilayered cuboidal and columnar, or micropapillary glandular epithelium (85) (Fig. 28.7). Rarely, small areas of parathyroid tissue will be seen (79), and even more uncommonly, salivary glandular epithelium can be observed (88,89). In the latter circumstances, the terms “third pharyngeal pouch cyst” or “mixed multilocular thymic cyst” can be used diagnostically. Abundant lymphocytes, granulation tissue, hemorrhage, and cholesterol granulomas are constant constituents in the fibrous walls and cyst cavities of multilocular thymic cysts (85,87,88) (Fig. 28.8). The cause of the inflammatory process that is putatively the underlying cause of multilocular thymic cysts is presently unknown. Identifiable Hassall corpuscular remnants and identifiable thymic tissue are seen in up to one-half of all cases with careful scrutiny, but specialized mesenchyme such as cartilage and smooth muscle is never observed.
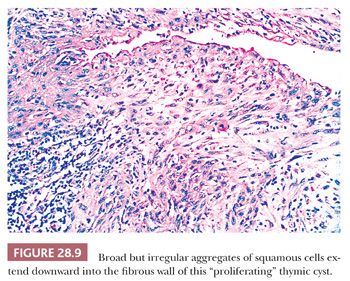
A peculiar variant of the multilocular thymic cyst is the “proliferating” subtype (90,91). It has significant histologic similarities to the cutaneous lesions known as “proliferating epidermoid cyst” and “proliferating trichilemmal cyst,” in that it exhibits narrow, often interconnecting tongues of squamoid epithelium that extend deeply into the fibrous cyst wall from its luminal aspect (Fig. 28.9). One can rightly consider this change to represent pseudoepitheliomatous hyperplasia of the cyst-lining cells. Close inspection shows that they have cytologic attributes like those seen in “hyperplasia with reactive atypia” of mucosal epithelia rather than truly dysplastic or overtly malignant features (90). Mitoses are indeed present in the proliferating epithelium, but they do not assume pathologic shapes. The importance of the proliferating thymic cyst is not that of an altered prognosis or risk of recurrence. Rather, it resides in the danger that exists for the pathologist to overinterpret the lesion as a squamous cell carcinoma. It should be remembered that true malignant change in a thymic cyst is a rare phenomenon indeed, with only a few well-documented examples of intracystic cancers in the English-language literature (92–97).
On the other hand, Moran et al. (98) have described three examples of low-grade serous ovarian adenocarcinoma that presented with metastases to the anterior mediastinum, simulating multilocular thymic cysts. This eventuality could be addressed with immunostains for CA-125, a marker that is expected in ovarian epithelial tumors but not in thymic proliferations.
Other potential causes of acquired thymic cysts concern changes that occur in thymuses involved by Hodgkin lymphoma (HL) and some that have been “traumatized” surgically. It is now well known that mediastinal HL may coexist with, or be succeeded by, the appearance of a thymic cyst (99–104). This observation was originally thought to be a reflection of thymic degeneration brought about by thoracic irradiation or systemic chemotherapy (91). Nevertheless, examples have also been reported wherein untreated HL was complicated by thymic cyst formation (97). It is likely that the lymphomatous infiltrate compromises the integrity of thymic epithelial nests, potentially leading to acquired cystic transformation. Admittedly, the addition of cytotoxic therapy—with subsequent lysis of intrathymic lymphoid cells—may accentuate this phenomenon through an “ex vacuo” mechanism (79). Curiously, thymic cysts have not been documented to date in association with non-Hodgkin lymphomas of the mediastinum. The other putatively acquired type of thymic cyst is that which is seen following open thoracotomies performed for nonneoplastic diseases. Jaramillo and colleagues (105) have reported three such cases in which these lesions were detected postoperatively. Those authors speculated that mechanical trauma to the thymus by the surgeon initiated the cystic glandular epithelial transformation, perhaps through microscopic vasodisruption.
Bronchogenic Cysts
Bronchogenic cysts may potentially arise in a broader anatomic distribution than those of parathyroid or thymic cysts; the first of these lesions can be seen in the anterior, superior, middle, or posterior mediastinum, and intrapericardial examples have been documented as well (106,107). They are likewise seen over a wide age range, extending from childhood to middle life (78). Whereas intrapulmonary cysts of this type often communicate with large airways, and therefore may present with symptoms and signs of superinfection, mediastinal bronchogenic cysts are usually asymptomatic (106,108). The radiographic appearance is that of a round or oval mass that molds itself to contiguous structures; the internal aspect of the lesion is uniformly hypodense, and the wall may contain linear calcification (109). Angiography may demonstrate an independent vascular supply to bronchogenic cysts, in contrast to other mediastinal cysts (108).
Gross examination of excised bronchogenic cysts may show them to be unilocular or multilocular with internal septation. Their contents are variably turbid or viscous (79).
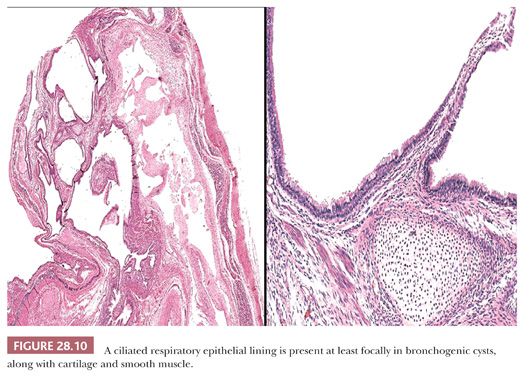
Histologically, the key features of such lesions are the presence of respiratory-type pseudostratified columnar epithelium, which is often ciliated, as well as small islands of predominantly mature cartilage (75) (Fig. 28.10). Smooth muscle also may be observed in small fascicles that are admixed with the chondroid elements, but cholesterol granulomas are uniformly absent. A possible resemblance to mature teratomas (discussed later in the chapter) must be acknowledged, but the overall organization of constituent tissues in bronchogenic cysts (resembling that of normal bronchi) should suffice to distinguish the two lesions.
Pericardial Cysts
Pericardial cysts of the mediastinum are typically found in the cardiophrenic angle (78,110,111). They may occur in patients of all ages and are generally asymptomatic; nonetheless, rare cases present with dyspnea or substernal chest pain (112). Radiographically, these cysts abut the cardiac contours and are irregularly shaped, with a uniform water-like internal density (111). Like bronchogenic cysts, they can arise in any of the mediastinal subcompartments (79).
The gross appearance of excised pericardial cysts is banal, being represented by thin fibrous-walled structures that collapse completely when opened. The internal surfaces are uniformly smooth, and the fluid contents are serous in nature (79,112).
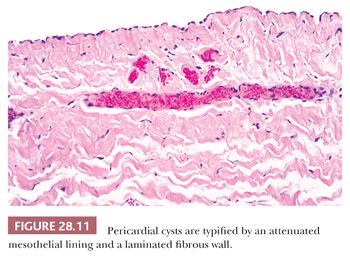
Microscopically, this configuration is recapitulated. One observes laminated fibrous tissue that is usually mantled by a single layer of bland mesothelium (Fig. 28.11); uncommonly, the latter tissue may undergo focal papillary hyperplasia (78). Smooth muscle, cartilage, specialized epithelium, and cholesterol granulomas are lacking.
Enteric Cysts
Enteric cysts are almost totally confined to the posterior mediastinum and characteristically occur in children and adolescents (113–121). Those individuals with paraesophageal cysts present with dysphagia or subnormal weight gain (113–115), whereas gastroesophageal cysts are associated with a wider array of symptoms; cough, vomiting, fever, and pneumonia or empyema have all been reported in conjunction with such lesions (116,118,119,121). Also, it has been shown that enteric mediastinal cysts may be linked to concurrent vertebral anomalies including hemivertebrae and spina bifida (117,120).
Radiographically, enteric cysts range in size from 2 to 10 cm; they are rounded or irregular in shape and may demonstrate obvious loculation on computed tomography. In those cases where leakage of cyst contents has occurred clinically, evidence may be seen of pleural effusions or pulmonary consolidation in the posterior lobe of either lung (118,119).
Macroscopically, one observes a variably thick fibromuscular wall and a smooth mucosal lining. In accord with the previously cited radiologic attributes, multiloculation may be seen (78). Cystic contents are variably mucoid, and an extremely acidic pH is seen in some examples on simple testing with litmus paper.
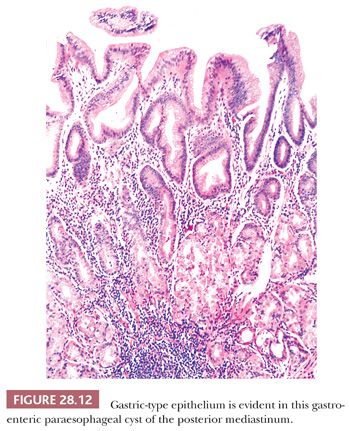
Histologically, both paraesophageal and gastroesophageal mediastinal cysts are bounded by a double layer of smooth muscle, in recapitulation of that seen in the remainder of the gut (78,79,116,119). The epithelial lining may be squamous, simple columnar, pseudostratified columnar, or mixed, but gastroesophageal lesions typically contain at least some specialized gastric glandular mucosa (potentially with chief and parietal cell differentiation) as well (78,79) (Fig. 28.12). Acid production by the latter tissue accounts for those cases that show spontaneous cystic rupture. Cartilage and cholesterol granulomas are absent.
“Cystic Hygromas” (Lymphangiomas)
Cystic lesions that represent lymphangiomas may be seen in the anterior, middle, and posterior mediastinum as well as the soft tissues of the neck, usually in pediatric patients (122–128). They may become large enough to produce respiratory compromise, cardiac embarrassment, or nerve compression but are just as often found radiographically in asymptomatic individuals (123–125,127–129). Radiographically, these cystic hygromas are often seen to be extensively infiltrating the mediastinal soft tissues. They have a more variegated internal appearance than many other cysts of the thorax, owing to their potential content of solid tissue (127,130). Discernible septation between cystic spaces may be observed on computed tomography.
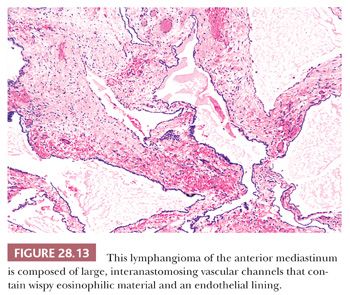
Macroscopically, these lesions are edematous-looking, unencapsulated white-gray masses with variably sized internal cavities and watery contents. The tissue lining the internal spaces is smooth and glistening. Histologically, lymphangiomas are characterized by large, irregularly shaped vascular spaces mantled by bland, flattened endothelial cells embedded in a fibroblastic and collagenous stroma (122,131). Small collections of lymphocytes are often scattered throughout the lesion; they may be present within the vascular spaces as well, together with flocculent eosinophilic precipitates of proteinaceous lymphatic fluid (122) (Fig. 28.13). Other specialized mesenchyme and epithelium are uniformly absent, as are cholesterol granulomas.
Cystic Meningoceles
In infants or children, the posterior mediastinum may harbor cystic meningoceles. These are thin-walled paraspinal malformations that demonstrate a communication with the meninges, usually through a defect in the vertebral bodies (132). Meningoceles are filled with clear to amber cerebrospinal fluid, and microscopic examination of such lesions demonstrates a thick fibrous wall lined internally by mature, flattened arachnoidal cells (110,133). Focal calcification and admixed neural tissue also may be apparent. Meningoceles are typically “incidental” surgical pathology specimens, owing to the sophisticated state of modern radiologic imaging techniques and the fact that the diagnosis is usually suggested clinically by concomitant neurologic deficits. Therefore, these lesions do not represent interpretative histologic problems.
Cystic Teratomas of the Mediastinum
Notwithstanding their highly organized and differentiated nature, most cystic teratomas of the mediastinum are true neoplasms rather than developmental malformations. These lesions have been classically defined by the presence of at least two tissue derivatives of the three germinal cell layers—endoderm, ectoderm, and mesoderm—although “monodermal” teratomas have been recognized as well (134).
Teratomatous and germ cell tumors make up 10% to 20% of all mediastinal lesions (132,135), and they are largely, but not exclusively (136), restricted to the anterior subcompartment. These are predominantly neoplasms of children, adolescents, and young adults, with an average age of approximately 20 years at presentation (135). Roughly one-half of the patients with such tumors are asymptomatic, whereas the remainder complain of chest pain, dyspnea, or cough (137). A minority of cases are associated with Klinefelter syndrome (138), but the distribution of cystic teratomas between the sexes is approximately equal (137).
Radiologically, one observes a predominantly cystic or solid and cystic mass with variable attenuation on computed tomography. Internal septation can reliably be considered a radiographic marker of benignancy in teratomas; likewise, calcification is usually an indicator of a biologically innocuous lesion (76,139).
