(1)
Faculty of Life Sciences, Bar-Ilan University, Ramat-Gan, Israel
Keywords
NADH monitoring fluorometersHemodynamic artifactsNADH calibration procedurePreparation for NADH monitoringProbe fixation to organs4.1 Introduction
As described in Chap. 3, NADH could be measured by utilizing its absorption spectrum in the UV range, as well as by the blue fluorescence spectrum emitted under UV illumination. In the early stages, NADH monitoring was based on the difference in the absorption of NADH and NAD+. At the range of 320–380 nm, only the reduced form, NADH, absorbs light, whereas NAD+ does not (Fig. 3.3c). Therefore, when a mixture of NADH and NAD+ is illuminated in a cuvette by 320–380 nm, only NADH will affect the absorption spectrum peak at 340 nm. This property of NADH was used in the early 1950s by a number of investigators, as reviewed in Sect. 3.1. Chance and collaborators utilized this technique to measure NADH in muscle homogenates or intact cells [1] and published a large number of papers concerning the unique absorption spectrum of NADH.
The absorption approach is not practical for measuring NADH in a thick tissue; hence, another property of NADH was used. Since the early 1950s, fluorescence spectrophotometry of NADH has been employed in various in vitro and in vivo models. The emission of NADH fluorescence, under illumination at 320–380 nm, has a very wide spectrum (420–480 nm) with a peak at 450–460 nm (Fig. 3.3d). NADH fluorescence has been identified by Chance and his collaborators as a good indicator of the intramitochondrial oxidation–reduction state [2].
The main technology used for NADH excitation involved one-photon excitation supplied by the 366-nm strong band of the mercury (Hg) lamp. It was necessary to avoid damage to the tissue by the UV light as well as to correct the signals for light scattering and absorption by the cellular environment. All the papers cited in this book reported the use of one-photon excitation technology. Nevertheless, in the past 20–25 years, another developed technology enabled using the two-photon excitation light for the measurement of NADH fluorescence [3]. Huang et al. in 2002 [4] applied this technology to measure NADH and Fp fluorescence in isolated single cardiomyocytes. The importance and significance of the two-photon technology was discussed in the next few years [5–7]. Takano et al. in 2007 [8] and Carlson et al. in 2012 [9] used the two-photon technology to study metabolic responses to cortical spreading depression. Tiede et al. [10] measured cochlear bioenergetics using the two-photon technology. Yu and Heikal [11] measured NADH concentration by two-photon technology in normal and cancer breast cells. The effect of intestinal ischemia was measured by in vivo imaging [12]. Multiphoton imaging was tested in vivo during acute kidney injury [13]. The effect of hemodynamic artifacts on brain NADH fluorescence measured by two-photon technology was tested by Baraghis et al. in 2011 [14]. A few more studies used the two-photon technology to study brain metabolic activity under various conditions such as hypoxia [15–17]. The group working with Bruce Tromberg used the two-photon technology in studying melanocytes [18] or keratinocytes of human skin [19]. Recently, Drozdowicz-Tomsia used two-photon fluorescence lifetime to study the ratio between free and bound NADH under metabolic inhibition [20].
4.2 Old Types of NADH Fluorometers
Since the early 1950s, different groups have developed a number of fluorometers adapted to their specific experimental protocols. At present, most of the groups using NADH fluorometers construct the devices in house because no suitable commercial products are available. During the past four decades, the workshop of the Johnson Research Foundation at the University of Pennsylvania Medical School (Philadelphia, PA, USA), headed by Prof. Britton Chance, has manufactured a few types of laboratory instruments purchased and used by various investigators [21].
The basic features of NADH fluorometers consist of the following components:
1.
A light source (including appropriate filters)
2.
An optical path to the preparation and back to the detection unit
3.
Detection and signal processing units
4.
Signal recording and storage units
In our earlier review published in 1984, we extensively specified the light guide-based fluorometry used in our studies [22].
The review article on in vivo NADH fluorescence monitoring, published in 1992 by Ince et al. [23], included many other technical aspects of the methodology. Nevertheless, here we elaborate on the historical development of the various models of NADH fluorometers.
In the present chapter, we do not recommend the use of any type of fluorometer–reflectometer but rather provide the reader with extensive reference information, gathered during the years, on the construction of fluorometers. Although a new type of NADH fluorometer based on a very small and stable UV light source, 375-nm LED, was reported [24], the chronological development is given here.
