RECOGNITION OF PSEUDOSARCOMAS
Table 5.2 lists the lesions that mimic sarcomas. Knowledge of the histologic appearance of these lesions is critical to prevent incorrect diagnoses of sarcoma. The following discussion provides some insight into this group of lesions.
Some of the most rapidly growing but relatively small mesenchymal lesions are benign. Any tumor with a tissue culture–like character, as is found in fasciitis and myositis, should be considered benign until it is proven otherwise. Circumscribed and encapsulated fatty lesions with either spindle cells or highly pleomorphic “floret” cells but without increased vasculature should also be considered benign. Bizarre nuclear features may be encountered in a host of benign soft tissue lesions, both reactive and neoplastic; for example, leiomyomas and neurofibromas may contain these features. Any circumscribed vascular lesion or a vascular lesion with a lobular architecture, such as that seen in pyogenic granuloma, should be considered benign. Last, a great majority of sarcomas are highly vascular, even if this fact is less than completely appreciated at the light microscopic level. For that reason, relatively avascular lesions can be considered benign, as the intramuscular myxoma exemplifies.
RECOGNITION OF OTHER TUMORS MIMICKING SARCOMAS
Tumors occurring primarily in organs should be considered carcinomas until they are proven otherwise. After all, a variety of carcinomas may mimic a spindle cell sarcoma or UPS with osteoclast-like giant cells; these may occur in the breast, pancreas, bladder, and other sites (2,3). Tumors resembling UPS often turn out to be pseudosarcomatous carcinomas, as frequently occurs in the larynx (4). Renal cell carcinoma may have a sarcomatoid appearance, a possibility that is well known; therefore, a clear cell component in a possible sarcoma should alert one to this possibility. Stains for epithelial markers should be initiated.
Lymphoma may not only mimic the small round cell tumors but may also simulate UPS. Only careful attention to nuclear detail (the majority of lymphomas contain irregular indentations in nuclear outline) alerts one to this possibility. Also, tumors of histiocytic and dendritic cell origin, such as histiocytic sarcoma (5), follicular dendritic cell sarcoma (6), and interdigitating dendritic cell sarcoma (7), as well as anaplastic large cell lymphoma (8), may also be mistaken for soft tissue sarcomas.
SPINDLE CELL CARCINOMA
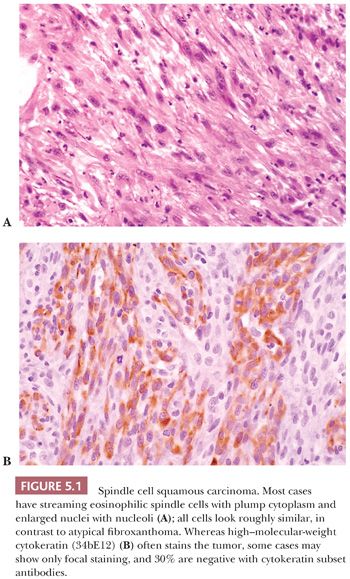
Spindle cell carcinoma is deceptive, and because some routine cytokeratin (CK) markers are limited in extent or entirely absent, it continues to be misdiagnosed as sarcoma. The transformation of a squamous cell carcinoma into a spindle cell carcinoma is a recognized phenomenon in a variety of mucosal sites, such as the larynx, the oral and nasal cavities, and the skin. Several studies note that this change may be accompanied by a gain of vimentin and smooth muscle actin (SMA) along with the complete or nearly complete loss of CK reactivity (9–12). The histologic appearance of a brightly eosinophilic tumor at low power is typical. Furthermore, most cases are not highly pleomorphic but, instead, relatively uniform. The spindle cells are elongated, with open oval nuclei and prominent nucleoli, and they have “plump” cytoplasm, meaning they are wide with some cytoplasm on the sides of the nuclei (Fig. 5.1). These features are not seen in smooth muscle and many “fibrohistiocytic” tumors. Usually, only a focal storiform pattern is seen. A reticulin stain may demonstrate elongated groups of cells surrounded by reticulin. These features should raise the suspicion of a possible spindle cell carcinoma. Studies continue to find a frequent absence of standard CKs AE1/3 and CAM5.2, but if a series of CK subsets is used, approximately 70% of cases are positive (13). Wide-spectrum screening of CK has been shown to be more useful in some situations (14,15). In skin and mucosal sites, this diagnosis should be considered before that of mesenchymal tumors, such as atypical fibroxanthoma.