The gross characteristics of mediastinal teratomas are also important reflections of their behavioral potential. Those tumors that demonstrate dense adherence to contiguous lung, pericardium, or blood vessels are almost certainly malignant, with the exception being those mature teratomas that have spontaneously ruptured and are associated with a fibroinflammatory response in the surrounding tissue (137). Mucoid cyst contents, cheesy keratinous debris, or obvious osteocartilaginous foci are most often seen in benign lesions, whereas spontaneous necrosis is a worrisome finding with regard to possible areas of malignancy in a teratoma (134). As Dehner (137) has indicated, one must address the question of adequacy in the sampling of large cystic teratomas; accepted procedure currently dictates that one block be submitted for every centimeter of tumor diameter or for every 10 g of neoplastic tissue. Obviously, the samples should be taken from as broad an area as possible.
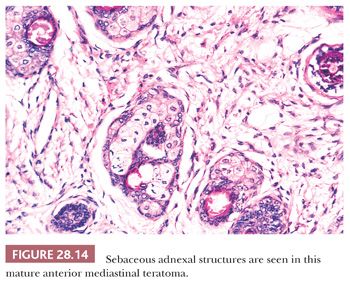
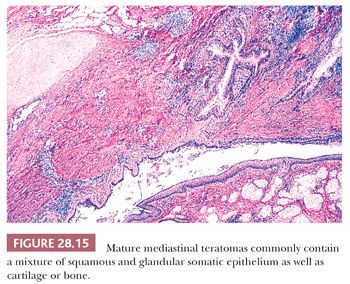
The microscopic complexity of teratomas is an immediate clue to their germinal nature, and it virtually eliminates all other differential diagnostic considerations among cystic mediastinal lesions. One commonly observes a mixture of mature squamous epithelium (often with cutaneous appendages), foregut-type columnar epithelium, neuroglial elements, bone, cartilage, fat, and fetal or mature striated muscle tissue (Figs. 28.14 and 28.15). Other potential histologic components include choroid plexus, hepatocytic islands, pancreas, and pigmented neuroepithelium resembling retinal tissue (138–140).
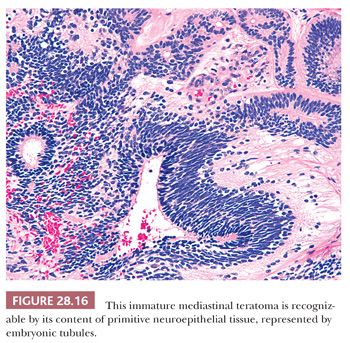
The definition of “immature” teratoma (with that term implying possible recurrence or even metastatic potential) is still somewhat problematic for some observers. In short, it is our belief that the presence of immature neuroepithelial tissue is all that is necessary for the diagnosis of this pathologic entity (140) (Fig. 28.16). It would be unwise to broaden the spectrum of immature teratomas to include those lesions containing fetal (but cytologically benign) mesenchyme or somatic epithelium. This opinion stems from the fact that immature neuroepithelium appears to be the sole potentially adverse marker in such tumors from a biologic point of view. In fact, even those neoplasms that do contain this tissue type fail to behave in an aggressive fashion in patients under 15 years of age, as shown by Carter and colleagues (141). In light of this information, it is likely that broadening the definition of immaturity for these neoplasms would only incur needless worry and possible overtreatment.
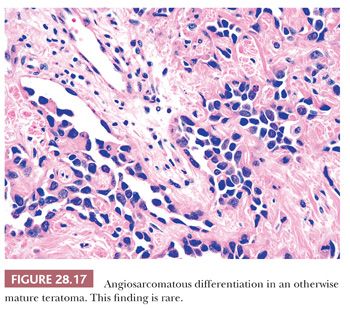
The foregoing comments notwithstanding, one should also recognize, however, that exceptionally rare examples of otherwise mature mediastinal teratomas exist wherein overtly sarcomatous, gliomatous, or carcinomatous somatic tissue makes up a portion of the lesion. For example, rhabdomyosarcoma, fibrosarcoma, chondrosarcoma, liposarcoma, angiosarcoma, enteric-type adenocarcinoma, carcinoid tumor (neuroendocrine carcinoma), squamous cell carcinoma, and glioblastoma multiforme have all been reported in this setting (139–147) (Fig. 28.17). The proper diagnostic classification of such tumors is that of “mature teratoma with sarcomatous/carcinomatous transformation.” Their prognosis is much more guarded, and specific treatment measures directed at the malignant somatic components are necessary.
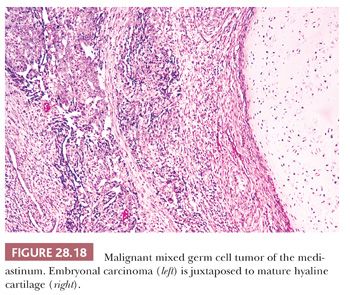
Still other teratomatous neoplasms contain obvious foci of seminoma/dysgerminoma, embryonal carcinoma, yolk sac tumor, or choriocarcinoma, as seen in the gonads (134,137,139,140) (Fig. 28.18) (see Chapter 47). The commingling of a mature or immature teratomatous component with the latter elements qualifies the lesion as a “mixed malignant germ cell tumor,” the biology of which is largely determined by the nonteratomatous germinal tissues. One should nevertheless clearly inform attending physicians of the presence of mature somatic elements in such cases because the latter tissues may well fail to involute following chemotherapy. In this circumstance, treatment converts a mixed malignant germ cell neoplasm into an apparently pure mature teratoma; the latter commonly requires subsequent surgical removal for complete extirpation (148).
Last, it should again be mentioned that occasional teratoid tumors of the mediastinum can be associated with antecedent, concomitant, or ensuing hematolymphoid malignancies, with acute myelogenous leukemia predominating (61,149,150). This association is seen even in the absence of potentially mutagenic chemotherapy for the germinal lesion, and common karyotypic abnormalities have been demonstrated in both the teratomatous cells and the aberrant hematopoietic elements (151). Hence, it appears that leukemia in such cases is derived from the germ cell tumors.
Cystic Thymomas and Seminomas
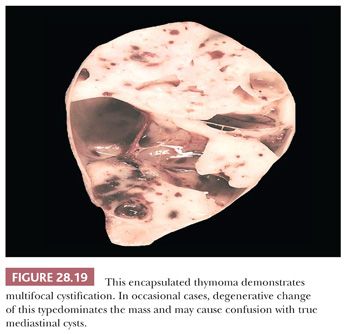
Among all other tumors of the mediastinum, only thymoma (Fig. 28.19) (defined as a cytologically bland proliferation of thymic epithelium) and intrathymic seminoma have any reproducible capacity for cystic change. Indeed, some of these lesions may be so extensively cavitary that numerous microscopic sections are necessary to detect their basically neoplastic nature.
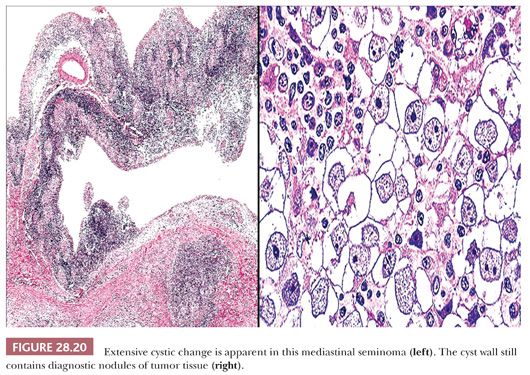
In many respects, cystic thymomas (152) may closely resemble thymic cysts microscopically, as described earlier. The two are distinguished from one another histologically in that nodular proliferations of thymic epithelial cells define the former entity but are not “allowed” in the walls of thymic cysts. Parenthetically, cystic thymomas also may exhibit extensive spontaneous necrosis and hemorrhage, neither of which is expected in thymic cysts. Other microscopic characteristics of thymomas in general are presented in the section “Differential Diagnosis of Selected Thymoma Variants.” Extensively cystic seminomas—which are presumably formed by spontaneous degenerative change—likewise may contain only small mural nodules of residual viable tumor cells (Fig. 28.20), which are overtly malignant with prominent nucleoli (103,153). The periodic acid-Schiff (PAS) stain is helpful in delineating the glycogen content that typifies seminomas and may be used to screen for the neoplastic cell aggregates in this particular setting.
Additional clues to the identity of cystic thymomas may be gleaned from the clinical history, if an appropriate paraneoplastic syndrome is evident; similarly, concomitant hematologic abnormalities or Klinefelter syndrome would be valuable evidence suggesting the existence of a mediastinal germ cell tumor. High-resolution computed tomography and magnetic resonance imaging of the thorax are also valuable. The latter techniques are capable of delineating the aforementioned mural nodular densities that would not be expected in thymic cysts (64,65,73,154). The most salient facts on cystic mediastinal lesions discussed thus far are summarized in Table 28.1.
DIFFERENTIAL DIAGNOSIS OF SELECTED THYMOMA VARIANTS
General Comments
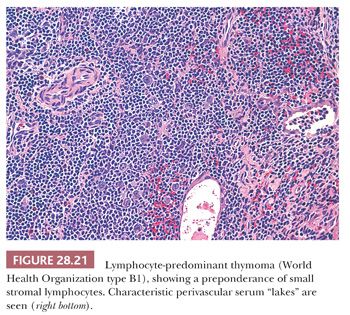
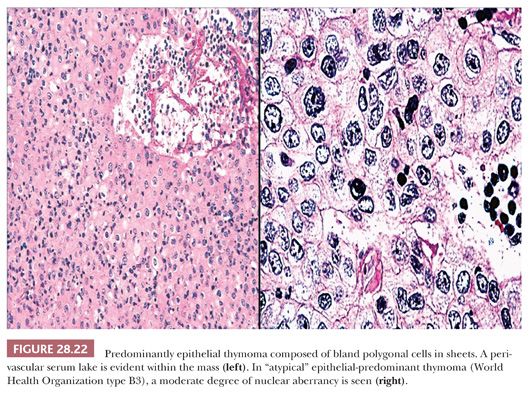
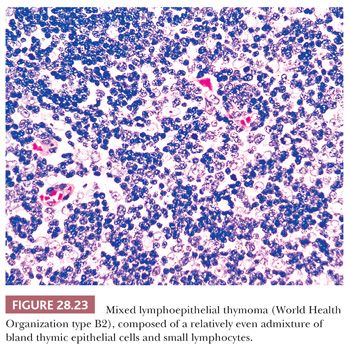
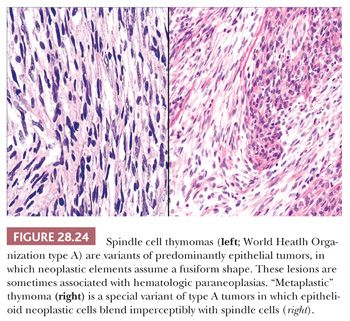
The microscopic variability of thymomas is considerable, perhaps second only to that of teratomas among all mediastinal neoplasms. There are, however, several nuances in the histologic traits of thymomas that play important roles in differential diagnosis with other lesions. These are outlined in a systematic fashion here using the simple classification scheme of Bernatz and colleagues (155). The latter nosologic construct divides thymomas into four discrete categories based on cross-sectional microscopic morphology: lymphocyte predominant (>66% lymphocytes) (Fig. 28.21), epithelial predominant (>66% epithelial cells) (Fig. 28.22), mixed lymphoepithelial (34% to 66% epithelial cells) (Fig. 28.23), and spindle cell (a subtype of epithelial-predominant thymoma featuring a nearly exclusive composition by fusiform tumor cells) (Fig. 28.24). It merits emphasis that thymoma must first be defined as a cytologically bland epithelial neoplasm in order for this scheme to have any histopathologic use (156). Moreover, the authors hold the opinion that the Bernatz system does not represent—and was not devised as—a prognostic classification. Rather, its usefulness is in serving as a cue mechanism for well-defined histologic differential diagnostic problems concerning thymomas.
In fact, there is currently no means of microscopically subcategorizing thymomas that correlates perfectly with behavior (157–159), including a scheme proposed by Marino, Muller-Hermelink, and colleagues (157,158). The latter system (usually abbreviated as the MMH classification) is based on the putative resemblance of thymomatous epithelial cells to those of the normal thymic cortex (“cortical” thymomas—usually equating to Bernatz lymphocyte-predominant, mixed lymphoepithelial, and most epithelial-predominant lesions) or medulla (“medullary” thymomas, which are largely synonymous with spindle cell lesions in the Bernatz system) (160–164). It is contended that medullary thymomas have a good prognosis, whereas cortical lesions (including those with modest cytologic atypia, termed well-differentiated thymic carcinoma by Kirchner and colleagues [165]) have a relatively unfavorable evolution and “mixed cortical/medullary” tumors manifest an intermediary behavior.
