(1)
Faculty of Life Sciences, Bar-Ilan University, Ramat-Gan, Israel
Keywords
DPNHPyridine nucleotidesNADH fluorescence spectraUV light absorption by NADHState 4–3 transitionIsolated mitochondriaIn vivo NADH spectrum3.1 Introduction
It is now 60 years since the significant work of Chance and Williams on mitochondrial metabolic state in vitro (Fig. 3.1) was published [1–4].

Fig. 3.1
The definition of mitochondrial metabolic state, in vitro, in 1955, by Chance and Williams, opened up a new era in spectroscopic measurements of the respiratory chain enzyme redox state in vitro as well as in vivo. Detailed explanation of the table is given in Sect 7.1. (© John Wiley and Sons, reprinted with permission [33])
The discovery of the pyridine nucleotides was made by Harden and Young almost 110 years ago [5, 6] and was followed by the description of its full structure by Warburg and collaborators 30 years later [7]. All those historical studies led to the first detailed experiments, by Chance et al. [8], in which NADH (nicotine amide adenine dinucleotide) fluorescence, which was recognized to be a component of Keilin’s respiratory chain, was used as a marker of mitochondrial function of the brain and kidney in vivo in anesthetized animals.
3.1.1 Historical Introduction: Written by Prof. Britton Chance in 2006 (The next three Paragraphs)
We start by with a citation from Chance et al. in 1973 [9], “The accumulation of evidence since the pioneer work of Otto Warburg an ‘Atmungsferment’ [10] and David Keilin on cytochromes [11, 12] as the keystones of cellular oxygen utilization led us to the study of the redox states of electron carriers in isolated mitochondria as a function of oxygen concentration and to develop techniques to measure the states of anoxia and normoxia in living tissues.”
Although Warburg isolated reduced diphosphopyridine nucleotide (DPNH) and reduced triphosphopyridine nucleotide (TPNH), he was apparently reluctant to imagine that those molecules were part of the respiratory chain. He also studied their optical properties and identified the 340-nm absorption band as being characteristic of the reduced form. The idea that these might also belong to Keilin’s respiratory chain [12] was not considered and, in fact, it was to some extent abhorrent to Warburg to imagine that he should support Keilin’s views.
Thus, the observation made spectroscopically in Chance’s difference spectrum of the respiratory chain showing prominent peaks of what was soon to be called NADH and a trough resulting from flavoproteins, and finding that they were, respectively, more than 40 and more than 10 times greater concentration than Keilin’s cytochrome c was, in a sense, an overwhelming blow to Keilin. In my (Britton Chance) close contact with him, he never wished to discuss these two components as new and overwhelmingly present members of the “respiratory chain” (taking note that the Keilin and Hartree preparation [13] was carried out in such a way that no trace of NADH survived the rigorous sand grinding and acid extraction). In fact, personal contact with Keilin subsequent to his award of the Copley Medal evoked no response whatsoever about these novel components, which certainly must have been prominent in his mind. Nor in any conversation with him did he discuss Otto Warburg’s discovery of these components.
Table 3.1 presents the historical milestones in the development of NADH monitoring after its discovery in 1906 by Harden and Young. As can be seen, most of the milestones were achieved by Prof. Britton Chance. The collaboration with the physiologist, Prof. Frans Jobsis, led to many studies in which various organs in vitro or in vivo were monitored. Most of the studies published in this field were expanded by students and postdocs who worked with Prof. Chance in Philadelphia and then moved to other universities around the world.
Table 3.1
Historical milestones in NADH monitoring
Year | Discovery/Activity | Author(s) |
|---|---|---|
1906 | Involvement of adenine-containing nucleotide in fermentation by yeast | Harden and Young [6] |
1935 | Description of full structure of hydrogen-transferring coenzyme in erythrocytes, naming the two cofactors DPN and TPN | Warburg et al. [7] |
1951 | A shift in the absorption spectrum of DPNH with alcohol dehydrogenase | Theorell and Bonnichsen [17] |
1951 | Development of a rapid and sensitive spectrophotometer | Chance [21] |
1952 | Monitoring of pyridine nucleotide enzymes | Chance [24] |
1954 | Development of double-beam spectrophotometer | Chance [30] |
1955 | The definition and characterization of five mitochondrial metabolic states under in vitro conditions | [3] |
1957 | The first detailed study on NADH using fluorescence spectrophotometry | Duysens and Amesz [44] |
1958 | Measurement of NADH fluorescence in isolated mitochondria | Chance and Baltscheffsky [46] |
1959 | Measurements of muscle NADH fluorescence in vitro | Chance and Jobsis [53] |
1962 | In vivo monitoring of NADH fluorescence from brain and kidney | Chance et al. [8] |
1966 | Comparison between NADH fluorescence in vivo and enzymatic analysis of tissue NADH | Chance et al. [84] |
1968 | Monitoring tissue reflectance in addition to NADH fluorescence | Jöbsis and Stansby [93] |
1971 | The first attempt to monitor the human brain during a neurosurgical procedure | Jöbsis et al. [106] |
1973 | The first fiber-optic-based fluorometer–reflectometer used in the brain of an awake animal | |
1979 | 2-D monitoring of mitochondrial NADH and Fp | Chance et al. 1979 [63] |
1982 | Simultaneous monitoring of NADH in vivo in four different organs in the body | Mayevsky and Chance [108] |
In Table 3.2, I list most of the papers on NADH monitoring published by the investigators in this field: this is not a complete list but gives the reader the main players in the field of NADH monitoring. The contributions of Prof. Chance to most of the studies were significant and appeared in the papers published by other investigators.
Table 3.2
The main investigators that used and published papers describing NADH monitoring in vivo (in addition to Prof. Chance group and Prof. Mayevsky)
Group of authors | Reference number |
|---|---|
Jobsis, O’Connor, Rosenthal, LaManna | |
Williamson | |
Kovach, Dora, Gyulai | |
Barlow, Harken, Harden, Haselgrove | |
Anderson, Meyer, Sundt | |
Reiviich | |
Kedem, Sonn | |
Ince, Coremans | |
Balaban | |
Renault, Duboc | |
Rump, Rosen, Klaus | |
Vern, Lewis, Schuette | |
Lubbers, Kessler | |
Welsh, O’Connor | |
Kohen | |
Ji, Chance, Kessler | |
Thurman, Gosalvez | |
Fink, Rex | |
Hassinen | |
Moravec | |
Masters | |
Thorniley | |
Osbakken | |
Brandes, Bers | |
Shiino, Matsuda | |
Li, Glickson, Nioka |
The blue fluorescence of cells and tissues was recognized from the first studies of its fluorescence microscopy and was termed “autofluorescence.” There was no attempt to characterize it or to do what was obvious to later researchers, which was to see whether oxygen or nitrogen affect the fluorescence, a study that Keilin relied upon to identify his cytochromes (the transition from trivalent to divalent caused a significant change in the electron configuration of the cytochromes).