SPINDLE CELL MELANOMA
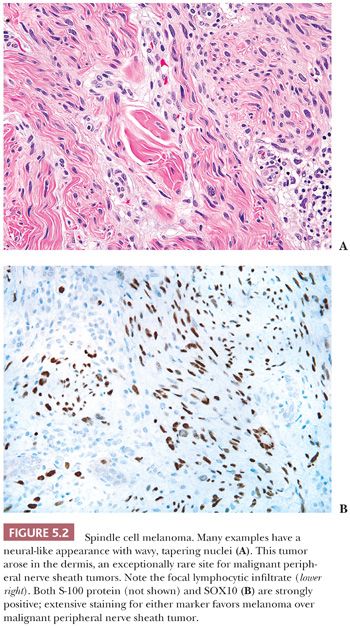
When a melanoma mimics a sarcoma (16–18), its recognition may be aided by the presence of separated cords (streaks of spindled tumor cells separated by fibrous tissue on either side—a kind of incomplete nest), a nesting pattern, or the failure to exhibit a completely idealized pattern (e.g., failure to exhibit a well-defined alternating fascicular pattern for leiomyosarcoma, failure to exhibit a storiform pattern or extensive sclerosis for UPS). Only a high index of suspicion helps one eliminate melanomas from the ranks of sarcomas. In particular, spindle cell or desmoplastic melanoma, whether in a skin primary tumor or a metastasis, may simulate a nerve sheath tumor (Fig. 5.2). A key helpful feature is the common diffuse S-100 and SOX10 positivity that is present, in contrast to MPNST. Even in the skin, an intraepidermal melanocytic component may be absent, signifying the lack of this important clue (19). Furthermore, HMB-45 and MART-1, which are present so frequently in ordinary melanoma, are also lacking in up to 75% of cases of desmoplastic melanoma (17,20). Thus, as in spindle cell carcinoma, caution must be used in the interpretation of negative results. Newer markers, such as microphthalmia transcription factor and the neuroectodermal transcription factor SOX10 (21–23), may assist in difficult cases.
RECOGNITION OF THREE MAJOR CATEGORIES
The statement that the two major sarcoma categories are the round cell sarcomas and the spindle cell sarcomas has been made. However, such a formulation is too simplified. The categories are not necessarily pure; for example, rhabdomyosarcoma (RMS), a “round” cell tumor, may have a spindled histology, and it may be included in the differential diagnosis of spindle cell sarcomas, such as leiomyosarcoma. Dividing sarcomas into three categories, with the third category labeled “other” or “odd,” is better; such tumors may show an epithelioid appearance or mixed spindle cell and epithelioid morphology. A variety of unusual sarcoma types, each with its own natural history and behavior, are encountered. For example, angiosarcoma, epithelioid sarcoma, clear cell sarcoma, malignant extrarenal rhabdoid tumor, and alveolar soft part sarcoma would all belong to the third group. Clearly, these do not fit neatly into the round cell and spindle cell categories, and just as clearly, the biologic behavior of each differs; furthermore, each requires an individualized therapeutic approach. On the contrary, the therapy for particular adult spindle cell sarcomas (those of fibroblastic, nerve sheath, and smooth muscle lineages) remains similar, although newer chemotherapy agents are beginning to target specific types such as leiomyosarcoma. Such histology-specific therapies will be increasingly important in the future. Critically important in dealing with this group of tumors is the enumeration of the pathologic characteristics of prognostic significance. Round cell sarcomas often have a rapid course, but as a group, they are also chemosensitive; a large fraction of these sarcomas is potentially curable.
IMPORTANCE OF LINE OF DIFFERENTIATION (“HISTOGENESIS”)
The line of differentiation is not particularly important, as yet, for the treatment of many adult spindle cell sarcomas, including most high-grade pleomorphic sarcomas, and it may not be discernible. Referring to the line of differentiation a tumor exhibits, rather than its histogenesis, is best because of lineage infidelity in tumor progression and the fact that most sarcomas likely arise from partially committed mesenchymal precursor cells. However, a pathologist who correctly identifies the tumor phenotype (the line of differentiation rather than the cell of origin) of a sarcoma and who renders a specific diagnosis performs a valuable service to the clinician. The treating physician may then refer to the literature on a specific sarcoma for guidance. Classifying RMS tumors correctly, for example, is particularly important because these are treated very differently from other tumors. A correct diagnosis of an unusual sarcoma (e.g., epithelioid sarcoma) likewise alerts the clinician to the likely natural history and potential prognostic factors. Thus, tumor phenotype or line of differentiation remains important, and it will become more so once effective targeted therapies are identified for each tumor.
ORIGIN AND ETIOLOGY OF SARCOMAS
SCOPE OF DIAGNOSTIC ISSUES
To give some perspective on sarcoma diagnosis, there are at least nine tumor phenotypes (smooth and skeletal muscle, adipocytic, vascular, etc.), each of which is complicated by multiple distinctive sarcoma subtypes, resulting in more than 100 sarcoma subtypes (24).