As well summarized by Shimosato and Mukai (14), it would appear that the MMH system is, in actuality, a derivative distillation of information that would already be made available from implementation of the Bernatz classification scheme, albeit using different terminology (166). In light of this, one could well question whether the former nosologic strategy can be regarded as progressive. Moreover, problems with interobserver reproducibility of the classification of thymomas have been reported (158,167), and Moran and Suster (168) have summarized the considerable effects of sampling bias on this process as well. Finally, it must be forthrightly stated that spindle cell (medullary) thymomas (169) have long been known to usually exhibit innocuous behavior, regardless of what specific adjectives were used to describe them. In light of that fact, it would seem advisable to reevaluate the claims of MMH proponents (18,170–178) after pure spindle cell neoplasms have been excluded from formal statistical analyses. In our personal experience with a large number of thymoma cases, the MMH system has failed to equal stage as a significant prognostic factor after this step is taken.
In 1999, Rosai published the second edition of Histological Typing of Tumors of the Thymus (179), under the auspices of the World Health Organization (WHO) International Histological Classification of Tumors project. In an effort to reconcile the Bernatz and MMH schemes, a hybrid system was advanced therein. It defines thymomas as:
• Type A (spindle cell or medullary)—composed of a population of neoplastic thymic epithelial cells having a spindle or oval shape, lacking nuclear atypia, and accompanied by few or no nonneoplastic lymphocytes.
• Type AB—foci having the features of type A thymoma admixed with foci rich in lymphocytes.
• Type B1—resembling the normal functional thymus in that it combines large expanses having an appearance practically indistinguishable from normal thymic cortex with areas resembling thymic medulla.
• Type B2—the neoplastic epithelial component appears as scattered plump cells with vesicular nuclei and distinct nucleoli among a heavy population of lymphocytes. Perivascular spaces are common and sometimes very prominent. A perivascular arrangement of tumor cells resulting in a palisading effect may be seen.
• Type B3—predominantly composed of epithelial cells having a round or polygonal shape and exhibiting no or mild atypia. They are admixed with a minor component of lymphocytes, resulting in a sheetlike growth of the neoplastic cells. This histotype is synonymous with so-called well-differentiated thymic carcinoma.
• Type C—thymomas that are outright thymic carcinomas, with obvious cytologic anaplasia.
Since the introduction of this paradigm, several publications have suggested that the WHO classification is a prognostically oriented system (180–183), even though it was not intended as such (184,185). The principal goal of the latter scheme was, in fact, to improve interobserver reproducibility in diagnosis. Another proposal appeared virtually simultaneously with the WHO monograph and was advanced by Suster and Moran. It contends that thymic epithelial neoplasms can be divided into “thymomas” (corresponding to WHO types A, AB, B1, and B2 tumors), “atypical thymomas” (WHO type B3 lesions), and “thymic carcinomas” (WHO type C tumors) (186–188); we strongly endorse that practical paradigm.
Our belief is that the best prognosticator in thymoma cases is whether the tumor invades through its capsule (189–195). Codification of the degrees of invasion was published by Masaoka and colleagues (196) in a formal staging scheme; it has recently been restructured by Moran et al. (197) based on studies at the MD Anderson Cancer Center (MDACC). Several publications attest to the value of staging for thymomas (14,166,198,199). For those conscientious practitioners wishing to use all-inclusive reporting practices, the Masaoka and MDACC stages can be provided to the surgeon as well as the Bernatz, Suster-Moran, MMH, and WHO histologic classifications of the tumor (200).
LYMPHOCYTE-PREDOMINANT THYMOMA VERSUS LYMPHOID HYPERPLASIA
Particularly in patients with myasthenia gravis, the pathologist is often asked to distinguish between true thymic hyperplasia (201) and thymoma in surgically resected glands. There are several straightforward histologic guidelines to make this separation.
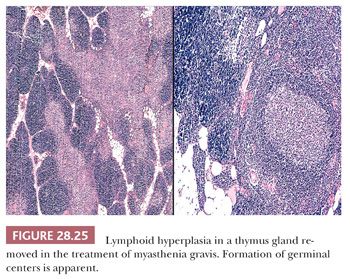
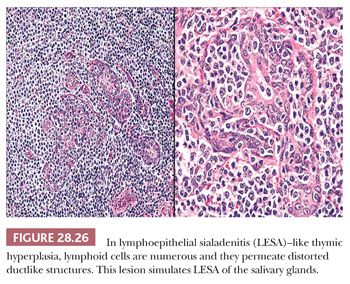
Thymic lymphoid hyperplasia is defined by the retention of a normal microscopic cortical and medullary glandular distinction, together with the presence of several well-formed intrathymic lymphoid follicles. The latter often contain germinal centers, complete with tingible body macrophages (Fig. 28.25). Small aggregates of proliferating thymic epithelial cells may be seen as well in hyperplastic glands, but they do not alter the normal architecture of the thymus and are not associated with the previously cited lymphoid follicles (202–205). An exception to the latter statement is represented by an unusual form of thymic lymphoid hyperplasia described by Weissferdt and Moran (206). It demonstrates a marked resemblance to “benign lymphoepithelial lesions” (lymphoepithelial sialadenitis) as seen in the salivary glands (see Chapter 20) (Fig. 28.26).
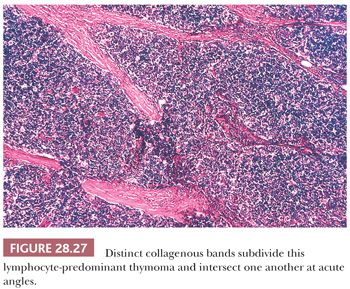
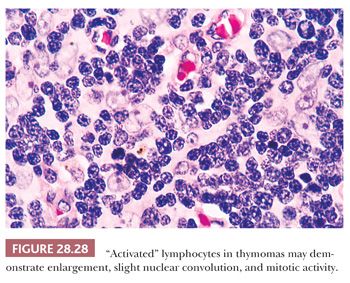
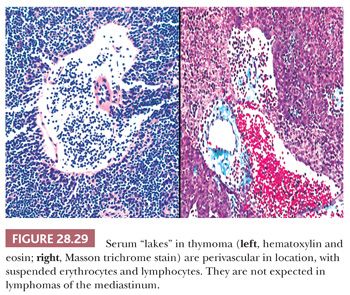
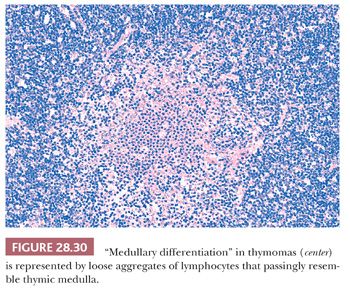
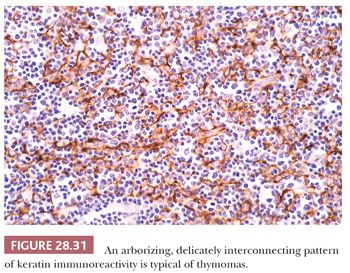
In contrast, lymphocyte-predominant thymoma (LPT) is characterized by effacement of most or all of the thymic substructure (8,155,156). Like all forms of thymoma, this neoplasm often features the presence of a thick fibrous capsule and internal fibrous stromal bands that intersect one another at acute angles (Fig. 28.27). Close inspection will disclose an admixture of bland thymic epithelial cells in admixture with “activated” lymphocytes (Fig. 28.28) that have folded nuclear contours and may be mitotic. The epithelial elements may be relatively inconspicuous, but they exhibit oval nuclei, fine dispersed chromatin, small chromocenters, and a moderate amount of amphophilic cytoplasm. Additional observations that aid in the recognition of LPT are the presence of intratumoral, perivascular serum “lakes” (Fig. 28.29); microcysts; numerous dispersed mast cells as seen with the chloroacetate esterase method; and “medullary differentiation” (multifocal loose aggregates of lymphocytes) (2,206,207) (Fig. 28.30). Medullary differentiation is particularly prominent in lesions that have been termed organoid thymomas by Pescarmona and colleagues (208). Immunostaining for keratin reveals a finely arborizing network of interconnecting epithelial cell processes in LPT, which is not seen in lymphoproliferative lesions (206,209,210) (Fig. 28.31).
A more difficult lesion to distinguish from thymic lymphoid hyperplasias, particularly in small biopsy specimens, is represented by “micronodular thymoma with lymphoid B-cell hyperplasia.” That tumor was described by Suster and Moran (211) and appears to represent a special form of spindle cell thymoma in which multiple small nodules of thymic epithelial cells are separated from one another by a prominent lymphoid stroma containing easily seen germinal centers, with a striking resemblance to those of Castleman disease (see next section) (212). Again, immunostains for keratin are very helpful in characterizing this tumor type.
LYMPHOCYTE-PREDOMINANT THYMOMA VERSUS CASTLEMAN DISEASE
Castleman disease (also known as angiofollicular lymphoid hyperplasia [AFLH]) is a peculiar hematolymphoid disorder that presents clinically with symptoms of a mass lesion or, alternatively, with systemic findings such as fever, weight loss, and anemia (13,213). AFLH commonly affects the mediastinum and, as such, may be confused with LPT (206). In fact, the seminal paper in the literature on AFLH was focused on this differential diagnostic problem (213).
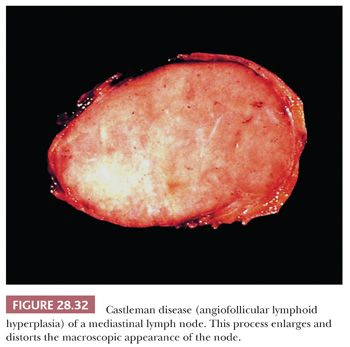
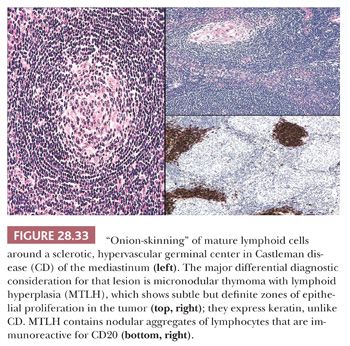
In contrast to the picture of LPT presented in this chapter, AFLH is rarely centered in the thymus. Instead, it affects mediastinal lymph nodes (Fig. 28.32). These cases demonstrate architectural distortion by follicular aggregates of small, mature lymphoid cells, with compression of normal nodal sinusoidal channels. The internal aspects of the lymphoid follicles often show an unusual proliferation of small-caliber blood vessels, surrounded by a variable amount of fibrohyaline eosinophilic matrix material. Many of the follicles also are surrounded by “onionskin” arrays of lymphocytes, simulating exaggerated mantles (12,13,213,214) (Fig. 28.33). In one subtype of AFLH—termed the plasma cellular variant—interfollicular zones of the affected node are replaced by sheets of mature plasma cells (215). None of these features is seen in thymomas, except for the “micronodular” (“Castlemanoid”) variant as described earlier.
LYMPHOCYTE-PREDOMINANT THYMOMA VERSUS LYMPHOMA
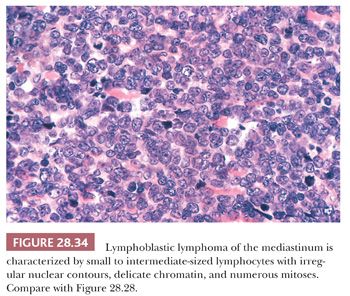
An important potential mistake in mediastinal pathology is the confusion of LPT with malignant lymphoma, particularly of the lymphoblastic type (lymphoblastic lymphoma [LBL]). Although the latter lesion is most often seen in the mediastinum in adolescents and young adults (12,216–219), patient groups that rather uncommonly develop thymomas (207), there is enough demographic and radiologic overlap between LBL and LPT to make a purely clinical distinction between them extremely difficult (220,221). Even the presence of an elevated peripheral white blood cell count offers no assurance of a diagnosis of LBL, because it has been reported in patients with thymoma as well (222). The mimicry of LBL in selected LPT cases is facilitated by the particular attributes of infiltrating lymphocytes in some thymomas; these cells may exhibit “convoluted” nuclear contours, increased nucleocytoplasmic ratios, and many mitotic figures (8,206), as typically seen in LBL (Fig. 28.34). Moreover, the immunophenotypic characteristics of LPT lymphocytes and LBL cells are closely similar. Both populations commonly express the CD1, CD2, CD3, CD99 (MIC-2) (223–225), and bcl-2 (225–227) antigens and terminal deoxynucleotidyl transferase (224,225,228–233); consequently, flow cytometric or immunohistochemical interpretations obviously must be made with extreme caution.
The most helpful ancillary studies in this differential diagnosis—and ones that the author, through sad experience, has made pro forma—are the immunohistologic detection of reactivity for p63 (an intranuclear transcription factor that is seen preferentially in squamous and transitional epithelium [234]) and keratin. As mentioned earlier, the interconnecting epithelial cells of LPT, which are absent in LBL, demonstrate a distinctive pattern of positivity for keratin, assuming a “lacy” staining configuration. This observation merits special emphasis because LBL and other lymphomas affecting the thymus may entrap scattered residual nonneoplastic thymic epithelial cells that are visible on keratin immunostains (235). p63 is also regularly seen in thymic epithelial cells but not in LBL (234).