Because the oxidized form, NAD+, does not absorb light in this range, it was possible to evaluate the redox state of the mitochondrion by monitoring the UV absorbance or blue fluorescence of NADH.
Lubbers in 1995 [14] concluded that “the most important intrinsic luminescence indicator is NADH, an enzyme of which the reaction is connected with tissue respiration and energy metabolism.”
Figure 3.2 presents pictures of the five cited scientists who affected significantly the development of the theoretical and experimental technology for the monitoring of mitochondrial NADH function in vitro and in vivo.

Fig. 3.2
Five scientists significantly affected our knowledge of mitochondrial function and NADH monitoring in vitro and in vivo
3.2 Monitoring of NADH UV Absorbance
The discovery of the optical properties of reduced nicotinamide adenine dinucleotide (NADH; earlier names, DPNH, diphosphopyridine nucleotide, or PN, pyridine nucleotide), has led to very intensive research since the early 1950s. The reduced form of this molecule, NADH (Fig. 3.3a, b) [15], absorbs light at 320–380 nm (Fig. 3.3c) and emits fluorescent light in the 420- to 480-nm range (Fig. 3.3d) [16].

Fig. 3.3
a Structures of NAD+. The nicotinamide group (broken ring) is the “functional” part of both molecules, i.e., the portion of the molecules where oxidation and reduction take place. b Transition between oxidized and reduced NADH. (© Springer Science, reprinted with permission [15].) c Absorption spectra of NAD+ and NADH. d Excitation and emission spectra of NADH. (© Reprinted with kind permission of Springer Science + Business Media [16])
As the oxidized form, NAD+, does not absorb light in this range, it was possible to evaluate the redox state of the mitochondria by monitoring the UV absorbance (this section) or blue fluorescence of NADH (Sect. 3.3).
In 1951, Theorell and Bonnichsen found a shift in the absorption spectrum of DPNH upon addition of alcohol dehydrogenase (ADH) [17]. In the same year, Theorell and Chance described a new spectrophotometric technique for measuring the formation and disappearance of the compound of ADH and DPNH (Fig. 3.4) [18]. This work was based on previous publications that also appeared in 1951 [19, 20]. Moreover, in 1951, Chance and Legallias described a rapid and sensitive spectrophotometer for the measurement of reaction kinetics [21]. A year later (1952), Chance and Castor showed the applicability of this new technique to the measurements of pyridine nucleotide enzymes of muscle homogenate or intact cells [22]. In this study they cast doubt about Warburg’s assumption that the Warburg effect was caused by suppressed mitochondrial function. Immediately afterward (1952), Chance used the same device to monitor respiratory pigments, including NADH, in metabolizing cells [23]. Chance and Neilands (1952) found that the interaction between NADH and lactic dehydrogenase (LDH) of the heart led to a shift in the absorption band [24]. In 1952 and 1953, Chance and Castor showed the dynamics of respiratory pigment behavior (including NADH) in ascites tumor cells [25, 26]. In 1954, Chance and Williams briefly described new sensitive differential spectrophotometric methods applied to the study of reduced NADH in isolated rat liver mitochondria [27]. The same approach was used by Connelly and Chance in monitoring NADH in stimulated frog nerve and muscle preparations [28]. The oxidation of NADH in the muscle was similar to its oxidation in isolated mitochondria upon addition of ADP. In a comprehensive paper on “Enzyme mechanisms in living cells,” Chance described in detail the measurements of the respiratory enzymes, including NADH [29].

Fig. 3.4
Block diagram of the double-beam system for measuring the difference of light absorption at 328 and 354 mμ as well as light absorption at 328 mμ only. All parts of the optical system, except the lamp bulb, are quartz. Mirror and switches vibrate at 60 cps. Symbol –⊳– represents an amplifier. (© Swedish Chemical Society, reprinted with permission [18])
A major milestone in NADH monitoring was the technique presented in 1954 by Chance [30] using a double-beam spectrophotometer to determine the appropriate wavelengths in measurements of respiratory enzymes.
The detailed descriptions of the respiratory chain and oxidative phosphorylation in the mitochondria, published in 1955 by Chance and Williams, established our basic knowledge of mitochondrial function [1, 3, 4, 31, 32]. In the latter five papers, Chance and Williams defined, for the first time, the metabolic states of isolated mitochondria in vitro, depending on the substrate, oxygen, and ADP levels (Fig. 3.1). In addition, they correlated those metabolic states to the oxidation–reduction levels of the respiratory enzymes. The physiological significance of those metabolic states was discussed in 1956 by Chance and Williams [33], and later by Chance and other collaborators [34–38]. In 1959, Chance et al. described a highly sensitive recording micro-spectrophotometer that enabled the monitoring of NADH absorbance in a single cell [39].
3.3 Monitoring NADH Fluorescence
The fact that NADH was monitored by the difference in the absorption spectrum of its reduced form limited the use of that technique to the study of mitochondria in vitro, and in very thin tissue samples (e.g., muscle) or in cell suspension. To provide a method more specific than absorption spectroscopy, fluorescence spectrophotometry in the near-ultraviolet range (UV-A) was applied for NADH measurement. The initial model of fluorescence recorder was described by Theorell and Nygaard in 1954 (Fig. 3.5) [40, 41] and Theorell, Nygaard, and Bonnichsen [42]. Using a modified device, Boyer and Theorell (1956) showed that the fluorescence of DPNH was shifted and the intensity was increased upon combination of DPNH and liver alcohol dehydrogenase-ADH [43]. The first detailed study using fluorescence spectrophotometry of NADH in intact Baker’s yeast cells and algae cells was published in 1957 by Duysens and Amesz [44] (Fig. 3.6).