Priority is given to articles mainly concerned with NADH monitoring in vivo, although in vitro fluorometer types are also considered. Duysens and Amesz [25] schematized the first fluorescence spectrophotometer used for intact cells. They utilized the “classical” light source, the mercury arc, providing a very strong band at 366 nm, although not at the maximal NADH absorption peak of NADH (340 nm). Using a monochromator, they were able to obtain the NADH fluorescence spectrum in baker’s yeast cells and photosynthesizing cells. They concluded that “the fluorescence excited by 366 nm can be used for measuring reduced pyridine nucleotide in vivo.”
In 1959, Chance and Legallais [26] described a differential fluorometer that heralded a new era in monitoring NADH fluorescence in vivo as an indicator of mitochondrial function. They used a microscope, serving as the fluorometer basis, with two light sources: tungsten and mercury lamps with appropriate filters. In 1959, Chance and Jobsis [27] and Chance [28] proved that mechanical muscle activity is associated with NADH oxidation measured in excised muscle. This study was the bridge from the subcellular (mitochondria) and cellular (intact cell) monitoring approaches toward actual in vivo applications.
The first in vivo NADH monitoring device was presented in the late 1950s and early 1960s. At that stage, the effects of scattered light and tissue absorption from blood were not taken into consideration when monitoring NADH fluorescence. The first detailed results of in vivo NADH fluorescence measurements were published in 1962 [29].
These “classical” papers described two micro-fluorometers that were modifications of previous designs [30, 31]. This micro-fluorometer (Fig. 4.1a, b) type employed Leitz “Ultrapack” illumination, which had been used for many years by various groups until the appearance of UV-transmitting optical fibers. To avoid movement artifacts, rats were deeply anesthetized and their heads were fixated in a special holder attached to the operation table. Numerous studies utilized the principles of the “Ultrapack” illumination system. The same instrumentation was used in other in vivo studies, including those of Chance’s group [30, 32–37], Dora and Kovach’s group [38, 39], Rievich’s group [40, 41], Jobsis and collaborators [42–47], Schuette et al. [48], Gosalvez et al. [49, 50], and Anderson and Sundt [51–53] (this is only a partial list).

Fig. 4.1
a Microspectrofluorometer developed by Chance et al., used in the 1960s. In addition to the interference filter, a Wratten type 2C filter is also placed in the back aperture of the objective. The wavelength range of the interference filter is 400–700 mμ, and the specification on its spectra interval is 30–40 mμ. Other features of the high-pressure mercury arc excite the fluorescence of the specimen at 366 mμ by means of an “Eppendorf” primary filter. Fluorescence excitation and emission pass through the Leitz Ultrapak objective (×11) and a ×3 ocular. (© Reprinted by permission from Macmillan Publishers Ltd. [30].) b Photograph of experimental setup used for simultaneous recordings of kinetics and fluorescence changes in kidney and liver of anesthetized rat. Fluorometer on left is employed for measurements of the kidney; that on right for measurements of the liver. Rat is anesthetized with urethane, with cannulation for trachea and femoral vein. Details of the fluorometer described in text. (© Reprinted with permission from Elsevier [149])
The same type of instrumentation was used by various groups for the measurement of NADH in single cells [54, 55] or in vitro preparations [56–59]. A new model of time-sharing spectrofluorometer and fluorometer was developed at the Johenson research foundation in 1971 [60]. In 1986, Hassinen reviewed the use of surface fluorometry for measuring the redox state of NADH in intact tissues [61].
4.3 Monitoring NADH Fluorescence and Reflectance
The effect of blood on NADH fluorescence was discussed early by Chance et al. [29]. To monitor NADH in vivo, Chance’s group had to avoid areas containing large blood vessels that interfere with the emission and excitation light. The monitoring of a second parameter in tissue fluorometry in vivo was reported by Chance et al. in 1963 [32]. They showed that “changes due to the deoxygenation of oxyhemaglobin do not interfere with measurement of the time course of fluorescence changes in the tissue studies.”
The addition of a second monitoring signal, namely, tissue reflectance at the excitation wavelength, was reported in 1968 by Jobsis and Stansby [62]. It was based on a previous model described by Jobsis et al. in 1966 [63]. In another two papers by Jobsis and collaborators [44, 45], the measurement of 366-nm reflectance was used for the correction of the NADH fluorescence signal from the brain (Fig. 4.2a). The reflectance signal was subtracted from the fluorescence signal [26]. The same type of fluorometer was used in by the Hungarian group (Fig. 4.2b) [64].