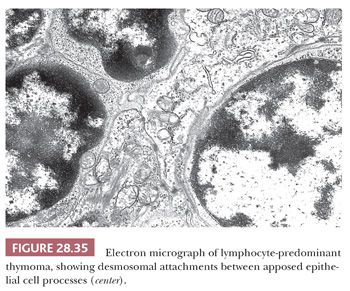
Electron microscopy may still play a role in this differential diagnostic context (236). In contrast to the ultrastructural appearance of LBL, thymoma shows well-formed intercellular junctions between apposed processes of epithelial cells (Fig. 28.35). In addition, the cytoplasm of these elements is usually rich in tonofilaments (8,9,206,207,237,238).
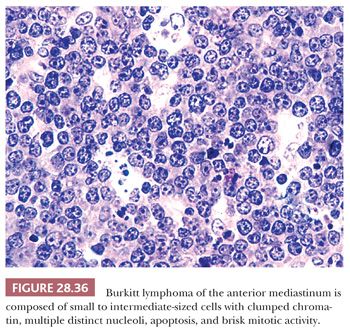
Only one other small cell, non-Hodgkin lymphoma besides LBL has any reproducible potential to present in the thymic region, thus raising the specified differential diagnosis. Several cases of Burkitt lymphoma have become manifest with mediastinal masses, in the absence of disease elsewhere (239,240). In our experience with such cases, most of the patients have had AIDS, in which Burkitt lymphoma has been reported with some frequency (241). The cells of this tumor have a characteristic coarsely clumped nuclear chromatin pattern, with prominent nucleoli and numerous mitoses (12,140) (Fig. 28.36). Nuclear detail is the most reliable discriminant between the lymphoid cells of Burkitt lymphoma and those of LPT. The “starry sky” pattern of dispersed tingible body macrophages that is often cited as a marker of the former lesion (see Chapter 17) may also be seen in thymomas (8,206).
The presence of still other forms of small cell lymphoma in the mediastinum is rare, but several examples of mucosa-associated lymphoid tissue (MALT)–type (marginal zone) lymphoma have been reported to affect the thymus (242,243). The proliferating lymphoid cells in this lesion also may be confused with those of LPT, but the methods of differential diagnosis just described are effective in this specific context as well. Skinnider and colleagues (244) have reported the rare but conjoint appearance of thymoma and lymphoma in the same patients, a situation which mandates that special studies be performed to distinguish one tumor type from the other.
THYMIC DYSPLASIA VERSUS “MICROSCOPIC” THYMOMA
On purely histologic grounds, the pattern of thymic epithelial growth that one observes in dysplastic glands (245) is somewhat similar to that described as “microscopic thymoma” by Pescarmona and colleagues (246). Both lesions feature the presence of nodular aggregates of cytologically bland cells that are subdivided by fibrous stromal bands and punctuated by admixed lymphocytes. However, the congruence ends there. Dysplasia occurs in smaller-than-normal thymuses. In addition, patients with this condition are children who almost invariably manifest severe clinical immunocompromise, such as the severe combined immunodeficiency syndrome (245). On the other hand, all reported cases of microscopic thymoma were seen in normally sized or enlarged glands removed in the treatment of myasthenia gravis in adults (246).
“ANCIENT” (SCLEROTIC) THYMOMA VERSUS FIBROSING MEDIASTINITIS
Moran and Suster (247) have described 10 examples of thymoma in which the tumor mass was dominated by hyalinized collagenous stroma. As such, the lesions microscopically resembled fibrosing mediastinitis (248), and thorough sampling was required to find diagnostic foci of thymoma.
SPINDLE CELL THYMOMA VERSUS FIBROUS HISTIOCYTOMA AND SOLITARY FIBROUS TUMOR
Predominantly epithelial thymomas (PETs) that are composed of plump or elongated fusiform cells may be difficult to distinguish from fibrous histiocytomas (FHs) or solitary fibrous tumors (SFTs) on purely histologic grounds (8,206). This resemblance is occasioned primarily on the basis of storiform cellular growth (FH-like thymoma) (Fig. 28.37) or the lobulated aggregation of bluntly spindled tumor cells punctuated by “staghorn”-like vascular channels (SFT-like thymoma) (Fig. 28.38). In the absence of biasing clinical information, such as the presence of pure erythroid aplasia or acquired hypogammaglobulinemia, which may be associated with spindle cell thymomas (156,206), one must rely on fine points in the microscopic appearance of each tumor type or on the application of discriminating special studies to make a proper diagnostic interpretation. However, it should be borne in mind that thymomas are far more common than anterior mediastinal mesenchymal tumors, and therefore the former lesions usually represent the final “answer” in such cases.
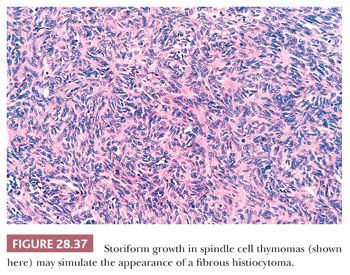
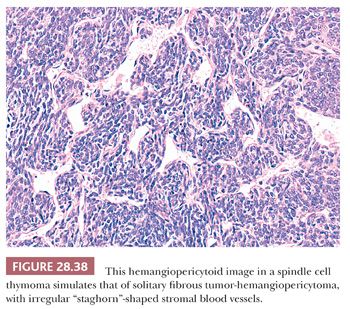
In most instances, thymic epithelial neoplasms that simulate FH or SFT still retain the microscopic hallmarks of thymomas in general. To reiterate, these include internal fibrous septa, intralesional microcysts, and perivascular serum lakes. Such findings are not seen in mesenchymal tumors. Reticulin stains are occasionally useful in differential diagnosis as well because they demonstrate delicate intercellular matrix deposition around individual tumor cells in true SFT but not thymomas.
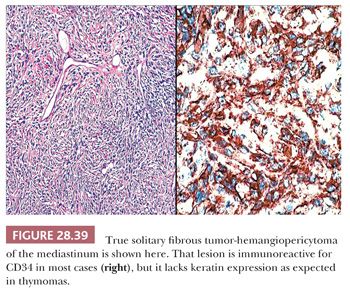
Immunohistochemistry or electron microscopy are the surest means of making a final interpretation in cases that are histologically indeterminate, particularly when only small biopsy specimens are available for evaluation. Pseudomesenchymal thymomas uniformly express keratin and lack vimentin (210), whereas FH and SFT show the converse of that immunoprofile. Moreover, the majority of SFTs are reactive for CD34 (Fig. 28.39), in contrast to thymomas. Similarly, the well-developed intercellular junctions and cytoplasmic tonofilaments of thymomas are not shared by the other two lesions (8,131,206,207).
DIFFERENTIAL DIAGNOSIS OF BENIGN MESENCHYMAL NEOPLASMS
In general, mesenchymal tumors of the mediastinum are uncommon lesions. Nonetheless, they are potentially distributed among all three anatomic subcompartments and may cause diagnostic consternation in selected cases. The following section considers selected problems that center on benign lesions in this group.
MEDIASTINAL LIPOMA VERSUS THYMOLIPOMA
Lipomas of the thorax may be seen in many sites, including the anterior, middle, and posterior mediastinum; the pericardium; and the deep soft tissues of the chest wall, including the parietal pleura (249). They are usually readily recognizable by radiographic means and hence are not excised in many cases. However, those in the anterosuperior mediastinum are similar clinicopathologically to an unusual neoplasm of the thymus known as thymolipoma (discussed later). One microscopic subtype of lipoma that may be unique to the mediastinum is that known as “elastofibrolipoma,” as described by DeNictolis et al. (250). This tumor manifests an admixture of ordinary lipoma with areas containing abnormal elastic fibers or dense zones of fibrosis. Verhoeff-van Gieson stains may be necessary to optimally visualize the elastic tissue in elastofibrolipomas.
Thymolipomas are generally asymptomatic tumors that are found radiographically in young to middle-aged adults (8,131,140,251,252). Only 10% are associated with systemic symptoms, which are like those of thymoma-related paraneoplasias (22,253–255). These tumors may attain a considerable size (up to 20 cm) (256,257) and characteristically show a uniform density on computed tomography that is essentially equivalent to that of adipose tissue (258,259).
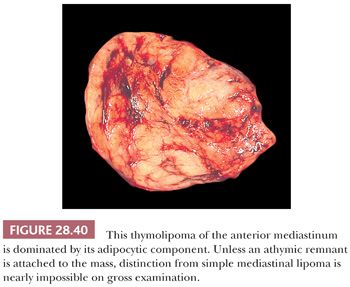
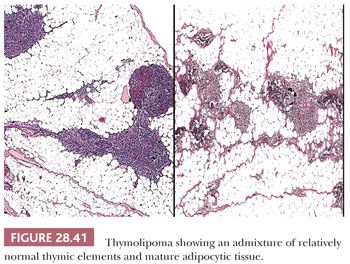
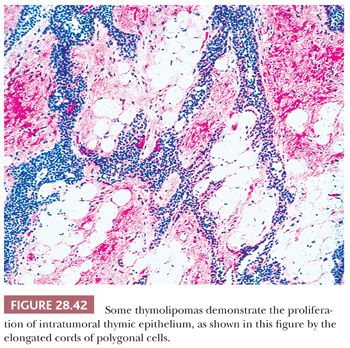
Macroscopically, thymolipomas have few if any traits that would dissuade one from a simple diagnosis of lipoma, except for the fact that they are often intimately associated with remnants of unremarkable thymus gland (260) (Fig. 28.40). Their histologic appearance, however, is distinctive. Lobules of unremarkable thymic tissue—complete with cortex, medulla, and Hassall corpuscles—are admixed evenly with mature adipose tissue and bounded by a thin fibrous capsule (8,131,140) (Fig. 28.41). The contention that the lesion is simply an infiltrative lipoma of the thymus is seemingly untenable; careful studies have shown that the absolute mass of thymic tissue in thymolipoma is increased far beyond that seen in normal glands (8). Furthermore, rare examples may demonstrate some degree of thymic epithelial proliferation as well (Fig. 28.42), with elongated cords of polygonal cells amid the fatty component; these variants have been termed proliferating thymolipomas by Hull and colleagues (260). Iseki and colleagues (261) have documented a remarkable thymolipoma in which obvious myoid cells were seen in the medullary areas of the admixed thymic tissue, and Moran and coworkers (262) have observed two cases in which broad zones of dense fibrosis were present. These last two cases were called “thymofibrolipomas” in recognition of their peculiar morphologic variation. Similarly, Ogino and colleagues (263) reported a thymolipoma with an extremely prominent vascular stromal component and designated it a “thymohemangiolipoma.”
BENIGN PERIPHERAL NERVE SHEATH TUMORS AND GANGLIONEUROMAS
The overwhelming majority of neoplasms encountered in the posterior mediastinum are neurogenic in nature (8–11,14,131). As such, they often show considerable gross and microscopic similarities and may challenge the pathologist to discern one from another. In particular, lesions with Schwann cell differentiation (peripheral nerve sheath tumors [PNSTs]) require further nosologic separation into neurofibromas and neurilemmomas (schwannomas), and they also must be distinguished from ganglioneuromas in this location.
NEURILEMMOMAS
Neurilemmomas are the most common neural tumors of the mediastinum (264–266). They are usually detected radiologically in asymptomatic patients, but occasional cases present with symptoms and signs of esophageal or spinal nerve root compression (266). Most affected individuals are young adults, but these lesions may be observed in all age groups (131,265).
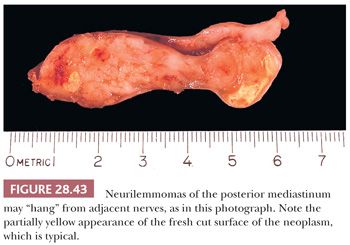
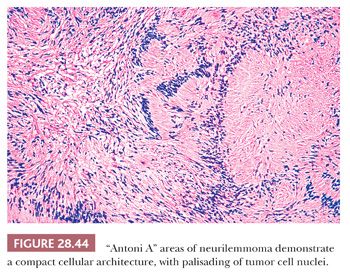
The macroscopic features of neurilemmomas are important clues to their distinction from other neural neoplasms. The latter lesion is typically encapsulated and sharply demarcated from adjacent soft tissue. An association with large nerves is common, and the tumor may appear to “hang” from the nerve like a pod on a tree branch (131) (Fig. 28.43). Histologically, one observes a biphasic arrangement of neoplastic fusiform cells; some are densely apposed and may exhibit nuclear palisading (Antoni A areas with Verocay bodies), whereas other foci show only loosely aggregated cells with a myxoid background (Antoni B areas) (8,131) (Figs. 28.44 and 28.45). Intralesional blood vessels are usually prominent, with thick walls. Division figures are typically sparse.