Fig. 3.5
The first fluorescence recording system developed by Theorell and Nygaard in 1954. (© Swedish Chemical Society, reprinted with permission [41])
Fig. 3.6
a Apparatus for measurement of “fluorescence spectra.” By means of a lens l 1 and a mirror in the mercury arc, a vessel was imaged containing the fluorescing solution or cell suspension. The fluorescing image was focused by lens l 2 on the entrance slit of the monochromator. Filter set l 1 isolated the spectral region of the exciting light; filter set l 2 absorbed the exciting radiation and transmitted light of longer wavelengths. The wavelength drum was driven by a synchronous motor. A sectioned disc d, mounted on another synchronous motor, modulated the incident light. The a.c. component of the current, caused by the light falling on the iP 21 multiplier, was amplified, rectified, and fed into the recorder. b Fluorescence spectra of DPNH. Open circles are for excitation by wavelength 313 mμ; black circles are for excitation by 366 mμ. The spectrum for 313 mμ has been multiplied by a certain factor to make its maximum of equal height as the maximum of the spectrum excited by 366 mμ. The spectra appear to be identical; the maximum is at about 462 mμ. (© Reprinted with permission from Elsevier [44])
In the next 5 years (1958–1962), the monitoring of NADH fluorescence was significantly expanded, led by Chance and collaborators. In a first preliminary study, Chance et al. [45] performed simultaneous fluorometric and spectrophotometric measurements of the reaction kinetics of bound pyridine nucleotides (PN) in the mitochondria. In the same year (1958) Chance and Baltscheffsky presented preliminary results of measuring the fluorescence of intramitochondrial PN (Fig. 3.8b) [46]. In this study, they proved the connection between the mitochondrial metabolic state and the redox state of NADH as measured by spectral fluorometry in mitochondria isolated from rat liver as seen in Fig. 3.8b [3].
The correlation between the enzymatic assay of PN and sensitive spectrophotometry was investigated by Klingeberger et al. using rat liver, heart, kidney, and brain [47].
In 1959, Chance and collaborators were able to expand the use of NADH fluorometry to various experimental models, from isolated mitochondria to intact tissue. Intramitochondrial pyridine nucleotides were analyzed in connection to the ADP-ATP cycle [48]. To monitor NADH localization in intact cells, Chance and Legallais developed a unique differential micro-fluorometer with a very high spatial resolution [49]. This approach was used, in various cells, to identify the intracellular localization of NADH fluorescence signals [50–52]. The next step was to apply the fluorometric technique to the higher organization level of animal tissues. Together with Jobsis, Chance measured in vitro changes in muscle NADH fluorescence following stimulation [53]. In another paper, published by Chance and Theorell [54], the authors came to the very significant conclusion that “The oxidation and reduction state of mitochondrial pyridine nucleotide without a measurable change of cytoplasmic fluorescence suggest that compartmentalization of mitochondrial and cytoplasmic pyridine nucleotide occurs in vivo, at least in the grasshopper spermatid.” In another article, Chance and Hollunger elaborated on the energy-linked reduction of the mitochondrial pyridine nucleotides [55].
Intensive use of the in vivo NADH monitoring approach started in 1962. The “classical” paper on in vivo monitoring of NADH was published in 1962 by Chance et al. [8]. They were able to simultaneously monitor the brain and kidney of anesthetized rats using two micro-fluorometers. In 1962, Chance and collaborators elaborated on this kind of in vivo monitoring and used it in other rat organs [56–58].
3.4 Fluorescence Emission Spectra of NADH
3.4.1 NADH Spectra in Solution
Several investigators have measured NADH fluorescence in solution. In most cases, the spectrum of NADH was compared to other biological preparations. For example, Cordeiro et al. compared the spectrum of NADH in solution to that of porcine myocutaneous flap and found a very significant correlation (Fig. 3.7a) [59]. The spectrum of NADH in solution and after binding to yeast alcohol dehydrogenase was published by Chance’s group [60]. In 1961, Theorell and McKinley-McKee measured the fluorescence emission spectra for DPNH (NADH) and its binary and tertiary complexes with enzyme and imidazole (Fig. 3.7b) [61].

Fig. 3.7
a Comparison of true excitation spectra from pure β-NADH in solution (0.05 mM) and from a representative porcine myocutaneous flap. Scale is arbitrary. Intensity of fluorescence is recorded at a wavelength of 450 nm. With the exception of a light shift in excitation maxima, the two spectra are nearly identical. (© Walters Kluwer Health, reprinted with permission [59].) b Fluorescence emission curves for LADH, DPNH, and imidazole. (1) Buffer (0.1 μ phosphate, pH 7, 23/5 °C); (2) buffer + 0/1 M imidazole; (3) buffer + 2.2 μN LADH; (4) buffer + 2.1 μM DPNH; (5) buffer + 0.1 M imidazole; (6) buffer + 2.2 μN LADH; (7) buffer + 0.1 M imidazole. (© Swedish Chemical Society, reprinted with permission [61].) c Spectra of major colonic tissue fluorophores. The fluorescence spectra were recorded on crystalline collagen, crystalline NADH, and FAD in solution. All spectra are system response corrected. (© John Wiley and Sons, reprinted with permission [62])
3.4.2 NADH Spectra in Isolated Mitochondria
The excitation and emission spectra of NADH (PN) and flavoprotein were measured in frozen samples of pigeon heart mitochondria [63].
Using rat liver mitochondria, Chance and Baltscheffsky in 1958 [46] measured the fluorescence spectra in the three metabolic states defined by Chance and Williams [33]. The 330-nm light excitation used resulted in a fluorescence peak at 440–450 nm. The same kind of spectra was obtained by other investigators using different fluorometers or mitochondria isolated from various organs. Eng et al. monitored the NADH emission spectrum from isolated rat heart mitochondria (Fig. 3.8a) [64]. The maximal fluorescence signal was measured at 455 nm using a 100-W mercury lamp for UV excitation. Chance el al. [58] compared the emission spectra emitted from pigeon heart mitochondria when using a micro-spectrofluorometer, shown in Fig. 4.1a to a macro-spectrofluorometer. They concluded that the micro-spectrofluorometer is adequate for in vivo monitoring of NADH fluorescence. Chance in 1962 showed the difference in spectra measured between starved and anoxic liver mitochondria (Fig. 3.8b) [65]. Galeotti et al. measured similar spectra from rat liver mitochondria [60]. Using rhodamine B as an internal standard for system calibration, Koretsky and Balaban found the same spectra emitted from isolated rat liver mitochondria (Fig. 3.8c) [66]. Koretsky et al. in another study (Fig. 3.8d) compared the emitted spectrum from heart homogenates (similar to isolated mitochondria) with that of dissolved heart homogenates (in vitro) [67].