Fig. 4.2
a Final form of regulated, differential fluorometer. The 366-nm line from the water-cooled mercury arc lamp is transmitted by the primary filter (F1) after previous removal of IR radiation by a heat filter within the arc lamp housing (not shown). A small fraction of this light passes to the regulating photomultiplier (PM 1). The excitation light is focused as a cone on the preparation. Diffusely reflected excitation light and emitted fluorescence enter the microscope optics, are divided by the beam splitter, and measured after passage through the secondary filter, F2 or F3, which allow passage of the 366-nm and the 465-nm regions, respectively. (© American Physiological Society, reprinted with permission [44].) b Schematic presentation of the fluoro-reflectometer used by the group established by Kovach in Hungry. (© American Physiological Society, reprinted with permission [64])
4.4 Fiber-Optic Fluorometer–Reflectometer
To enable the monitoring of NADH fluorescence in unanesthetized animals or other in vivo preparations, a flexible means was needed to connect the fluorometer with the tested organ, for example, the brain. This method was achieved in 1972, when UV-transmitting quartz fibers became available (Schott Jena Glass, Germany). We have used the light guide-based fluorometer for in vivo monitoring of the brain [2, 65] subjected to anoxia or cortical spreading depression. The historical development of light guide-based fluorometry–reflectometry is shown in Fig. 4.3. The original device functioned on the time-sharing principle (Fig. 4.3a) in which four filters were placed in front of a two-arm light guide. Filters 1 and 3 enabled the measurement of NADH fluorescence, and filters 2 and 4 were used to measure tissue reflectance at the excitation wavelength. The reflectance trace was used to correct the NADH signal for hemodynamic artifacts and to indicate changes in the blood volume of the sampled tissue.

Fig. 4.3
Structure of two main types of fiber-optic-based fluorometers: time-sharing (a) and standard DC (c) fluorometer–reflectometer. d Effect of monitored tissue volume (diameter of fiber-optic probe) was tested under anoxia as shown in upper three traces (2-mm diameter) and lower three traces (1-mm diameter). (© Reprinted with permission from Elsevier [22].) In b, the fiber-optic probe is connected to the brain of a small animal. (© Reprinted with kind permission of Springer Science + Business Media [66])
In this original system, only one photomultiplier tube was used for the detection of the two signals. Figure 4.3b presents one of the first in vivo brain monitoring time-sharing setups, connected to the brain of an anesthetized rat [66]. To simplify the monitoring system, the time-sharing approach (AC mode) was replaced by splitting the light emitted from the tissue into two unequal fractions for the measurement of fluorescence and reflectance signals. This model, named the DC-type fluorometer, had originally a three-way light guide that was later replaced by a two-arm light guide probe (Fig. 4.3c). In all the three configurations, the reflectance signal was used for the correction of the fluorescence signal (see details in Sect. 4.5). The model shown in Fig. 4.3a, was used to study the brain [65–70] and kidney [71]; the model shown in Fig. 4.4c was used to monitor the heart [72], brain [73–75], and kidney [76, 77].

Fig. 4.4
a The MitoViewer device uses 365-nm UV LED for excitation of NADH fluorescence (fluor signal). This excitation light is also utilized for the correction of hemodynamic artifacts by measuring reflection light intensity at the same excitation wavelength of 365 nm (Refl signal). Light is transmitted from and to device by flexible fiber-optic probe. Software displays reflectance, fluorescence, NADH-corrected fluorescence signal, and photodiode (PD) signal. b General view of the MitoViewer
Our group developed and used the model shown in Fig. 4.3c in the late 1970s. This model is still being used in our laboratory to monitor the brain [78–81], heart [82–84], liver [85], and kidney [86–89], and also in multisite or multiorgan monitoring [78, 90–94]. The responses to anoxia using different diameters of the fiber-optic bundle are shown in Fig. 4.3d [22].
Other groups used optical fibers to connect the monitored tissue to the fluorometer differently than the three models shown in Fig. 4.3. Renault and collaborators used a light guide fluorometer for monitoring a heart in vivo [95–99]. Fink, Rex, and collaborators used another type of light guide fluorometer for the brain and other systems [99–102]. Only micro-light guides were used for in vitro and in vivo studies [103–107]. Another approach was developed by Raman et al. [108], who constructed a noncontact method to monitor the kidney during ischemia.
4.4.1 The “MitoViewer”
A new fluorometer–reflectometer device (MitoViewer) was developed in 2007 by an Israeli company, Prizmatix (Fig. 4.4a, b.
The MitoViewer system contains the following main subunits:
1.