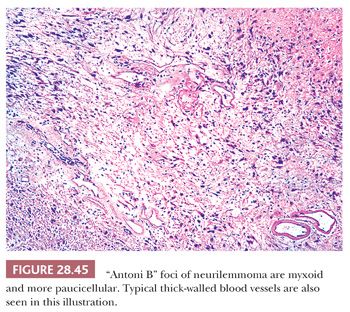
Some neurilemmomas exhibit specialized differentiation that can cause interpretative consternation. At the same time, this feature is useful in differential diagnosis because neither neurofibromas nor ganglioneuromas demonstrate such complexity. Neurilemmomas may show partial pigmentation (“melanotic” schwannoma) (267,268); alarming nuclear pleomorphism and hyperchromasia (“ancient” schwannoma) (131); foci with true epithelial differentiation (“glandular” schwannoma) (269); psammomatous calcification and pigmentation (“psammomatous-melanotic” schwannoma) (270); and dense cellularity with herringbone-like, storiform, or fascicular growth patterns, slight nuclear atypia, and brisk mitotic activity (“cellular” schwannoma) (271–273). In the last of these variations, the tumor may be mistaken for a sarcoma; however, unlike nerve sheath malignancies, cellular schwannomas are encapsulated and globally immunoreactive for S100 protein. They also lack foci of necrosis or pathologically shaped mitotic figures (273).
Ultrastructurally, neurilemmomas also are singular in the degree of neural differentiation that is observed. Elongated overlapping cell processes are present in such tumors; these may be attached to one another by primitive junctions, sometimes yielding structures that simulate embryonic mesaxons. Pericellular basal lamina is usually discernible and may be abundant (271).
NEUROFIBROMA
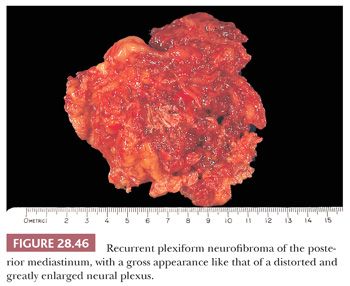
Mediastinal neurofibroma represents the principal diagnostic alternative to neurilemmoma. It has a similar demography and clinical presentation but also possesses the capacity for multiplicity or “plexiform” growth that macroscopically simulates a neural plexus (264–266) (Fig. 28.46). Tumors of this type with the latter characteristics are pathognomonic of von Recklinghausen disease, which is not associated with either mediastinal neurilemmoma or ganglioneuroma (274). Also in contrast to these lesions, neurofibromas often are centered on, or grow within, a large spinal nerve root; in fact, some examples have both intradural and extradural components, protruding through neural foramina of the vertebral column with a “dumbbell” configuration (131). They are not encapsulated.
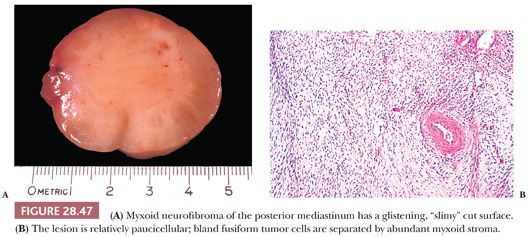
Microscopically, neurofibromas show a more uniform growth pattern than those seen in schwannomas. Bland spindle cells are arranged in fascicles, storiform arrays, or tactoids in neurofibromas. A myxoid stroma is common, but one does not observe biphasic Antoni A/B areas or thick-walled blood vessels as seen in neurilemmoma (Fig. 28.47). Mast cells often are numerous in neurofibromas as well (131).
Neurofibroma is potentially immunoreactive for S100 protein, CD34, CD56, CD57, and factor XIIIa. Its electron microscopic attributes are such that evidence of clear-cut schwannian differentiation is often lacking. Often, one observes only fibroblast-like tumor cells with relatively abundant rough endoplasmic reticulum, rudimentary cytoplasmic processes, and sparse formation of pericellular basal lamina (275).
GANGLIONEUROMA
Mediastinal ganglioneuroma is typically a tumor of childhood, but in some cases, it may be detected as late as the third or fourth decade of life (131,276). Patients with this tumor are usually asymptomatic. However, rare individuals present with watery diarrhea because of the ectopic synthesis of vasoactive intestinal polypeptide by the neoplasm, and other lesions may cause symptoms relating to spinal nerve root compression (277).
Similar to neurilemmoma, ganglioneuroma is an encapsulated tumor that is situated in the paraspinal soft tissue of the posterior mediastinum. Nevertheless, it also may show partially intradural growth and a dumbbell-shaped profile, as seen in some neurofibromas (131).
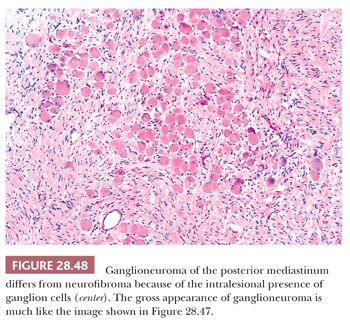
Attention to the presence or absence of a tumor capsule on gross and microscopic examination is particularly important in the recognition of ganglioneuroma. This is true because the basic pattern of spindle cell proliferation in this tumor is virtually identical to that seen in neurofibroma. The major histologic difference between the two lesions is the presence of well-formed ganglion cells in ganglioneuroma (Fig. 28.48), but these may be observed only regionally, and scrutiny of many tissue blocks may be required to document their presence (278). The use of immunostains for synaptophysin—a synaptic vesicle–related protein that is seen in neuronal lesions (279)—can assist in the rapid identification of widely scattered ganglion cells in ganglioneuromas.
FIBROGENIC AND MYOFIBROBLASTIC PROLIFERATIONS
Four cytologically bland spindle cell proliferations of the mediastinum may be histologically mistaken for one another. These are represented by SFT, desmoid-type fibromatosis, sclerosing mediastinitis, and inflammatory myofibroblastic tumor.
SOLITARY FIBROUS TUMORS
SFTs are best known as pleural neoplasms, but they also have been reported as primary lesions of the mediastinum (as well as many other locations) in adults (280–282). All three mediastinal subcompartments appear to be at equal risk. Some lesions are polypoid and protrude into the mediastinum from bases in the medial reflections of the pleura, but others appear to truly take their origin in the soft tissues between the lungs (280). As is true of their counterparts in the serosal investments of the lungs, mediastinal SFT have no pathogenetic relationship to asbestos exposure (280). These neoplasms are either found incidentally on chest radiographs, or they manifest themselves with nonspecific symptoms and signs of structural displacement (280,281).
As just mentioned, a subset of SFTs has a circumscribed and polypoid gross configuration and is covered by a smooth layer of mesothelium. The remainder are less well-defined macroscopically, although their demarcation from adjacent soft tissues is usually clear to the surgeon at thoracotomy. On cut section, these lesions are white-gray with a uniformly firm consistency. Occasionally, a whorled, fascicular gross appearance is appreciated, similar to common leiomyomas of the uterus.
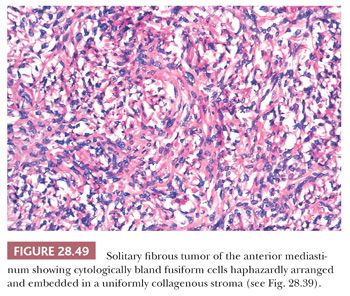
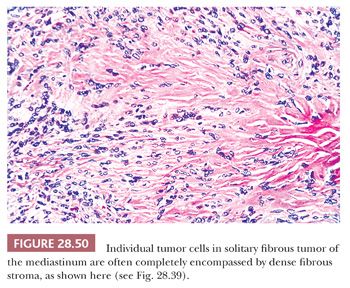
The histologic features of SFT include a constituency by cytologically banal spindle cells that are arranged haphazardly in a densely collagenous matrix (Fig. 28.49). The intercellular stroma has a distinctive fibrohyaline character—focally resembling that of keloids—and often circumferentially invests the individual cells in these neoplasms (Fig. 28.50). Tumor vascularity is typically rich, and some supporting blood vessels may assume an irregularly branched appearance (280,281). Mitotic activity is usually limited, but some SFTs show as many as 10 division figures per 10 high-power (×400) microscopic fields. Spontaneous necrosis is virtually unknown in such tumors, although areas of myxohyaline degeneration are not uncommon (131). Variant microscopic patterns include one featuring focal storiform growth, an epithelioid-predominant form (282), and another wherein considerable nuclear pleomorphism, or dense cellularity, or both, are observed focally or regionally (280). The last cited of these SFT subtypes may be termed “atypical” SFT; however, the “minimal” criteria for a diagnosis of malignant SFT have not yet been codified. With that having been said, it is wise to consider SFT as a “borderline” tumor instead of a completely benign lesion, in light of its potential for recurrence (283).
Immunohistochemical analysis of SFT demonstrates consistent reactivity for vimentin. CD34 is seen in approximately 85% of cases, along with potential labeling for CD99 and bcl-2 protein (284–286). The cells of this tumor lack keratin, epithelial membrane antigen, S100 protein, desmin, muscle-specific actin, and TLE1 protein (287).
Ultrastructural studies further support the fibroblastic nature of SFTs, in that they show nondescript spindle cells with notable profiles of rough endoplasmic reticulum and focal intrareticular collagen fibers (286). Features of myofibroblasts or myogenous cells, such as cytoplasmic thin filament skeins and dense bodies, pericellular basal lamina, and subplasmalemmal dense plaques, are generally not observed. Moreover, an absence of intercellular junctions militates against the presence of epithelial differentiation (288).
DESMOID-TYPE FIBROMATOSIS
In contrast to SFT, desmoid-type fibromatosis (DTF) is a poorly demarcated tumefactive proliferation that is centered in the soft tissue. It may affect the anterior or posterior mediastinum and is most often seen in children or young adults (289). These patients may present with the superior vena caval syndrome, a nerve-entrapment syndrome, or dysphagia, depending on the location of the lesion.
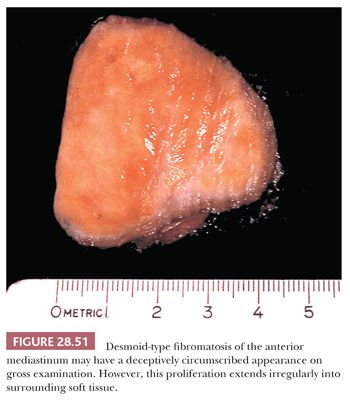
To the surgeon, the true boundaries of DTF are indistinct; it tends to blend imperceptibly with surrounding fibroadipose soft tissue. Consequently, the pathologist often receives an incompletely excised mass or a “morselized” specimen that reflects the technical difficulty of its resection. Like fibromatoses in general, DTF has a firm, almost gritty cut surface, with a vaguely fascicular white-tan appearance (290) (Fig. 28.51).
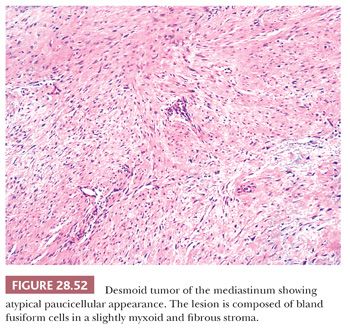
The microscopic features of this lesion include a variably fibromyxoid matrix in which parallel or interweaving fascicles of cytologically bland tumor cells are enmeshed. The latter may have either a stellate or fusiform shape, and their nuclei show dispersed chromatin and inconspicuous nucleoli (Fig. 28.52). The cytoplasm is eosinophilic or amphophilic, and close scrutiny may reveal a slightly fibrillary quality therein (291). One of the most characteristic histologic attributes of DTF is its vascularity; numerous venule-sized supporting vessels with thick walls are interspersed regularly throughout the mass (292) (Fig. 28.53). Despite the obviously fibrogenic nature of the proliferation, the vascular lumina are open and distinct rather than compressed. Nonetheless, staghorn-type blood vessels are not seen in this lesion. Similarly, areas with storiform growth or nuclear pleomorphism are regularly lacking in DTF, and mitotic activity is virtually nonexistent. In fact, the observation of even a few division figures in several high-power microscopic fields should raise the strong diagnostic suspicion of a low-grade fibrosarcoma rather than DTF. Intratumoral inflammation also is not expected in the latter lesion.
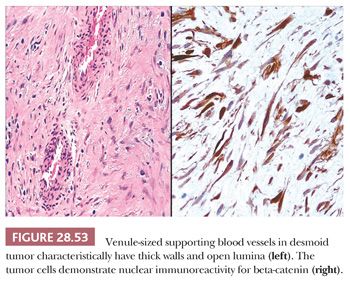
Ultrastructural studies of DTF and other fibromatoses have demonstrated that the tumor cells have myofibroblastic properties, including intrareticular collagen fibers, thin filament bundles, and cytoplasmic dense bodies (293). Otherwise, they resemble those seen in SFT. This same profile is reflected in the results of immunohistochemical analyses of DTF, which demonstrate reactivity not only for vimentin but actin and desmin as well (294). DTF is potentially reactive for nuclear beta-catenin, unlike most other spindle cell proliferations (295), and it is nonreactive for CD34 (296).