Fig. 3.8
a Autofluorescence emission spectrum of isolated rat heart mitochondria. (© Reprinted with permission from Elsevier [64].) b The fluorescence emission spectrum representing the difference between an aerobic and an anaerobic suspension of liver mitochondria (corrected data). Excitation wavelength 353 nm. (© John Wiley and Sons, reprinted with permission [65].) c Fluorescence spectrum obtained from a mitochondrial suspension incubated with 5 mM pyruvate. The two peaks seen are from mitochondrial NAD(P)H (440 nm) and rhodamine B (586 nm, 2 μM). The three spectra correspond to mitochondria respiring in (a) pseudo-state 4, (b) state 3 (which was initiated by injection of 700 μM ADP), and (c) state 4. The spectra are overlaid to illustrate that changes in NAD(P)H fluorescence occurred without any change in rhodamine B fluorescence. (© Reprinted with permission from Elsevier [66].) d Effects of diluting heart homogenates on fluorescence spectrum obtained using video fluorometer. A no dilution; B 1:3 dilution of heart homogenate with saline. Intestines are in arbitrary units and are scaled to allow comparison of three dilutions. (© American Physiological Society, reprinted with permission [67])
3.4.3 Intact Cells
The use of micro-fluorometry to study intact cell metabolism was described in a number of publications by Kohen and collaborators (see, for example, [15]). Figure 3.9a was published by Kohen et al. in 1976 [68].

Fig. 3.9
a Sequence of fluorescence spectra recorded from an EL2 cell following microinjection of glucose-6-P. The time interval between two consecutive scans is ~64 ms. Abscissa: (260–340) channels (608–426) nM; ordinate: counts. Spectrum extends from about 610 nm on the left to about 420 nm on the right. The scan number is shown for each curve. The EL2 cell was microinjected shortly before scan 155, which explains the relatively small difference in intensity between scan 155 and scan 1 and the gradual increase after scan 155. There was no further rise after scan 246. The emission maximum fluctuates between approximately 460 and 475. (© Reprinted with permission from Elsevier [68].) b Effect of metabolic state on the fluorescence emission spectrum of a dilute suspension of ascites tumor cells. The suspension, 5.6 mg dry wt/ml, was first treated with rotenone (13 μM) and its fluorescence spectrum recorded. Glucose (20 mM) + NaF (15 mM) were then added and the spectrum recorded again. After removal of the cell suspension, the fluorescence spectrum of a solution of NAD (0.6 mM) was recorded in the same cuvette. A portion of the latter is shown by the broken line, but it should be noted that the gain of the instrument was reduced several fold for this spectrum. (© John Wiley and Sons, reprinted with permission [69].) c Fluorescence emission spectra for the cyanide (A) and FCCP (B) transitions. The top spectrum of each set was acquired during a control period. The middle spectra were acquired 5 min after adding cyanide (G) or FCCP (B) to the surrounding medium. The bottom spectra are different spectra (middle minus top). (© Reprinted with permission from Elsevier [64])
The typical NADH fluorescence spectrum was measured in suspension of ascites tumor cells [69]. This study demonstrated that the spectrum of intact cells was similar to that of NADH solution (Fig. 3.9b).
Using isolated myocytes, Eng et al. compared the spectra measured under various conditions of the mitochondria [64]. They found that cyanide induced an increase in the spectrum difference, whereas FCCP, used as a typical uncoupler of oxidative phosphorylation, produced a marked decrease in the spectrum. These results are presented in Fig. 3.9c.
Terzuolo et al. [70] monitored the fluorescence spectrum in a single neuron and compared it to that recorded from NADH solution.
3.4.4 Tissue Slices and Blood-Free Perfused Organs
The next step in the development of NADH fluorometry, after its use in isolated mitochondria and single cells, was to apply it to tissue slices and isolated perfused organs in vitro.
The classical results of Chance et al. (seen in Fig. 3.10a) were obtained from DPNH in solution, in a suspension of mitochondria, and in a perfused kidney slice [8]. Only a very small spectral difference was found between the kidney slice, isolated kidney mitochondria, and the solution of NADH (DPNH). The same type of comparison was performed by Aubert et al. [71] using a slice of the electric organ isolated from an Electrophorus. The spectrum from the electric organ was very similar to pigeon heart mitochondria and NADH in solution. Chance published more spectra originating from perfused rat heart exposed to various metabolic perturbations (Fig. 3.10b) [72]. A large, significant increase in the emission spectrum was recorded between normoxic and anoxic conditions, and between normoxic and amytal-treated hearts. These results suggested that when O2 supply is eliminated (state 5 of the mitochondria) or when site I in the mitochondria is blocked by amytal, a large change in NADH levels is recorded.

Fig. 3.10
a Fluorescence emission spectra. Dashed-dotted curve, spectrum from a solution of DPNH; solid curve, spectrum from DPN reduction in a suspension of rat kidney mitochondria; dashed curve, spectrum from to an aerobic-anoxic transition in a slice of kidney cortex obtained from a perfused rat. The peaks of the emission bands, uncorrected for photocell sensitivity, are at 480, 463, and 472 mμ, respectively. (© Reprinted with permission from AAAS [8].) b Fluorescence emission spectra of the perfused rat heart under aerobic and anoxic conditions. (© Reprinted with kind permission of Springer Science + Business Media [72].) c Excitation (upper) and emission (lower) spectra of the perfused rat heart in the aerobic and carbon monoxide-inhibited states using 366-nm excitation light. (© Reprinted with kind permission of Springer Science + Business Media [74].) d Fluorescence emission spectra: comparison of skin slices, liver slices, and an NADH solution. Excitation at 366 nm; anaerobic conditions. These spectra are uncorrected for energy sensitivity. (© American Physiological Society, reprinted with permission [75])
The same kind of normoxic–anoxic transition was found by Jamieson and Van den Brenk [73] using isolated gastrointestinal mucosa.