Light source unit: provides UV light at 365 nm for NADH fluorescence excitation and tissue reflection measurements. Also included is a reference photodiode that enables correction of signals for intensity changes of the LED during measurement.
Fiber-optic probe: transmits the UV light from the light source unit to the measured tissue site and transmits the collected light (the reflection, Refl) and the fluorescence (Fluor) from the tissue to the detection unit.
2.
Detection unit: provides detection to transform the reflectance and fluorescence light into electrical signals that are transmitted to the electronics board for amplification.
Electronics unit: provides amplification for the reflectance and fluorescence signals as well as control of the detector gain and driver for the UV LED. The amplified signals are transmitted to an analog to digital convertor module. A USB-6009 module provides analog to digital (A/D) conversion for the two signals and digital to analog (D/A) conversion for control signals sent from a PC to control the function of the MitoViewer.
3.
PC: a personal computer enables control of the MitoViewer operation through the MitoViewer software.
4.
Power adaptor: provides the DC voltage for operation of the MitoViewer.
The fiber-optic probe of the MitoViewer was attached to the surface of the brain via an appropriate holder cemented to the skull with dental acrylic cement. We used rats (200–250 g) anesthetized and operated as discussed in the various papers that appear in the reference list.
Figure 4.5a presents typical responses, measured by the MitoViewer, of the brain to complete oxygen depletion by exposing the rat to 100 % nitrogen. The fluorescence signal (blue) is elevated by inhibition of respiratory chain activity. The reflectance signal (green) is decreasing, as expected under this condition, because of the elevation in blood volume in the monitored tissue. The corrected NADH signal (black) shows a symmetrical increase and decrease during the anoxic cycle.

Fig. 4.5
Typical responses to metabolic perturbations measured in the rat brain using the MitoViewer: responses to anoxia (a), responses to cortical spreading depression (b), and responses to hypoxia and hyperoxia (c)
The effects of the increase in energy consumption (Fig. 4.5b) were induced by exposure of the brain to cortical spreading depression (by a high level of potassium.). In the normoxic brain the oxygen supply is not limited, and accordingly the fluor (blue) and the NADH (red) decreased because of oxidation of NADH. Under this condition the ATP turnover was dramatically increased, and the extra oxygen supply was provided by an increase in microcirculatory blood flow. The effects of hypoxia (6 % oxygen and hyperoxia (100 % oxygen) are shown in Fig. 4.5c.
In the past few years, the MitoViewer was incorporated in studies in which the NADH was monitored in real time and compared to various parameters including tissue oxygen partial pressure. These studies were performed on rat skeletal muscle exposed to hypoxemia [109, 110] or graded hemorrhage [111, 112].
4.5 Factors Affecting NADH Fluorescence and Reflectance Signals
The excitation and emission spectra of NADH are affected by the redox state of this fluorochrome and by other factors, leading to artifacts in the fluorescence measurements. In this section, various NADH unrelated factors affecting the measured signal are discussed. Because most fluorometers involve the measurement of the total backscattered light at the excitation wavelength (i.e., 366 nm), the discussion concerns changes in NADH fluorescence as well as in tissue reflectance.
The following factors may affect the two measured signals: 366 nm reflectance (R) and 450 nm fluorescence (F):
1.
Tissue movement caused by mechanical or intracranial pressure changes in the brain.
2.
Extracellular space events, such as volume changes or ion shifts between intra- and extracellular spaces.
3.
Vascular and intravascular events, for example, oxy-deoxy Hb changes, and blood volume changes caused by autoregulatory vasoconstriction under pathological conditions.
4.
Intracellular space factors, such as O2 level, ATP turnover rate, substrate availability, and mitochondrial redox state, as discussed later in this chapter.
4.5.1 Movement Artifacts
Using the fiber-optic technique, we found that when there is a good contact between the bundle of fibers and the monitored tissues, such as the brain, all movement artifacts are eliminated. We also found that to obtain a good signal-to-noise ratio, as well as a reliable and repeatable measurement, good contact between the fibers and the tissue is required during the entire monitoring period. If a small space is left between the fibers and the tissue, the signal will not be greatly affected, but responses will not be observed. Pressure on the brain must be avoided, and we solved this problem by using a special light guide holder cemented to the skull [65, 67]. In monitoring other organs, a micromanipulator can be used to hold the light guide above the organ [90, 93, 94]. In the heart, this problem has been solved by using a light guide-holding cannula connected to the heart muscle by three sutures [82, 113–115]. In brain preparations, even while the animal was undergoing hyperbaric convulsions or decapitation, only very small artifact changes in the traces were measured from the brain, indicating that the movement has only a negligible effect on NADH measurements. The same approach was applied previously [38, 116–118], in anesthetized and/or artificially ventilated rats or cats, using the “Ultrapac” optics for brain monitoring. Another option is to glue a light guide holder by cyanoacrylate glue when monitoring exposed organ surfaces [85, 88].