OVERVIEW
Although the speculation has been that most sarcomas arise from a primitive uncommitted mesenchymal cell, this idea seems less than completely tenable when one considers that the vast majority of sarcomas are indeed of a single phenotype and that they remain true to that phenotype throughout their natural history. Occasional sarcomas do “dedifferentiate,” or transform, a phenomenon that is discussed later. True malignant mesenchymomas, with their multiple phenotypes, are exceedingly rare. Thus, sarcomas quite likely arise from an abnormal event occurring in a cell of an already partially committed mesenchymal phenotype, even though such a cell may seem primitive and without visible differentiation.
Do sarcomas arise from benign STTs? The visualization of this phenomenon is rare (25–27). Most sarcomas appear to arise de novo. However, one may occasionally encounter a low-grade or benign lesion associated with a sarcoma. In some gastrointestinal stromal tumors, an adjacent nodule has absolutely bland histology. Similarly, a nerve sheath lesion with a long clinical duration (both neurofibromas and much more rarely schwannomas) may give rise to malignant neoplasms. Other notable examples of this phenomenon include “fibrosarcomatous” transformation of dermatofibrosarcoma protuberans and dedifferentiated liposarcoma arising from atypical lipomatous tumor. However, the vast majority of soft tissue sarcomas have no recognizable benign precursor.
SARCOMA NOT OTHERWISE SPECIFIED
Even with technologic advances such as the electron microscope and monoclonal antibodies, occasional sarcomas (roughly 5%) may not be definitively characterized. Therefore, using the term sarcoma not otherwise specified (NOS) for these lesions is appropriate. However, some qualifier should be used as a clinical guide (e.g., spindle cell sarcoma NOS or round cell sarcoma NOS). If such a lesion is encountered, a listing of all the attempts to assign the lesion to a particular diagnostic category should be included within the pathology report.
The etiology of sarcomas remains elusive, except in postradiation tumors. Some sarcomas occur in the setting of familial genetic syndromes, such as Li-Fraumeni syndrome (TP53 germline mutation) (28) and RB1 (retinoblastoma) gene mutations (29). Weak associations have been made with polychlorinated biphenyls and polyaromatic hydrocarbons and with dioxin (30–32). However, the latter association is very weak, and it may not be borne out by future work. Diet has been investigated only rarely (33). In some cases, these tumors may develop in a prior injection site, a phenomenon supported by animal studies (34). A viral etiology of Kaposi sarcoma is now accepted, with the agent being the human herpesvirus-8 (HHV8), formerly known as Kaposi sarcoma herpesvirus (KSHV) (see “Kaposi Sarcoma”), and some leiomyosarcomas in patients with acquired immunodeficiency syndrome (AIDS) have been associated with the Epstein-Barr virus (35).
TISSUE SECTIONS
As a general rule, a minimum of one block per centimeter of greatest tumor dimension should be processed; more material may be submitted, depending on the variable nature of the sectioned surface. This general rule must be tempered by the size of the tumor. In small tumors, some sections could include both the marginal and peripheral areas, whereas the remaining sections are obtained from the center of the lesion; thus, in a small lesion of approximately 3 cm, four to five sections might be obtained. In large lesions (5 to 10 cm), 8 to 10 sections may be required. In very large lesions (10 to 20 cm), 10 to 15 sections may be obtained if the lesion is relatively uniform. When sections are obtained, documenting (a) necrosis (by including it in a section with viable tissue), (b) any unusual appearance seen at gross examination, (c) the margins, and (d) the relationships to surrounding structures is important.
For specimens resected after chemotherapy, it is now commonplace to evaluate a complete “slab” of the tumor—a slice taken along its largest length and width—using numbered blocks, in order to document the percent necrosis and fibrosis. Fibrotic areas lacking tumor cells are added to the percent necrosis to arrive at the total “percent necrosis/fibrosis” secondary to the therapy, information that is valuable to treating physicians.
COMMENT ON MARGINS
A thin, stretched fascial margin (even when it is 1 to 2 mm) is a complete surgical margin, and such areas do not generally give rise to recurrences. These may be described as “close,” but the tumor is really contained in that direction, and that area should be described as an intact fascial plane. In contrast, the cut surgical margins, if they are close, are a major concern, and in such situations, the margin should be described in a note or comment. Multiple ink colors are now commonly used, and reports should include distances to various margins in a properly oriented specimen.
EVALUATION OF PROGNOSTIC FACTORS
The gross and microscopic evaluation of a neoplasm is of critical importance to the clinician in determining the prognosis. In some cases, this information is even more vital than the exact line of differentiation of the neoplasm. The evaluation must begin with a gross assessment of the lesion. The entire lesion should be inked, the exterior should be dabbed with fixative to ensure that the ink adheres, and the margins should be adequately sampled. The presence and degree of necrosis should be recorded because this affects tumor grade. Communication with the surgeons is vital for an understanding of their impression of the closest margin. Furthermore, a diagnosis is incomplete without an assigned grade (when appropriate) and a comment on the status of margins. The grade of a sarcoma (see “Grade” under later “Evaluation of Prognostic Factors” section) is absolutely key to the subsequent clinical management of some tumor types, and its absence from a report may result in a call from clinicians.