Chance et al. studied the excitation and emission spectra from a perfused rat heart as seen in Fig. 3.10c [74].
To verify that the fluorescence emission under study originates from the pyridine nucleotide, it is desirable to obtain both excitation and emission spectra. These data obtained for the perfused heart, and the excitation spectrum corrected for the energy distribution of the light source, are indicated in Fig. 3.10c (upper part). The curve for the anoxic heart (perfused with saturated CO) shows a maximum at approximately 350 mμ, which resembles that of DPNH.
The emission spectrum for the perfused heart in both the aerobic and anaerobic (CO saturated perfusate) condition is shown in Fig. 3.10c (lower part). Here, a symmetrical peak with a maximum at 476 was observed for the anaerobic heart, which was close to the maximum observed with this equipment for mitochondrial suspensions
The studies of Chance and collaborators were further expanded to isolated rat skin [75]. The spectrum of the skin slice was very similar to that of the liver slice and NADH solutions (Fig. 3.10d). To test the responsiveness of the skin to metabolic manipulations, they compared spectra under normoxic and anoxic conditions (Fig. 3.11a) [75]. They also found a clear, significant increase in the skin spectrum under amytal treatment (response not shown).

Fig. 3.11
a Fluorescence emission spectra: comparison of skin slices during oxygen and nitrogen perfusion. Note different fluorescent scales in the two plots. Bottom solid line, before nitrogen perfusion; bottom broken line, after nitrogen perfusion. (© American Physiological Society, reprinted with permission [75].) b Emission spectra for reduced pyridine nucleotide component of rabbit cornea compared with oxidized (state 2) and reduced (state 4) rat liver mitochondria: 366 nm excitation at 77 K. (© Reprinted with permission from Elsevier [76].) c Fluorescence spectrum obtained from surface of a perfused heart using a rapid-scan video fluorometer (with excitation at 337 nm) and fluorescence from heart (maximum at 460 nm). (© American Physiological Society, reprinted with permission [67].) d Examples of NADH fluorescence emission spectra and hippocampal field potentials recorded during control conditions, 2 min after anoxic depolarization, and 60 min following reoxygenation. Failure of evoked potential recovery was related to hyperoxidation of NADH/NAD+. (© Reprinted with permission from Elsevier [77])
Another organ of interest tested by Chance and Lieberman was rabbit cornea (frozen) compared to rat liver mitochondria (state 2 and 4) (Fig. 3.11b) [76].
Other research groups adopted NADH fluorometry and tested the validity of its principles. For example, Koretsky et al. [67] employed rapid-scan video fluorometry and found the same NADH spectrum when using excitation light from a N2 laser (337 nm) (Fig. 3.11c). In 1997, the spectrum of the hippocampal slice in vitro was measured by Perez-Pinzon et al. [77] and a marked increase in NADH spectrum was recorded under anoxia (Fig. 3.11d).
3.4.5 Organs In Vivo
The use of NADH fluorometry for in vivo studies started at the end of the 1950s and has been presented in various publications. In a summative study, Chance compared spectra of NADH measured from rat kidney cortex to those of rat brain cortex (Fig. 3.12a, b) and found that the main effect of anoxic transition, both in the kidney and the brain, was a large increase in the fluorescence intensity with no detectable shift in the spectra [58]. Similar results were obtained by Chance [72] when intact sartorius toad muscle was stimulated (Fig. 3.12c). Stimulation of the muscle (state 4 to state 3 transitions) led only to a decrease in the intensity of the fluorescence emission spectrum.

Fig. 3.12
a, b Emission spectra of rat kidney (a) and rat brain cortex (b) under aerobic conditions (lower trace) and under anoxic conditions (upper trace). (© Reprinted by permission from Macmillan Publishers Ltd. [58].) c Emission spectra for the resting and stimulated sartorius muscle of the toad. Note that the spectrofluorometer is calibrated in a separate experiment by the emission of 4-methylumbelliferone (UBF) shown at the top of the chart. (© John Wiley and Sons, reprinted with permission [72].) d Emission spectral fluorescence curves for NADH in solution, in normal brain, in ischemic cortex, and in cortex 5 min after death. (© American Heart Association, reprinted with permission [78])
In 1975, Sundt and Anderson [78, 79] applied in vivo fluorometry to study the brain of the squirrel monkey (Fig. 3.12d), and recorded a greatly increased intensity in the dead brain as opposed to normal brain, with intermediate values in the ischemic brain. At the same time, Harbig et al. showed a clear increase in the NADH spectrum between normoxic and anoxic conditions in cat brain (Fig. 3.13a) [80].

Fig. 3.13
a Pyridine nucleotide fluorescence emission spectra from cat brain cortex in vivo in (1) normoxia and (2) nitrogen anoxia. Both spectra peak at 469–473 nm. The spectra are not corrected for photocell sensitivity. (© American Physiological Society, reprinted with permission [80].) b Representative excitation spectra from an experimental myocutaneous flap at baseline, after 6 h of ischemia, and after 4 h of reperfusion. Fluorescence is recorded at a wavelength of 450 nm, and the excitation energy ranges from 260 to 430 nm. Excitation maximum of all spectra is 330–340 nm. (© Walters Kluwer Health, reprinted with permission [59].) c Emission spectra of the brain under excitation of 366-nm light (A1, B2, B1, B2, C1) or laser 324-nm light (C2). C1 and C2 were measured from a dead brain. (© Reprinted with permission from Elsevier [81])
Corderio et al. compared the excitation spectra in porcine myocutaneous flap with those of NADH in solution shown in Fig. 3.13b [59]. Exposing the flap to 6-h ischemia dramatically increased the intensity of 450-nm light. Furthermore, Mayevsky compared two sources of excitation light: the usual mercury arc and laser excitation at 324 nm [81]. The main problem in applying such spectra, under in vivo conditions, is the effect of hemodynamic changes under anoxia. To overcome this problem, we induced repetitive cycles of anoxia that suppress the autoregulatory compensation mechanisms [81].
Figure 3.13 (part C) shows six scans obtained from the normoxic (A1, A2) and the anoxic brain (B1, B2) using a Hg arc, and comparative scans using Hg (C1) and the laser source (C2) in the dead brain. The results indicate that the location of the emission spectrum peak is identical in normoxia and anoxia, and is close when the laser is used. We also used the laser source in several in vivo studies and found that the light intensity did not harm the brain during several hours of measurement. Under anoxia, a clear increase in NADH fluorescence spectra was recorded.