To avoid movement artifacts in monitoring patients, we used diverse approaches. In the neurosurgical intensive care unit, we used a metal holder screwed to the skull of comatose patients [33]. In the operating room, we used a floating light guide probe fixed to the head holder in neurosurgical procedures [119–121], or a ring is used to hold retractors during abdominal operations or kidney transplantations [89].
4.5.2 Intracellular and Extracellular Space Events
The second factor that can be a potential source of error in NADH fluorescence measurement is a change in the absorption properties of the tissue, during various perturbations, at the monitored site. This artifact has been mainly recognized in brain studies. Very little is known and published about this factor because it is impossible to separate it from other factors affecting the NADH fluorescence readings. It seems that under physiological or pathological conditions involving ions and water movement between the intracellular and the extracellular space, this factor may have a greater effect on NADH fluorescence measurements. We have earlier published indirect evidence for the involvement of this factor in our measurements and a possible correction method
(a)
When the blood was eliminated from the brain, using a fluorochemically perfused brain preparation [122], only very small, if any, changes in the reflectance signal were measured during the anoxic cycle. As shown in Fig. 4.6a, the uncorrected NADH fluorescence and the corrected fluorescence (CF) had similar kinetics. Furthermore, the CF response of the perfused brain to anoxia was similar to that of the blood-perfused brain in the same animal before the initiation of perfusion. However, when a spontaneous spreading depression (SD)-like response was recorded in several perfused brains, the reflectance tracing showed a change typical of the initial increase phase of the regular response to SD found in a normoxic blood-perfused brain (for details, see [122]). The second phase of the decrease in R during SD did not occur in the perfused brain because of the lack of blood in the system (Fig. 4.6b). We believe that this initial increase in R results from water and ion movement during the SD event in the perfused brain. The same type of R response (an increase) was obtained when a brain slice in vitro was stimulated [123].

Fig. 4.6
Responses to “ischemia” measured after perfusion with fluorochemical emulsion (a, b). In the perfused brain, anoxia was induced by closing the perfusion pump. The time interval between a and b was 5 min. The perfusion pressure was not calibrated and served as a qualitative parameter. (© Maney Publishing; reprinted with permission [122])
(b)
An interesting correlation was found between the R trace and pO2 monitored from the gerbil brain subjected to spreading depression [124]. As seen in Fig. 4.7a, in the awake state the biphasic change in R, under SD, is recorded as described here. However, under deep anesthesia, the two phases were separated, and the initial increase was not correlated to pO2. The second decrease in R was directly correlated with the changes in pO2 that resulted from blood volume alterations during SD. It seems that this initial increase phase of R is also caused by water and ion shifts during the initiation and propagation of the SD wave and is not correlated to the recovery phase.

Fig. 4.7
a Responses to cortical spreading depression measured in normal brain, perfused brain, and anesthetized rat brain. b Effects of ischemia induced in rat brain monitored by multiparametric monitoring system. (© Reprinted by permission from Macmillan Publishers Ltd. [150])
(c)
In experiments where complete cerebral ischemia was induced by decapitation in rats and gerbils [66, 125, 126], bilateral carotid occlusion in rats [127], or four-vessel occlusion in rats [125], a large reflectance increase was recorded after NADH reached its maximum level (pO2 = 0). This increase was termed secondary reflectance increase (SRI). In recent experiments, we have monitored the DC potential as well as K+ from the extracellular space, together with NADH fluorescence and 366 nm reflectance. In all cases, we found a very significant correlation between the complete depolarization occurring during complete ischemia (as identified by the DC and K+ changes) and the SRI phenomenon (Fig. 4.7b). It seems that the SRI is partly caused by the water and ion movements during complete depolarization, and it may be also caused by a decrease in tissue blood volume as a result of a spasm of blood vessels. In several cases, the SRI was very large (significantly) and the regular correction technique was insufficient. We have found that under partial ischemia in gerbils, with SRI occurring, a decrease in blood flow and pO2 was recorded, indicating a possible massive vasoconstriction response that might be responsible for the SRI.