SCLEROSING MEDIASTINITIS, INFLAMMATORY MYOFIBROBLASTIC TUMOR, AND CALCIFYING FIBROUS TUMOR
DTF is most likely to be confused with sclerosing mediastinitis, which occurs almost exclusively in the anterosuperior mediastinum (297), or with inflammatory myofibroblastic tumor (IMT [298]). Sclerosing mediastinitis or IMT may manifest clinically with the superior vena caval syndrome or with symptoms of cardiorespiratory compromise, and both may be observed at any age (297,299–302). All of the cited lesions are characteristically represented by asymmetric widening of the mediastinum on chest films, usually with projection of a mass into an upper lung field (300).
The gross features of sclerosing mediastinitis and IMT are like those of dense but banal fibrosis occurring in other settings. Firm, white tissue is seen on cut section, without any suggestion of fascicular growth, and the junction of the periphery of the process with adjacent adipose tissue is clear cut (298,301).
Microscopically, sclerosing mediastinitis demonstrates the deposition of dense, fibrohyaline, relatively avascular tissue that is extremely paucicellular (297,301). Variably sized collections of mature lymphocytes and plasma cells are entrapped within the fibrotic zones and probably play a pathogenetic role in their formation (301) (Fig. 28.54). Caseating or noncaseating granulomas are sometimes observed as well, relating to the fact that sclerosing mediastinitis is, in a substantial number of cases, an idiosyncratic response to infection.
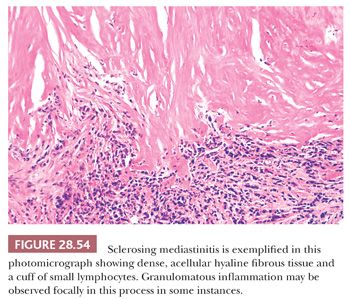
The most common causative organism in these instances is Histoplasma capsulatum, which may be detectable with silver impregnation stains in the granulomatous foci (300). Fungal infection accounts for no more than half of all examples of sclerosing mediastinitis; in addition to histoplasmosis, aspergillosis, cryptococcosis, and mucormycosis have been documented as potentiators of this condition. Other cases have been reported in association with sarcoidosis, nocardiosis, actinomycosis, syphilis, therapy with methysergide (an antimigraine medication), and trauma to the chest (301). The remainder are of indeterminate etiology, but the postulated common pathogenetic thread is that of a delayed cell-mediated hypersensitivity reaction (301), with a possible relationship to the family of IgG4-related diseases. Immunostains for that immunoglobulin subclass may show many reactive plasma cells in the lesions (303). It would appear that a process similar to sclerosing mediastinitis may rarely involve the thymus itself and be relatively limited to it (302,304).
Other examples of mediastinal “pseudotumor” were reported by Brachet and colleagues (305) and by Coffin and coworkers (298), but these lesions had the histologic features of IMT rather than those of sclerosing mediastinitis as noted previously. The former of these two proliferations is regarded as a true neoplasm (298), and it is more cellular than the latter lesion. Indeed, IMT is composed of bland fusiform elements that are arranged in variably dense arrays—often being fascicular or randomly configured—admixed with lymphocytes and a scattering of other inflammatory cells. The proliferating spindle cells in most IMTs are immunoreactive for vimentin and α-isoform actin (298), overlapping with the immunophenotypes of DTF and smooth muscle tumors. Approximately 40% of IMTs also are immunoreactive for the ALK-1 protein, in contrast to other diagnostic possibilities (306).
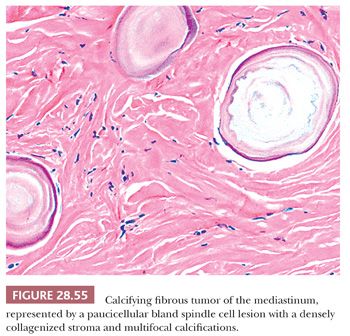
Nascimento and colleagues (307) have documented the presence of calcifying fibrous tumor (CFT) in the mediastinum in examining the possibility that the latter entity was a form of IMT. They instead concluded that the two lesions were distinct from one another. CFTs show a composition by bland spindle cells in a densely hyalinized collagenous stroma, with focal calcification (Fig. 28.55). Variable immunoreactivity is present in such tumors for CD34 and alpha-isoform actin, but ALK-1 is absent.
An important caveat must be remembered in the evaluation of putative cases of sclerosing mediastinitis. It is the ability of the tumor cells in selected malignant lymphomas (e.g., “obliterative total sclerosis” HL [103,308]), metastatic carcinomas, or desmoplastic mesotheliomas to incite a brisk and obfuscating fibrotic response in the mediastinal soft tissue or lymph node groups. The neoplastic cells in such lesions are rather sparse and may be deceptively bland cytologically. Accordingly, they may be easily overlooked, leading to diagnostic misadventures (302,309,310). Stains for keratin, CD15, CD20, CD30, and CD45 are a prudent part of the workup of such lesions (see “Mediastinal Large Cell Non-Hodgkin Lymphoma” and “Sarcomatoid Mediastinal Mesothelioma”).
DIFFERENTIAL DIAGNOSIS OF CYTOLOGICALLY ATYPICAL MEDIASTINAL NEOPLASMS
Because of the diversity of cytologically atypical and overtly malignant proliferations that are capable of arising in the mediastinum, differential diagnosis of these lesions can be daunting. However, as mentioned earlier, this process can be simplified significantly by considering generic groupings of tumors that bear reproducible similarities to one another. Accordingly, the following material is organized in that fashion. Each pathologic entity in the respective groups is synopsized briefly from a clinicopathologic perspective, and differential diagnostic summaries are provided thereafter.
SMALL CELL NEOPLASMS OF THE MEDIASTINUM
The primary considerations among those tumors with a uniform small cell appearance encompass lesions with epithelial, lymphoid, neuroectodermal, and mesenchymal characteristics. This group includes small cell neuroendocrine carcinoma (both primary and metastatic), basaloid carcinoma, neuroblastoma (and congeners), primitive neuroectodermal tumor, rhabdomyosarcoma, and malignant lymphomas comprising small tumor cells.
MEDIASTINAL SMALL CELL NEUROENDOCRINE CARCINOMA
The overwhelming majority of small cell neuroendocrine carcinomas (SCNCs) that present as anterior, middle, or posterior mediastinal masses are metastatic in nature (311), usually from primary sources in the lungs or esophagus. Patients with such lesions will often have symptoms referable to intrabronchial disease (cough, hemoptysis, dyspnea) or enteric mucosal abnormalities (dysphagia), but this statement is not absolute. Also, a minority of individuals have evidence of a paraneoplastic endocrinopathy, such as Cushing syndrome or SIADH (52). Because it is well known that primary pulmonary SCNC may not be detectable on conventional chest radiographs, computed tomography, or even magnetic resonance scanning, one should not accept negative results from such studies as providing definitive evidence against the existence of these tumors in cases where a mediastinal biopsy shows small cell carcinoma. Indeed, the diagnosis of primary mediastinal (thymic) SCNC is one to be made with extreme circumspection in light of the fact that there are less than 150 well-documented cases in the English-language literature (51,93,94,311–318).
The gross characteristics of SCNC are identical, regardless of their anatomic origins. These tumors have a fleshy, white-gray, partially hemorrhagic and necrotic appearance, simulating that of malignant lymphoma in some respects (311,313).
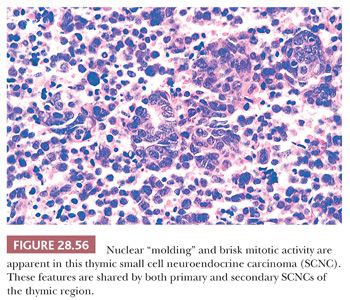
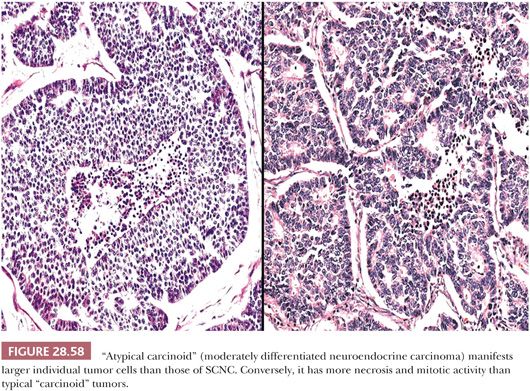
Microscopically, particular attention should be given to whether or not the neoplasm is actually centered in the thymus gland or rather represents a nodal metastasis. Even in the former of these circumstances, a secondary lesion still cannot be excluded; however, thymic involvement could provide at least provisional support for the primary nature of such a tumor. The histologic attributes of SCNC are familiar to all surgical pathologists. They include a vaguely clustered or organoid architecture, regional spontaneous necrosis (often in the central areas of tumor cell nests), extremely high nucleocytoplasmic ratios with scanty cytoplasm, dispersed or homogeneously dark nuclear chromatin with inconspicuous nucleoli, “molding” of adjacent nuclei against one another, and numerous mitoses (Fig. 28.56). “Crush” artifact is common in small endoscopic biopsy specimens of SCNC, and one may also observe the “Azzopardi phenomenon,” wherein deeply basophilic rings of nuclear material are encrusted on intratumoral blood vessels (37) (Fig. 28.57). Another characteristic of SCNC, be it primary or metastatic in the mediastinum, is its capacity for divergent differentiation. Examples of this tumor showing an admixture of obvious squamous cell carcinoma or adenocarcinoma have been seen in the thymus, lung, gut, and other sites (93,94,314).
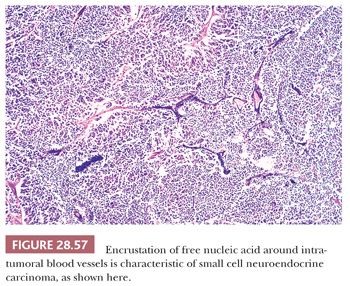
A long-standing problem concerning SCNC is its distinction from so-called “atypical carcinoids.” The latter tumors have traditionally been defined histologically by a larger cellular size, with more cytoplasm than that of SCNC, and a more distinctly organoid growth pattern with potential spindle cell foci (37) (Fig. 28.58). From a practical point of view, the authors prefer the term grade 2 neuroendocrine carcinoma to atypical carcinoid to avoid associating the lesion in clinicians’ minds with low-grade tumors (“classic” or “conventional” carcinoids [which are synonymous with grade 1 neuroendocrine carcinomas]). More important, the distinction between the small cell type and other forms of neuroendocrine carcinoma does not appear to be particularly important prognostically in the context under discussion here. All of these tumors, whether metastatic or primary, are capable of an aggressive clinical evolution in cases with mediastinal involvement (94).
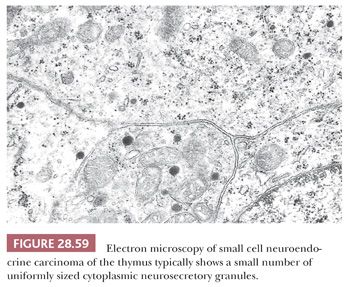
Ultrastructurally, SCNC is defined by the presence of primitive junctional complexes between apposed tumor cells as well as the presence of neurosecretory (neuroendocrine) granules in the cytoplasm (37,93,94,312,317). The latter organelles are small (80 to 250 nm) and relatively uniform in appearance (Fig. 28.59). In order to be assured that they do not represent lysosomes, a clear-cut peripheral “halo” should be seen within each granule around a central, electron-dense core; moreover, clusters of granules are more convincing than isolated inclusions in a minority of cells.
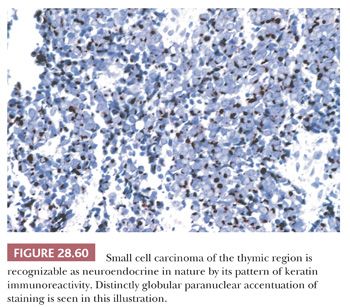
On an immunohistochemical level, SCNC is recognizable either by a distinctive pattern of intermediate filament expression, with perinuclear “globules” of keratin protein (210) (Fig. 28.60), or by its potential reactivity for one of several neuroendocrine markers. These include chromogranin A (a matrix protein of neurosecretory granules), synaptophysin, CD56, CD57, and selected neuropeptides such as adrenocorticotrophic hormone (37). Thyroid transcription factor-1 (TTF-1) is present in the majority of pulmonary SCNCs (319,320), but it is seen in only 8% of primary thymic neuroendocrine carcinomas (320). The converse of that statement applies to PAX8, albeit with lesser sensitivity (320).
BASALOID CARCINOMA OF THE MEDIASTINUM
Basaloid carcinoma (BC) shares the same potential as SCNC to involve the mediastinum either primarily as a thymic tumor (93–96) or by metastasis (Fig. 28.61). Potential primary sites in the latter instance include the oropharynx, hypopharynx, larynx, esophagus, lungs, and anorectal region (96). Hence, the same cautions pertain to rushed conclusions that such neoplasms originate in the mediastinum, as emphasized previously in connection with mediastinal SCNC. Patients with BC of the mediastinum are adults who present with generic symptoms and signs of an intrathoracic mass (94,321) and, potentially, other findings that may be referable to the sources of metastatic lesions.