3.5 Comparison Between Fluorescence Monitoring and Biochemical Analysis of the Pyridine Nucleotides
To verify the source of NADH fluorescence signals monitored in vivo, it was necessary to freeze the monitored tissue and to biochemically measure its pyridine nucleotide content. Initially, Avi-dor et al. [82] compared the fluorescence signal in isolated mitochondria to the concentration of pyridine nucleotides. Chance et al. (Fig. 3.14a) compared the fluorescent and analytical measurements of DPNH and TPNH using in vivo liver [83]. It became clear that ischemia, limiting the availability of oxygen, led to a large increase in the fluorescent signal. At the same time, DPNH showed a marked increase whereas TPNH remained unchanged. These results suggested that the source of the in vivo fluorescent signal is mainly from NADH.

Fig. 3.14
a Fluorometric trace of an ischemic cycle together with analytical values of AMP (bars), DPNH (crosses), and TPNH (circles) of several analogous experiments. (© Springer Science, reprinted with permission [83].) b Comparison of the direct fluorescence assay of pyridine nucleotide reduction measured from the beating heart (a) and assays for increases of pyridine nucleotide reduction under similar metabolic conditions (b). (© John Wiley and Sons, reprinted with permission [72])
In 1966, Chance summarized the studies comparing the fluorescence signals with the biochemical analysis of pyridine nucleotides [72]. It was found (Fig. 3.14b) that DPNH was the main source of fluorescence change in the in vitro beating heart. This observation held true under anoxia and under treatment with amytal. Chance et al. showed a clear correlation between the fluorescence signal measured in the heart and the biochemical analysis of NADH in the tissue.
To test conditions where NADH becomes oxidized, the hyperbaric oxygenation effect was tested in the rat liver in vivo [84]. NADH was found to be the main source for the increase of in vivo signal under amytal treatment. Under hyperbaric hyperoxic conditions, the contribution of TPNH (NADPH) was larger. In the same study, Chance et al. showed that in the brain cortex, under anoxic or hyperbaric conditions, NADH was the main source of the fluorescence signal (Fig. 3.15a, b) [72].
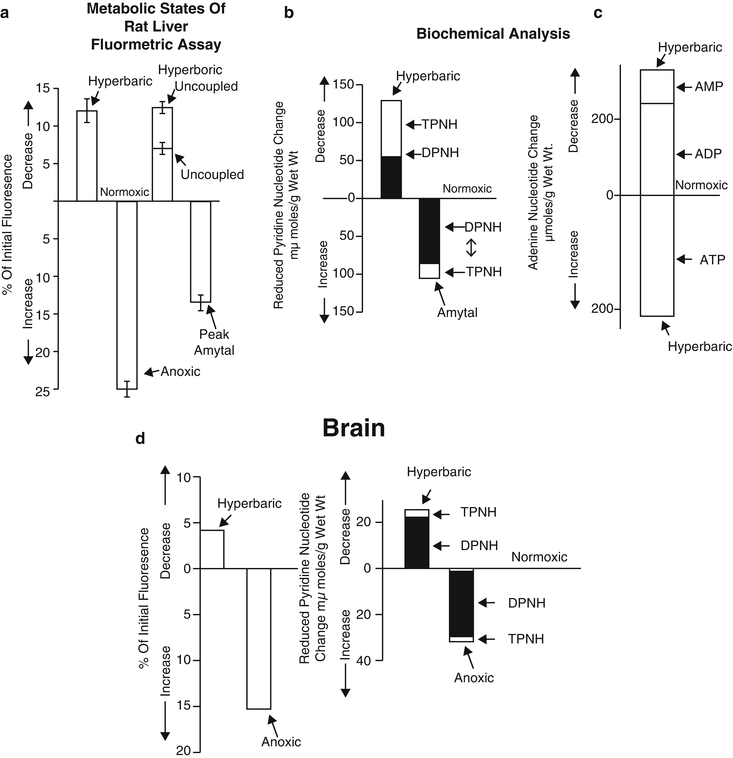
Fig. 3.15
Comparison of fluorometric assay of metabolic states of the rat liver (a) with biochemical analysis of pyridine nucleotides (b) and adenine nucleotides (c). (© Prof. Britton Chance, reprinted with permission.) (d) Comparison of the fluorescence recordings of pyridine nucleotide changes on brain cortex of the anesthetized rat (left) with metabolite assays under similar conditions (right). (© Prof. Britton Chance, reprinted with permission [72])
In 1971, Jobsis et al. [85] compared the corrected NADH fluorescence signal (after subtracting the reflectance signal) to its concentration measured in a brain tissue sample after induction of convulsions or anoxia (Fig. 3.16a). A very clear correlation was found, but it was concluded that “a quantitative interpretation of the changes of fluorescence in terms of nanomoles of NADH and NADPH is not truly warranted at this point.” Other investigators performed similar comparisons between the fluorescence signals and the analytical measurements. Shimazaki et al. showed a good correlation between the fluorescence of NADH and its concentration in control cornea as well as in cyanide-treated cornea [86].