4.5.3 Vascular Events
These events include changes in blood oxygenation, namely, in the saturation level of HbO2, as well as changes in the blood volume in the monitored microcirculation.
4.5.3.1 Blood Oxygenation
Because hemoglobin (Hb) is a strong light absorber at various wavelengths, the measurements of NADH are affected by the amount of Hb in the monitored tissue. Kramer and Pearlstein [128] attempted to use the hemoglobin isosbestic point (448 nm) to correct the NADH measurement for changes in the [Hb]/[HbO] ratio. However, the attempt was unsuccessful and without any continuation, as their paper only presented preliminary results. To test the effect of Hb concentration in the tissue on NADH fluorescence spectrum, Rahmer and Kessler used perfused rat liver, and found that higher concentrations of Hb corresponded to a lower intensity of the fluorescence spectrum emitted from the liver [129].
Coremans et al., in their measurements of NADH fluorescence and light reflectance (365 nm), tested the effects of NADH and hemoglobin on the two signals in a tissue phantom model [130]. They concluded that the ratio between the two signals (F/R) provides a good corrected signal.
In transition from oxygenated blood (HbO2) to deoxygenated blood, the absorption spectrum is different and may affect the NADH signal. The oxy-deoxy hemoglobin transition and its effect on the measured signals have been discussed previously [2], and this effect has been shown to be negligible. Indirect studies produced similar results:
(a)
In decapitation (Fig. 4.8a), only a small change, if at all, was measured in the reflected light (R), although the hemoglobin present in the measured field was rapidly losing O2. This finding indicates that the oxy-deoxy Hb transition has only little effect on NADH fluorescence and on the reflected light.

Fig. 4.8
Effects of oxygen depletion induced by decapitation (a) and anoxia (b–d) in the rat brain. (© Reprinted by permission from Macmillan Publishers Ltd. [150])
(b)
In comparing the normoxic–anoxic transition to the anoxic–normoxic change (induced by breathing N2), the different kinetics in the R traces suggests that blood oxygenation does not have a significant effect on the CF, when using 366-nm reflectance changes for correction (Fig. 4.8b). The decrease in R trace after initiation of anoxia has very fast kinetics (it takes less than 1 min to reach the minimum R level). However, during reoxygenation, the CF has very fast kinetics, and it takes 5 min or more for R to reach the baseline, indicating that the oxy-deoxy transition has only little effect on the two measured signals (R, F).
(c)
In our previous publications, we have shown that during anoxia in a normal rat, a two-step decrease in reflectance occurs [65, 67]. First, there is a small decrease in R, which is followed by a very large decrease when NADH reaches its maximum level. This secondary decrease in R during anoxia did not appear (Fig. 4.8c) if the brain had previously been made partially ischemic by bilateral carotid artery occlusion for 24 h [81]. The corrected fluorescence (CF) responses, however, were about the same. Furthermore, when N2 cycles of 1 min were applied to the same rat every 10 min (Fig. 4.8d), we found that the reflectance responses decreased in time, whereas the corrected fluorescence showed the same response to anoxia [65]. This response occurred even though the oxy-deoxy hemoglobin change probably took place in all N2 cycles.
4.5.3.2 Blood Volume Changes
Blood volume changes may occur as a response to various physiological and pathological conditions. This is the main artifact in monitoring tissue NADH fluorescence, and it has been discussed by many investigators.
During the past 30 years, we have used fiber-optic surface fluorometry to monitor the brain exposed to various conditions, as well as other organs such as the heart [82, 131]. Hence, evidence for the involvement of blood volume artifacts and their correction have been drawn from various published experiments.
(a)
The typical decrease in R during brain anoxia can be corrected to a level of 90–100 % depending on the microcirculatory pattern of the site under observation. When hemoglobin was eliminated from the brain (perfused with a fluorochemical), no blood volume changes could occur, and indeed no changes in R (Fig. 4.6a) were observed [122]. In the heart, anoxia did not have a great effect on R and the observed change was in the same direction as in the brain [82]. In a partially ischemic brain [132] or when N2 cycles were repeated many times [65], the typical decrease in R during anoxia was also minimal (Fig. 4.8d) as a result of the low capacity of the brain to increase its blood volume to compensate for low pO2.