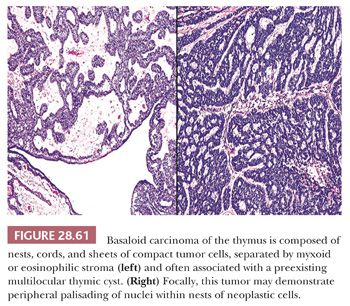
Histologically, BC is composed of organoid clusters of small polygonal cells with high nucleocytoplasmic ratios, scanty cytoplasm, uniformly hyperchromatic round nuclei, and abundant mitoses. However, there is no nuclear “molding.” In addition, this lesion may show foci of stromal mucin-containing glandlike profiles that may recall the appearance of adenoid cystic carcinomas, globular eosinophilic intercellular deposits of basement membrane material as seen in cylindromas of the skin, and areas of overt squamous differentiation with keratin “pearls” (93,94,206,207). The few cases reported as primary in the thymus have shown a tendency for association with—and probable origin in—antecedent multilocular thymic cysts (93–96).
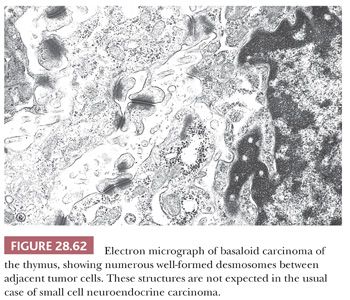
The electron microscopic attributes of this neoplasm are basically those of a poorly differentiated squamous proliferation (93). Therefore, limited numbers of cytoplasmic tonofilaments are expected, together with well-formed desmosomal-type intercellular junctions (Fig. 28.62). Neurosecretory granules are not observed. In addition, the globular accumulations of basement membrane seen by optical microscopy are reflected in the presence of focally redundant basal lamina on an ultrastructural level.
Keratin is uniformly detected immunohistochemically in BC with a diffuse intracellular distribution, and reactivity for p63 and epithelial membrane antigen is common (322). However, we have detected no staining in mediastinal BCs for neuroendocrine determinants.
NEUROBLASTOMA OF THE MEDIASTINUM
In its usual form, neuroblastoma is a disease of young children (323). Intrathoracic examples of this tumor are largely—but not totally (38,39,323)—restricted to the posterior mediastinum (324), where they are typically primary and originate in association with nerve roots in a paraspinal location (266). The latter region is only exceptionally involved by metastases of a neuroblastoma arising at another anatomic site.
Symptoms of the mass most often relate to the compression of spinal nerves or interference with esophageal motility. Some children also may have the paraneoplastic neurologic syndrome known as opsoclonus-myoclonus (“dancing feet and dancing eyes”) (60). Erosion of contiguous vertebral bones may be seen as well (325). Interestingly, when neuroblastomas involve the thymus and anterior mediastinum, they appear to show an inordinate association with production of the SIADH (38,39). Laboratory evaluation of the urine or blood often reveals elevated levels of homovanillic acid and vanillylmandelic acid, representing metabolites of tumoral catecholamine products (313). In accord with information previously mentioned, marked hyponatremia also may be apparent in connection with those tumors synthesizing antidiuretic hormone.
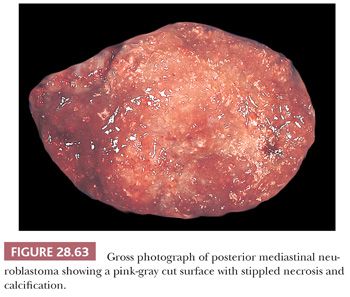
The resectability of neuroblastoma is predicated on its relative circumscription, and, in the mediastinum, most cases do demonstrate partial or complete encapsulation (131). Cut surfaces are fleshy and pink-gray, often with foci of necrosis, hemorrhage, and punctate calcification (Fig. 28.63).
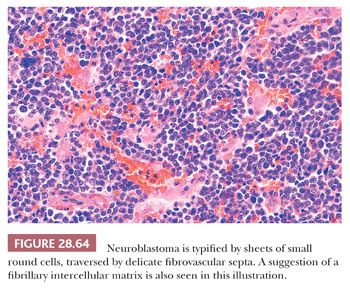
Neuroblastoma is potentially one of the prototypical, undifferentiated, small round cell tumors of childhood (326); in its primitive form, this lesion is composed of sheets of monomorphic elements with uniformly dispersed chromatin, inconspicuous nucleoli, and a scant amount of eosinophilic cytoplasm (Fig. 28.64). Mitotic activity and karyorrhexis vary considerably but may be striking. Regional necrosis and superimposed dystrophic calcification also are observed rather frequently, and an arborizing network of delicate stromal blood vessels is a consistent finding (131). Differentiating examples of this entity betray their neuroblastic nature through the formation of a fibrillary eosinophilic intercellular matrix, and even more mature tumors may demonstrate the presence of scattered primitive ganglion cells. If the latter are numerous, the diagnostic term ganglioneuroblastoma is more appropriate (327) (see Chapter 14).
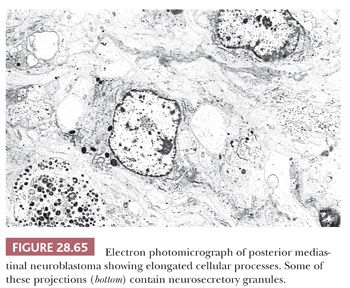
Electron microscopy discloses variable neuritic differentiation in the form of complex, long, interdigitating cytoplasmic processes that contain microtubules (Fig. 28.65) (38,323). Some also show synaptic vesicles or scant numbers of neurosecretory granules, but the latter structures are not necessary diagnostically (328). Basal lamina is scarce to absent, intercellular junctions are few in number and primitive, and only a minority of cases exhibit the presence of intracellular glycogen granules (329). Cytoplasmic intermediate filaments are usually not seen; when present, they are inconspicuous and widely separated.
Immunohistochemical assessment of neuroblastoma reveals variable reactivity for vimentin and neurofilament protein; many tumors lack both of these polypeptides altogether (330). Evidence of neural differentiation is manifest by positivity for CD56, CD57, and synaptophysin, which are seen in the majority of cases (131). Many neuroblastomas also react with NB84, a monoclonal antibody recognizing a 57 kd tumor-selective moiety that is preserved in paraffin sections (331,332). Markers of muscular lesions (desmin, muscle-specific actin), lymphoid tumors (CD45), and epithelial neoplasms (keratin, p63, epithelial membrane antigen) are absent (38,285).
MEDIASTINAL PRIMITIVE NEUROECTODERMAL TUMOR
Primitive neuroectodermal tumor (PNET) was formerly considered to be synonymous with “peripheral neuroblastoma” or “extraskeletal Ewing sarcoma” (333). The pathologic spectrum of PNET is now well established; it is predominantly a tumor of childhood, but it can indeed be seen throughout life. Patients with PNET complain of a rapidly growing mass, which, when located in the mediastinum (a rare occurrence), may produce symptomatic structural displacement (334). We have observed examples in both the anterior and posterior mediastinum in adolescents or young adults. Similar cases have been reported by Glick and Page (335) and Grosfeld and colleagues (336). For statistical purposes, primary mediastinal PNET should be separated from the Askin tumor (primitive small cell thoracopulmonary tumor), which takes its origin in the soft tissues of the chest wall (337). However, the lineages of these two entities are identical, in that both demonstrate neuroectodermal differentiation (318).
Mediastinal PNET is a bulky, poorly delimited, necrotic and hemorrhagic, white-gray fleshy mass on macroscopic examination. It may show adherence (or apparent origin) from a spinal nerve root when located in the posterior subcompartment.
The microscopic characteristics of this neoplasm are again those of a primitive round cell sarcoma in most cases. Nonetheless, some may exhibit a vaguely organoid growth pattern or focal formation of primitive rosettes, suggesting the identity of the lesion as neuroectodermal. The overall histologic appearance is very much like that of “undifferentiated” neuroblastoma, including a prominent network of delicate intratumoral blood vessels, but, unlike the latter, PNET does not demonstrate any attempt at cytoplasmic neurofibrillary maturation (329,333) (Fig. 28.66). In other sites, PNETs have been shown to manifest divergent differentiation, with areas resembling rhabdomyosarcoma or even primitive glands (307,309). Although this phenomenon has not yet been reported in mediastinal lesions, it is potentially possible.
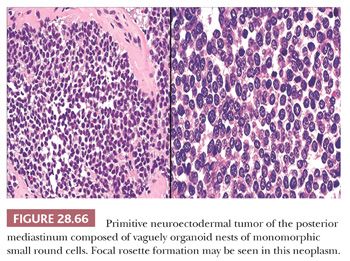
The ultrastructural attributes of PNET are somewhat similar to those of neuroblastoma, except that those cytoplasmic processes which are formed are blunt and rudimentary. There are no intracellular microtubules in the tumor cells, and neurosecretory granules or synaptic vesicles are sparse (247,329,338). Intercellular junctions assume a primitive nature. Examples of PNET with divergent differentiation may manifest foci wherein the cells show cytoplasmic thick and thin filaments or form nascent glandular lumina (338,339).
Immunohistochemical characteristics of PNET are again like those of neuroblastoma, but the former of the two lesions exhibits more uniform reactivity for vimentin, less often shows NB84 positivity (332), and only exceptionally expresses neurofilament protein (247). Moreover, the CD99 and MB2 antigens, FLI-1, and β2-microglobulin are reproducibly present in PNET but not in neuroblastoma (340–344). Synaptophysin, CD56, and CD57 are detectable in a proportion of cases, and divergent neoplasms also may show focal reactivity for keratin, desmin, and actin (247,340).
The most certain method for securing a firm diagnosis of PNET is by cytogenetic analysis. A characteristic and distinctive reciprocal chromosomal translocation (t[11;22][q24;q12]) serves as a definitive marker of PNET among all small cell tumors of the mediastinum (345). With the benefit of suitable nucleotide primers and the polymerase chain reaction, this abnormality can now be identified using paraffin-embedded tissue.
RHABDOMYOSARCOMA OF THE MEDIASTINUM
Rhabdomyosarcoma (RMS) of the mediastinum also may arise in either the anterior or posterior subcompartments, and, in pure form, it almost exclusively affects children and adolescents (131,289,334,335,346). The latter demographic point is an important one, because another posterior mediastinal neoplasm that can be seen in adults—the so-called malignant triton tumor (see Chapter 5)—may demonstrate partial rhabdomyoblastic differentiation. Childhood RMS produces manifestations of structural displacement but is associated with no other distinctive clinical syndromes (289,334,346).
The macroscopic findings in RMS are also relatively nondescript, being those of a poorly delimited, bulky lesion of the soft tissues with a fleshy, white-gray cut surface and frequent areas of spontaneous hemorrhage or necrosis (289). There is no reproducible association with particular anatomic structures, such as the paraspinal muscles.
RMS is one small round cell neoplasm of the soft tissue with histologic features that may serve as a clue to the lineage of the lesion on conventional microscopic examination. Although some of these tumors are indeed monomorphic proliferations of primitive cells, more of them demonstrate discernible nuclear and cellular pleomorphism that is not observed in differential diagnostic alternatives. Scattered large cells with multinucleation, eccentric nuclei, fusiform features, or deeply eosinophilic cytoplasm strongly suggest the diagnosis of RMS, as does the presence of focal myxoid stromal change in an otherwise uniform small cell lesion (Figs. 28.67 and 28.68). Likewise, areas of alveolar growth are distinctive when present (131,329). Begin and colleagues (347) also have drawn attention to the ability of mediastinal RMS to manifest clear cell change when the glycogen content of the lesion is high; this variation increases the overall size of the tumor cells, potentially causing confusion with large cell neoplasms of other lineages.
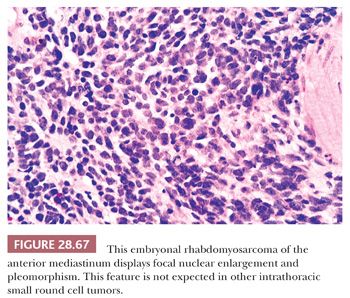
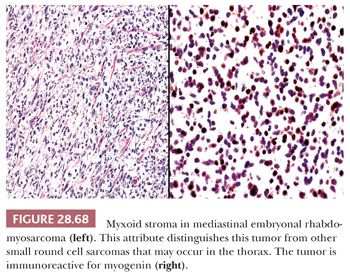
Electron microscopically, RMS is recognized by the tight aggregates of intermediate filaments that are present in the cytoplasm of tumor cells. These may occasionally be organized with thick (myosin) filaments into primitive sarcomeric structures (Fig. 28.69), complete with Z bands. Glycogen is usually present, together with short segments of pericellular basal lamina. Intercellular junctions, cytoplasmic processes, microtubules, synaptic vesicles, and dense-core granules are lacking (247,343).