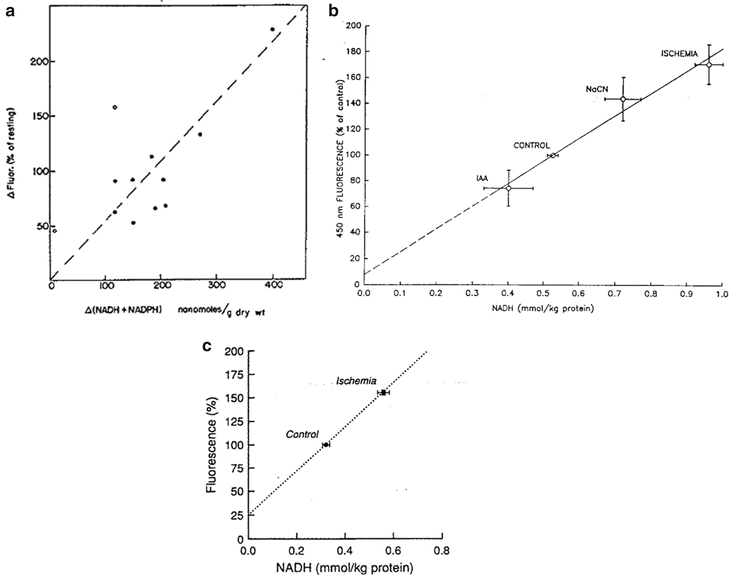
Fig. 3.16
a Correlation between fluorescence changes (compensated for vascular quenching) and differences in cortical, reduced pyridine nucleotide content (NADH + NADPH) determined analytically in the convulsive and anoxic states. Open circles indicate two experiments in which the occurrence of movement artifacts is suspected (see text). (© American Physiological Society, reprinted with permission [85].) b Comparison of NADH tissue concentrations obtained by enzymatic method applied to quick-frozen samples of spinotrapezius muscle with tissue fluorescence at 450 nm determined by in vivo microfluorometry; Values are means ± SD. IAA iodoacetate. (© American Physiological Society, reprinted with permission [88].) c Comparison of total tissue NADH determined enzymatically and NADH fluorescence in control conditions and after 5 min of ischemia. Fluorescence rise during ischemia represents combined steady-state values from arteriolar and venular sites, weighted equally. Dotted line represents line of best fit. Error bars represent means ± SE. (© American Physiological Society, reprinted with permission [89])
NADH fluorescence and enzymatically determined NADH levels were similarly compared in the brain subjected to ischemia [87]. Toth et al. found a clear correlation between the two parameters under various metabolic perturbations of the spinotrapezius muscle (Fig. 3.16b) [88]. In the in vivo muscle, cyanide treatment and ischemia caused an increase in both parameters whereas the glycolysis inhibitor IAA induced a decrease in both. The same effects have been recently found by Toth et al., testing cat sartorius muscle in vivo [89]. A significant correlation was found between the fluorescence signal change in ischemia and the amount of NADH determined enzymatically (Fig. 3.16c). In 2005, Uppal et al. developed a technique to estimate the concentration of NADH in human blood samples [90]. They compared the fluorescence signals to the biochemical analysis results.
3.6 Intracellular Origin of NADH Fluorescence Signal
The intracellular localization of the signals measured from NADH had been discussed even before NADH fluorescence was measured.
It is important to understand that the excitation and emission wavelengths of NADH (350 and 460 nm) are well separated from other endogenous chromophores, as described by Anderson-Engels and Wilson [91], and that the 460-nm fluorescent emission originates primarily from NADH bound to mitochondria.
Klingenberg et al. [47], who used mitochondria from various organs, found that the sum of [DPN] and [DPNH] was of the same order of magnitude in the liver, heart, kidney, and brain. Using mitochondria of rat liver and kidney, Avi-Dor et al. [82] came to the following conclusions:
The average fluorescence yield of reduced pyridine nucleotide in mitochondria is six to eight times the yield of DPNH and TPNH in aqueous solution.
The yield of mitochondrial DPNH is substantially higher than the yield of mitochondrial TPNH.
The significance of these conclusions is that the in vivo fluorometric technique provides information about the redox state of NADH in the mitochondria with a negligible contribution of the cytoplasm. Estabrook arrived at the same conclusion using rat liver mitochondria by comparing the fluorometric technique with the spectrophotometric approach [92]. The same conclusion was drawn by Chance et al. [8, 65] for rat brain and kidney in vivo, and by Jöbsis et al. [93] for cat brain induced to epileptic activity. Jöbsis and Duffield [94], using the fluorometric technique in intact toad sartorius muscle, concluded “that cytoplasmic NADH does not interfere materially with these measurements.” A similar conclusion was reached by Chapman [95], positing that “mitochondrial NADH is the sole significant origin of labile fluorescence under the condition used.” The same was reported later by Jöbsis and Stainsby [93] for the mammalian skeletal muscle. O’Connor [96], who used in vivo monitoring of cat brain in combination with biochemical assays, stated the following: “The cortical fluorescence recorded with in vivo fluorometric techniques originated from mitochondrial NADH.” The same results and conclusions were obtained by Nuutinen [97] using isolated perfused rat heart. Accordingly, “The NADH + NADPH fluorescence of the intact tissue originates largely from the mitochondria.” In our own studies summarized in a review paper [81], we also confirmed the notion that most of the labile NADH signal in the brain originated from the mitochondria. Our conclusion was based on studies of the rat brain.
In preliminary experiments, we used a time-sharing fluorometer to monitor both NADH and flavoproteins (Fp) fluorescence from a small brain area free of large blood vessels [98–100]. As seen in Fig. 3.17 (right), exposure of the brain to anoxia led to NADH reduction (increased fluorescence signal) simultaneously with the reduction of Fp (a decrease in fluorescence signal). These similarities in the kinetics of response to anoxia suggest that the signal originates from the same cellular compartment, that is, the mitochondria. Such similarities were also found in a decapitation model (data not shown). Additional indirect evidence comes from other decapitation model studies performed by us [100, 101]. We found, in the awake rat, that the NADH fluorescence increase starts within 1 s and reaches its maximal level within less than 1 min (Fig. 3.17, right). The same timing pattern has been described previously [102] when effects of decapitation were studied in an anesthetized mouse brain. It has been reported that after decapitation, a large enhancement of glycolysis occurs, but this may have only started several minutes after the decapitation [103]. Lowry’s findings also suggest that if NADH increases dramatically several minutes after the decapitation, it may derive from the cytosolic source. In no decapitated animal (more than 200 animals) did we ever find such a secondary increase in NADH fluorescence after the initial maximal increase. The initial increase is correlated to the decrease in pO2 as measured by a multiparametric technique (see Fig. 3.17, left) [104]. Eng et al. [64], studying single rat cardiac myocytes, asserted that “These data are consistent with the notion that the blue autofluorescence of rat cardiac myocytes originated from mitochondrial NADH.” Coremans et al. [105] confirmed that “The results show that the NADH fluorescence/UV reflectance ratio can be used to monitor the mitochondrial redox state of the surface of intact blood perfused myocardium.”

Fig. 3.17
Two records of experiments used to support the evidence that the main origin of the NADH signal in vivo originates from the mitochondria. a The response of the rat brain to complete ischemia induced by decapitation (Decap.). PO2 partial pressure of oxygen. b Effects of anoxia on brain NADH and Fp (flavoproteins). (© Reprinted by permission from Macmillan Publishers Ltd. [104])
References
1.