(b)
In another set of experiments, we monitored NADH during hyperbaric oxygenation of the brain [133, 134]. We found that when the animal was exposed to compressed 100 % O2, a large increase in R was recorded. This finding was explained by a decrease in blood volume in the brain caused by the vasoconstrictive response of the blood vessels to high pO2. When the compression mixture contained 1.5 % CO2 at 5–6 ATA, the reflectance trace (Fig. 4.9b) showed a sharp decrease from the vasodilation response occurring under high pCO2. The CF showed the same level of oxidation. These findings suggest that CF can be corrected for blood volume changes induced by high pO2 or high pCO2 in the brain, using the 1:1 subtraction technique.
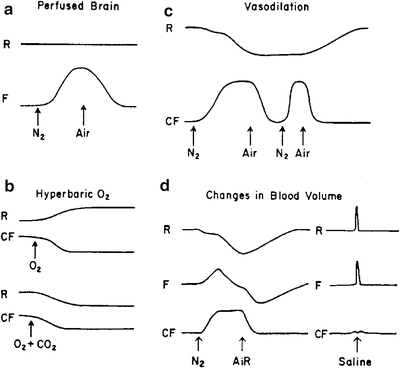
Fig. 4.9
Effect of changes in blood volume on responses of parameters monitored by NADH fluorometer. a Effect of anoxia in the perfused brain. b Responses to hyperbaric oxygen. c Effect of repeated anoxia event. d Changes in blood volume induced by anoxia or saline flush to the brain. (© Reprinted by permission from Macmillan Publishers Ltd. [150])
(c)
In studying the effects of unilateral or bilateral carotid artery occlusion in gerbils, the observed R changes were in the same direction [124]. Under unilateral ischemia induction, the R trace showed a small decrease (or no change) because of the increase in blood volume through the open artery. However, when complete ischemia was induced (by additionally occluding the other artery), an increase in R was recorded in the preparations, indicating that a decrease in blood volume may lead to the expected increase in R.
(d)
A decrease in blood volume was induced by saline injection into the ipsilateral common carotid artery (Fig. 4.9d, left), or into the brachial artery (Gyulai and Mayevsky, unpublished results). The results showed that the increases in R and F were similar and varied in the range of 10–20 % between various rats or in the same animal.
4.6 Principles of NADH Artifact Correction
To compensate for the various factors affecting the NADH fluorescence signal, various approaches have been developed. In the paper published by Ince et al. [23], the various correction techniques were listed and discussed in detail (Fig. 4.10). The reference numbers that appear in the table itself are listed here in the following order (from top to bottom): [41, 69, 90, 39, 45, 136, 105, 137, 95, 128, 138, 139].

Fig. 4.10
Overview of compensation methods employed for assessment of NADH fluorescence from measured blue fluorescence. F λ and R λ , fluorescence and reflectance intensities at wavelength λ; CICF 5(6)-carboxy-2′,7′-dichlorofluorescein, k, k 1 constants. (© Reprinted with kind permission of Springer Science + Business Media [23])
It appears that the correction technique is also dependent on the instrument configuration [41, 140]. Most of the published materials are based on the 1:1 correction factor or ratio, when subtracting the 366-nm reflectance signal from the fluorescence signal.
To determine the “correction factor” value, the use of saline flushes toward the brain was the main approach. In Fig. 4.11 the results obtained in the cat brain are shown [41]. When a bolus of saline is injected into the brain via the carotid artery a fast response in the reflectance and fluorescence signals are recorded (Figs. 4.11Aa, Ab). The details appear in the legend to the figure. In part B, the correlation between the percent change in the reflectance and fluorescence signals is shown. The same approach was used by the group in Hungary that performed a systematic study on this issue [116].
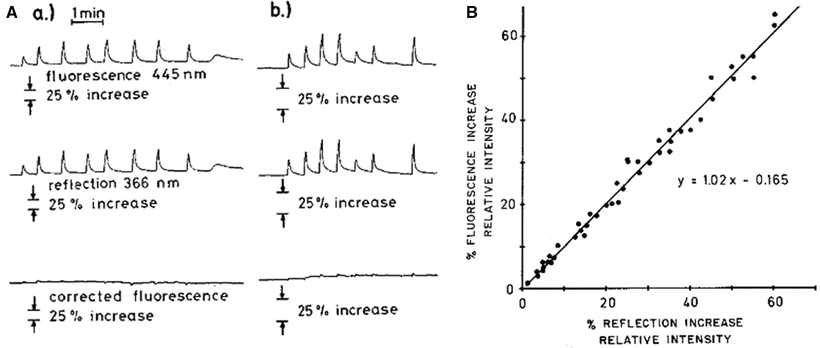
Fig. 4.11
(A) Saline flushes into carotid artery on fluorescence and reflectance signals measured in cat brain. Every spike represents one flush. Hemoglobin concentration in the field of observation is decreased, which results in increase of both fluorescent and reflected light. The third channel gives the subtraction of the first two channels, the ratio between the two being one. (B) Relationship between reflected ultraviolet light and emitted fluorescent light during 75 different saline flushes in same animal used for a: gain is double that of a. (© American Physiological Society, reprinted with permission [41])
A similar study was performed in the rat model by our group (Fig. 4.12) [141]. The saline flushes were done into the left carotid artery and the response was recorded in the left hemisphere (Fig. 4.12a). The correlation between the changes in the R and the F signals measured in five rats are presented in Fig 4.12b.