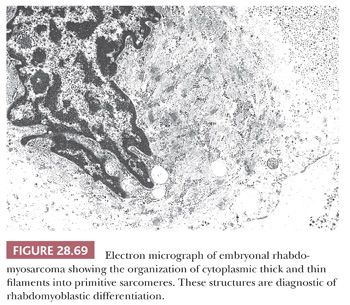
Virtually all RMSs are immunoreactive for desmin and muscle-specific actin, together with vimentin. Myoglobin is observed only in large maturing rhabdomyoblasts and is therefore not a particularly useful marker of RMS in its purely small cell form. Synaptophysin is regularly absent, but some examples of RMS may demonstrate focal positivity for CD56 or CD57 (247,285,343). The specificity of desmin and actin for the diagnosis of RMS has been questioned by some because these determinants also are seen in smooth-muscle proliferations. Nonetheless, because a small cell variant of leiomyosarcoma does not exist, that argument seems irrelevant in this context. In any event, other markers that are apparently restricted to striated muscle—particularly MyoD1 and myogenin (348)—can be applied to the differential diagnosis in question. As a word of caution, it should be also be remembered that both RMS of the alveolar subtype and PNET share potential reactivity for CD99 (342,343,348); hence, that determinant should not be employed diagnostically in isolation.
SMALL CELL MALIGNANT LYMPHOMAS OF THE MEDIASTINUM
As mentioned earlier, there are three small cell non-Hodgkin malignant lymphomas (SCML) that may affect the mediastinum as primary processes. These are LBL (216,217), Burkitt lymphoma (BL) (239,240), and marginal-zone lymphoma of the thymus (242,243,349). The first two of these tumors are seen predominantly in children and adolescents, but LBL also has a second incidence peak in the seventh to eighth decades (220) and we have observed examples of mediastinal BL in adults as well. Either LBL or BL is capable of peripheralization, with the appearance of acute lymphoid leukemia (218,219,240). However, that complication is observed at presentation in only a minority of cases with mediastinal masses.
The gross characteristics of all lymphomas are roughly similar and have been described earlier. Similarly, the light microscopic attributes of LBL and BL have been documented earlier in this discussion. Mediastinal marginal zone lymphomas (MZLs), on the other hand, show a constitution by clusters of differentiated, centrocyte-like lymphoid cells that are commingled with mature-appearing lymphocytes as well as lymphoid follicles containing reactive germinal centers. Plasma cells also may be observed. All of these elements must be centered in the thymus, virtually by definition. The centrocyte-like cells diffusely expand the gland and permeate the thymic epithelium in small groups (yielding so-called “lymphoepithelial lesions”); this phenomenon may cause microcystic change as well (242,243). MZLs also demonstrate admixed monocytoid cells (349). With the electron microscope, small cell non-Hodgkin lymphoma is a diagnosis of exclusion. One should not observe intercellular junctions, cytoplasmic filament bundles, neurosecretory granules, synaptic vesicles, microtubules, basal lamina, cytoplasmic processes, or primitive sarcomeres (350). In addition, most lymphoma cells demonstrate the formation of nuclear “blebs,” which are irregular protrusions of the nuclear membranes (140) (Fig. 28.70).
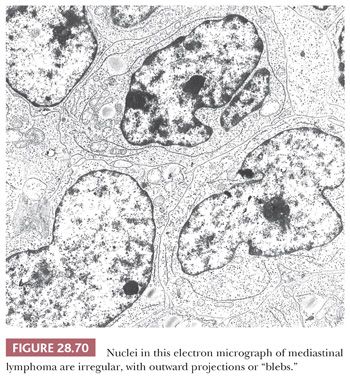
Immunohistochemical analysis is helpful in solidifying the diagnosis of LBL, BL, and MZL of the mediastinum. These lesions express the CD45 (leukocyte common) antigen as well as show reactivity with other lymphoid-selective monoclonal antibodies. LBL commonly is labeled with CD43 reagents (L60, Leu-22, MT-1) (Fig. 28.71), CD99 antibodies, and others to bcl-2 protein and terminal deoxynucleotidyl transferase (TdT) (225). BL is reactive for CD10 and CD20 but typically lacks all of the other determinants just cited, with the possible exception of bcl-2 protein (225). In MZL, reactivity for monotypic immunoglobulin light chain can be detected by ribonucleic acid in situ hybridization methods in the centrocyte-like cells (243). That tumor type lacks CD10, CD43, CD99, bcl-2, and TdT (351). Positivity for keratin, CD57, synaptophysin, desmin, and actin is not observed in any of these three lymphoma morphotypes, but some examples of all of them may exhibit immunoreactivity for vimentin.
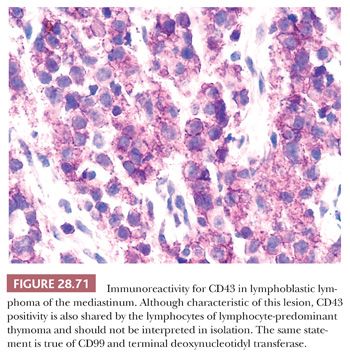
OTHER SMALL CELL MEDIASTINAL NEOPLASMS
In addition to those small cell tumors presented in the foregoing sections, others of a metastatic nature also may involve the mediastinum. These include small cell osteosarcoma and small cell malignant melanoma. Except for the second of these possibilities, the primary lesion in such cases is typically obvious and there is no question of whether the intrathoracic neoplasm might have arisen there. However, it is common knowledge that melanomas have the capability of presenting with distant metastasis in the face of an undetectable primary source, and rare examples are thought to have arisen in the thymus (352,353). Moreover, the potential for melanomas to assume the guise of a small cell lesion also has been well documented (354).
Ultrastructural analyses may be helpful in these circumstances in demonstrating the presence of cytoplasmic premelanosomes in small cell melanomas (343,350). Also, the immunohistochemical characteristics of such lesions feature reactivity for S100 protein and the MART-1 antigen (354), neither of which is expected in other small cell neoplasms of the mediastinum.
SPECIFIC DIFFERENTIAL DIAGNOSTIC CONSIDERATIONS AMONG SMALL CELL TUMORS OF THE MEDIASTINUM
It should be apparent that the foregoing discussion of mediastinal small cell neoplasms was structured in part to set the stage for a consideration of particularly vexing differential diagnoses in this group of lesions. These are discussed in the sections immediately following.
Rhabdomyosarcoma versus Primitive Neuroectodermal Tumor versus Neuroblastoma
Among all soft tissue tumors, those in the small round cell category in children are, perhaps, the most challenging diagnostically. As mentioned earlier, mediastinal RMS, PNET, and neuroblastoma all may assume an undifferentiated appearance that is closely similar. Thus, histologic examination alone is unlikely to provide a definitive answer and other, more specialized techniques must be employed to reach a final interpretation. The pathologic features of greatest value in this context are outlined in Table 28.2.
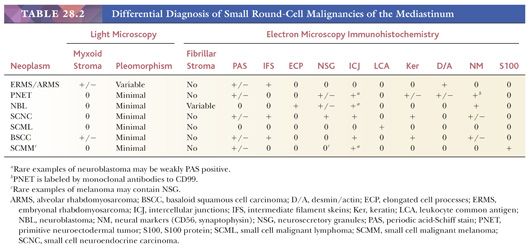
Small Cell Malignant Lymphoma versus Small Cell Carcinomas
Especially in small biopsy specimens of mediastinal tumors, it may be difficult to distinguish SCMLs from small cell carcinomas. If a distinctly clustered growth pattern is observed, despite obscure nuclear features, the latter of these diagnoses should be favored. Likewise, the presence of the Azzopardi phenomenon suggests the presence of a carcinoma rather than lymphoma. Nonetheless, most cases will require specialized studies to reach a final conclusion. Electron microscopy is not very productive when specimens have been distorted badly during procurement; however, it is fortunate that CD45 and keratin immunostains often can still be performed successfully even on crushed tissue samples. These should be sufficient for making the general diagnostic separation under discussion, but more specific interpretations may well require that additional specimens be obtained.
Obtaining additional specimens is important because the ultrastructural and immunohistologic markers separating the types of carcinoma or lymphoma (discussed previously) usually are optimally seen only in well-processed tissue samples. As a practical solution to this problem—mindful of the fact that clinicians are often loath to undertake another surgical procedure without much debate—fine-needle aspiration biopsy of the mediastinal mass is suggested (2,3,5). This technique typically yields sufficiently preserved material to allow for more detailed study of a lesion that is already known to be carcinomatous or lymphomatous in nature.
Small Cell Neuroendocrine Carcinoma versus Basaloid Carcinoma
The distinction between carcinoma morphotypes, as just mentioned, is often an important step in the planning of therapy for a mediastinal tumor. In particular, primary small cell neuroendocrine thymic carcinoma is usually managed, at least in part, with chemotherapy that would be appropriate for SCNC of the lung. In contrast, primary basaloid thymic carcinomas should be resected surgically (95).
To reiterate selected points made earlier in this chapter, the finding of dispersed nuclear chromatin, inconspicuous nucleoli, and nuclear molding should suggest a diagnosis of SCNC. On the other hand, association with remnants of a thymic cyst, cellular nucleolation, and intercellular deposits of eosinophilic basement membrane material (potentially highlighted with a PAS-diastase stain) militate in favor of basaloid carcinoma. Cases that are histologically indeterminate do exist in this context, but they usually yield themselves to electron microscopy and immunohistochemistry (see earlier sections in this chapter).
Primary versus Metastatic Carcinoma of the Mediastinum
In cases where a nonmelanocytic lineage of a small cell malignancy of the mediastinum is observed, the question arises as to the primary or secondary nature of the proliferation. Although the incidence of certain mesenchymal tumors is skewed in one direction or another in this regard (see previous discussion), the origin of most other neoplasms is questionable.
Unfortunately, there are no reliable pathologic indicators that allow for a definitive resolution of this problem. Accordingly, one must require that the clinician undertake appropriate radiologic or endoscopic procedures to address the potential existence of primary lesions outside of the mediastinum before assigning a final diagnostic interpretation to the tumor under consideration. This stipulation has more than mere academic significance because, in many cases, it ultimately determines the surgical or nonsurgical management of the lesion.
LARGE POLYGONAL CELL NEOPLASMS OF THE MEDIASTINUM
Malignant large polygonal cell tumors of the mediastinum comprise a heterogeneous group of lesions with respect to cellular lineage. These include variants of thymic carcinoma, germ cell tumors, carcinoid tumor of the thymus, large cell lymphoma, granulocytic sarcoma, plasmacytoma, syncytial HL, paraganglioma, parathyroid carcinoma, malignant mesothelioma, and metastatic carcinoma or melanoma.
PRIMARY THYMIC CARCINOMAS
Thymic carcinomas are neoplasms that have been well recognized as distinctive entities only in the recent past (63,93,186,191,202,317,318,355–362). Before their current characterization, literature on tumors of the thymus often stated that their biologic potential could not be determined by histologic appearance; that view is only partially correct. It is true that the regional recurrence of conventional thymoma is predicated most on the presence or absence of gross invasion (188) rather than on its cytologic attributes. Moreover, a small number (approximately 7%) of invasive thymomas do show extrathoracic (but idiosyncratic) spread (“metastasizing thymoma”). Thus, these biologic events do not necessarily equate with a diagnostic label of “carcinoma.” In this specific context, it is notable that the entity advanced by Kirchner and colleagues (165) as “well-differentiated thymic carcinoma” (WHO type B3 thymoma) shows a clinicopathologic synonymity with that subset of epithelial-predominant thymomas which manifests moderate nuclear atypia (156).
Outside of the foregoing remarks, there is a distinct group of thymic epithelial lesions that shows overt cellular anaplasia and aggressive clinical evolution. These do deserve the designation of carcinoma because of such features.
Thymic carcinomas also differ clinically from thymomas in that they are typically not associated with paraneoplastic syndromes such as myasthenia gravis or pure red cell aplasia (93,94,317,363,364). The former lesions most often present themselves with symptoms of structural displacement, although a minority are discovered incidentally on screening chest radiographs (316,317). Patients with thymic carcinomas are typically middle-aged or older adults, with a slight predominance in men. Nevertheless, a few cases have been observed in children. Because some of these lesions may resemble metastases to the thymic region histologically, one must strictly require that clinical attention is given to the possibility of an occult malignancy elsewhere before a definitive diagnosis is made.
Grossly, thymic carcinomas usually lack the encapsulation or internal fibrous septation that is seen in thymomas (317). They are firm to hard (sometimes gritty) and white-gray on the cut surface, with frequent foci of necrosis and hemorrhage. A particular variant of thymic carcinoma (the basaloid type, discussed earlier) may demonstrate a close association with remnants of a multilocular thymic cyst on macroscopic examination (93,94). Other types—mucoepidermoid and mucinous thymic carcinomas—often have a mucoid cut surface and a “slimy” consistency (93) (Fig. 28.72).
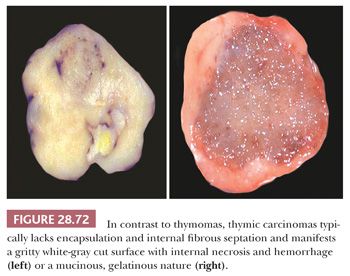
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