Chance B, Williams GR, Holmes WF, Higgins J (1955) Respiratory enzymes in oxidative phosphorylation (V-A mechanism for oxidative phosphorylation). J Biol Chem 217:439–451PubMed
2.
Chance B, Williams GR (1955) A method for the localization of sites for oxidative phosphorylation. Nature (Lond) 176(4475):250–254
3.
Chance B, Williams GR (1955) Respiratory enzymes in oxidative phosphorylation. III: The steady state. J Biol Chem 217:409–427PubMed
4.
Chance B, Williams GR (1955) Respiratory enzymes in oxidative phosphorylation. IV: The respiratory chain. J Biol Chem 217:429–438PubMed
5.
Harden A, Young WJ (1905) The influence of phosphates on the fermentation of glucose by yeast-juice: preliminary communication. Proc Chem Soc (Lond) 21:189–191
6.
Harden A, Young W (1906) Alcoholic ferment of yeast-juice. Part ΙΙ. Co-ferment of yeast-juice. Proc R Soc B Lond 78:369–375
7.
Warburg O, Christian W, Griese A (1935) Wasserstoff-Ubertragendes Co-ferment, seine Zusamensetzung und Wirkungsweise. Biochem Z 282:157
8.
Chance B, Cohen P, Jobsis F, Schoener B (1962) Intracellular oxidation-reduction states in vivo. Science 137:499–508PubMed
9.
Chance B, Oshino N, Sugano T, Mayevsky A (1973) Basic principles of tissue oxygen determination from mitochondrial signals. Adv Exp Med Biol 37A:277–292PubMed
10.
Warburg O (1949) In: Lawson TA (ed) Heavy metal prosthetic groups and enzyme action. Clarendon, Oxford
11.
Keilin D (1966) The history of cell respiration and cytochrome. Cambridge University Press, Cambridge, UK
12.
Keilin D (1925) On cytochrome, a respiratory pigment, common to animals, yeast, and higher plants. Proc R Soc Lond B Biol Sci 98:312–339
13.
Keilin D, Hartree EF (1945) Purification and properties of cytochrome C. Biochem J 39:289PubMedCentralPubMed
14.
Lubbers DW (1995) Optical sensors for clinical monitoring. Acta Anaesth Scand Suppl 39(104):37–54
15.
Kohen E, Kohen C, Thorell B (1969) Use of microfluorimetry to study the metabolism of intact cells. Biomed Eng 4:554–565PubMed
16.
Mayevsky A, Barbiro-Michaely E (2013) Shedding light on mitochondrial function by real time monitoring of NADH fluorescence: I. Basic methodology and animal studies. J Clin Monit Comput 27:1–34. doi:10.1007/s10877-012-9414-5 PubMed
17.
Theorell H, Bonnichsen R (1951) Studies on liver alcohol dehydrogenase I. Equilibria and initial reaction velocities. Acta Chem Scand 5:1105–1126
18.
Theorell H, Chance B (1951) Studies on liver alcohol dehydrogenase II. The kinetics of the compound of horse liver alcohol dehydrogenase and reduced diphosphopyridine nucleotide. Acta Chem Scand 5:1127–1144
19.
Chance B (1951) Enzyme–substrate compounds. Adv Enzymol 12:153–190
20.
Chance B (1951) Rapid and sensitive spectrophotometry. I. The accelerated and stopped-flow methods for the measurement of the reaction kinetics and spectra of unstable compounds in the visible region of the spectrum. Rev Sci Instrum 22:619–626
21.
Chance B, Legallias V (1951) Rapid and sensitive spectrophotometry. II. A stopped-flow attachment for a stabilized quartz spectrophotometer. Rev Sci Instrum 22:627–638
22.
Chance B (1952) Spectra and reaction kinetics of respiratory pigments of homogenized and intact cells. Nature (Lond) 169:215–221
23.
Chance B (1952) Respiratory pigments of metabolism cells. Fed Proc 11(1)
24.
Chance B, Neilands JB (1952) Studies on lactic dehydrogenase of heart. II. A compound of lactic dehydrogenase and reduced pyridine nucleotide. J Biol Chem 199(1):383–387PubMed
25.
Chance B, Castor LN (1952) Some patterns of the respiratory pigments of ascites tumors of mice. Science 116(3008):200–202PubMed
26.
Chance B (1953) Dynamics of respiratory pigments of ascites tumor cells. Trans NY Acad Sci 16(2):74–75
27.
Chance B, Williams GR (1954) Steady-state of reduced pyridine nucleotides in phosphorylating rat liver mitochondria. Am Soc Biol Chem 13 (abstr. 633)
28.
Connelly CM, Chance B (1954) Kinetics of reduced pyridine nucleotides in stimulated frog muscle and nerve. Am Physiol Soc 13(1):29
29.
Chance B (1954) Enzyme mechanisms in living cells. In: McElroy WD, Glass B (eds) The mechanism of enzyme action. Johns Hopkins Press, Baltimore, pp 399–453
30.
Chance B (1954) Spectrophotometry of intracellular respiratory pigments. Science 120:767–775PubMed
31.
Chance B, Williams GR (1955) Respiratory enzymes in oxidative phosphorylation. I: Kinetics of oxygen utilization. J Biol Chem 217:383–393PubMed
32.
Chance B, Williams GR (1955) Respiratory enzymes in oxidative phosphorylation. II: Difference spectra. J Biol Chem 217:395–407PubMed
33.
Chance B, Williams GR (1956) The respiratory chain and oxidative phosphorylation. In: Nord FF (ed) Advances in enzymology, XVIIth edn. Interscience, New York, pp 65–134
34.
Chance B, Hollunger G (1957) Succinate-linked pyridine nucleotide reduction in mitochondria. Fed Proc 1 (abstr. 703)
35.
Chance B, Connelly CM (1957) A method for the estimation of the increase in concentration of adenosine diphosphate in muscle sarcosomes following a contraction. Nature (Lond) 179:1235–1237
37.
Chance B (1957) Techniques for the assay of the respiratory enzymes. In: Colowick SP, Kaplan NO (eds) Methods in enzymology, vol 4. Academic Press, New York, pp 273–329
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