Fig. 4.12
(a) Effects of saline bolus injections (arrow) into left carotid artery on reflectance (R), fluorescence (F), and corrected fluorescence (CF) measured from the two hemispheres of rat. (b) Effects of saline bolus injection into common carotid artery on percentage change in reflectance (R) and NADH fluorescence signals (F) in five rats. Each symbol represents a different animal. (© Maney Publishing; reprinted with permission [141])
As of today, a new approach is still lacking to compensate for non-NADH factors affecting the NADH fluorescence signal. Bradley and Thorniley [142] published a review article covering the various correction techniques for tissue fluorescence. They summarized their review by the following conclusion: “even though research has been conducted into correction techniques for over 30 years, the development of a successful and practical correction technique remains a considerable challenge.”
4.7 Calibration of NADH in Solution
The effect of NADH concentration in solution on the intensity of the fluorescence signal was studied by various investigators. It is important to run such a study when a new monitoring system is introduced to run in vivo protocol. Two examples published in the past are shown in Fig. 4.13.

Fig. 4.13
a Fluorescence intensity versus NADH concentration plot: time-averaged result and result after normalization to laser intensity. (© Walters Kluwer Health, reprinted with permission [151].) b Fluorescence intensity as a function of concentration of β-NADH solution. Photomultiplier tube output at 450 nm: λ ex = 340 nm. (© Walters Kluwer Health, reprinted with permission [152])
This type of calibration is necessary when a new commercial device is developed for patient monitoring and needs clearance by the FDA.
CritiSense Ltd. developed such a device (the CritiView) and was able to get the FDA clearance a few years ago. The details of this procedure are described in the next few paragraphs.
4.7.1 Aims of the Study
To demonstrate the efficiency of the CritiView device for the measurement of NADH concentration, in two steps:
Demonstrate the correlation between the NADH concentration in solution and the fluorescence intensity measured by the CritiView.
Compare the correlation between NADH fluorescence in solution measured in this study by the CritiView to that measured by a clinical fluorometer approved by the FDA
(Victor2 1420 DA).
4.7.2 Basic Experimental Approach
The ability of the CritiView device to quantitatively measure NADH fluorescence comparable to a control NADH solution and to a clinical fluorometer (“Victor2”1420) was demonstrated by direct fluorescence measurements in aqueous NADH solutions.
An aqueous solution of NADH is a suitable simulation of fluorometric analysis of tissue because the principles of in vitro NADH monitoring are identical to in vivo monitoring.
A universal agreement exists on the characteristics of NADH spectra and their biochemical significance when measured both in vivo and in vitro. The intensity of the fluorescence band is independent of the organizational level of the environment and is proportional to the concentration of NADH (reduced form). As such, the ability to measure a NADH fluorescence signal of aqueous NADH solutions at various concentrations is presented here.
4.7.3 Experimental Methodology
The experimental methodology is straightforward. Various solutions of NADH were measured with the CritiView. The in vitro comparison was performed using NADH aqueous solutions.
Insofar as the measurement with the CritiView involves placing its probe tip into the solution, one might ask if this measurement is the same as placing the tip of the probe against a tissue surface. The equivalence of these two measurements can be understood if one considers that although immersion in aqueous solution and placement adjacent to tissue are, clearly, two different physical situations, nevertheless, the process and principle of measurement that takes place is the same for both. Aside from their contiguity, there is no interaction between the probe and the solution or any adjacent tissue. The probe merely collects emitted light from its immediate environment, that is, a hemispherical volume adjacent to the probe tip, and transfers the collected light to detectors for measurement. As previously mentioned, the principles of in vitro NADH monitoring are identical to in vivo monitoring, and the intensity of the fluorescence band is independent of the organizational level of the environment and is proportional to the concentration of NADH (reduced form).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