RECOMMENDATIONS FOR THE FINAL PATHOLOGY REPORT
The final pathology report should contain all relevant gross and microscopic descriptive information, and it should be able to stand alone if all of the blocks and slides from a case are lost. In this day of frequent second opinions and detailed therapeutic protocols, standardizing reports of sarcomas is even more important. To that end, the ideal pathology report contains a detailed gross description, particularly of the following: (a) size—in three dimensions—of the lesion, (b) the overall color and texture of the lesion, (c) the variable areas in the lesion, and (d) the extent of visible necrosis. How closely the lesion approaches any of the margins should be specified with precise measurements. Within the gross description, a guide or diagram of labeled sections may be helpful in reconstructing the lesion later. The value of a microscopic description (or comment) is enhanced when it includes information concerning the shape and size of cells, the characteristic patterns that were noted, and any ancillary studies that were performed. The actual wording of the diagnosis should include tumor size, grade (when relevant), and modifiers such as “superficial,” “deep,” and “intramuscular.” When preoperative chemotherapy has been administered, the percent necrosis/fibrosis should be recorded, supplying the clinician with valuable data on the effectiveness of therapy. A comment on the margins should be incorporated into the report, and a tumor template is recommended.
CONSULTATION
Because of the overall rarity of STTs and the tremendous variation in histologic patterns, even within a given sarcoma phenotype, the suggestion is that consultation be sought in difficult cases, particularly when the original pathologist receives only a handful of cases per year. Sending a case for consultation is also important when immunohistochemistry or molecular studies may provide important information regarding the patient’s management. To save time, recut sections should be accompanied by at least one representative block or 5 to 10 unstained slides.
LIMITATIONS OF PROCEDURES
FROZEN SECTION
One should not try to make a specific diagnosis (unless it is obvious) from intraoperative frozen sections, but instead, one should attempt to decide whether the lesion is malignant. If this determination cannot be made with confidence, one can resort to terms such as “spindle cell tumor of unknown malignant potential”; even this noncommittal diagnosis guides the surgeon, who may then elect complete excision or await the final report.
ASPIRATION BIOPSY CYTOLOGY
The use of aspiration biopsy cytology in the diagnosis of sarcomas has been successful in the hands of expert cytopathologists, particularly when ancillary techniques, such as immunohistochemistry and molecular genetic analysis, are applied. Indeed, both the overall sensitivity and specificity have been estimated to be 95% (36). However, this high diagnostic yield is undoubtedly related to experience, and those who see only an occasional cytologic aspirate of a sarcoma are unlikely to match this rate. Also, unless small chunks of tissue are available, the pathologist does not have the added benefit of visualizing the morphologic pattern of the lesion. Therefore, a certain amount of interpretive caution is mandated, particularly when a distinction must be made between benign and malignant lesions.
NEEDLE AND INCISIONAL BIOPSY
Because of the presence of variable patterns within any given STT, particularly malignant lesions, the application of a needle biopsy technique can give rise to difficulties. Once again, those who have a great deal of experience with the technique report excellent results in predicting the ultimate diagnosis. Kissin et al. (37), for example, report an overall diagnostic yield of 84% and an accuracy of 90%, and they highly recommend the use of this technique in planning a definitive primary surgical procedure. On the other hand, both they and other pathologists stress the possibility of a sampling error with incisional biopsy or needle biopsy. Underestimation of tumor grade is a recurring problem with needle biopsies as a result of sampling error. Therefore, samples obtained with this approach may not be representative of a neoplasm, and this possible shortcoming should be recognized and communicated to the clinicians planning further therapy.
EXCISIONAL BIOPSY
Material may also be received from an “excisional biopsy.” In this form, a rim of normal or marginal tissue may or may not be present, and occasionally, the surgeon may mention that the lesion was “shelled out.” Some sarcomas are sufficiently circumscribed to be shelled out, yet they are malignant lesions. Even so, the excisional biopsy should be treated as if it is the definitive surgical procedure, and all margins should be inked and well sampled. Experience with the excisional biopsy has shown that it is an inadequate approach to sarcomas because of the presence of tumor at or near marginal areas.
CURRENT SARCOMA THERAPY
Wide local excision (a minimum of 1- to 2-cm margins of normal tissue) has been the most common current approach to sarcomas, and a compartmentectomy is often performed whenever this is possible. In the hands of an experienced sarcoma surgeon, a biopsy is performed in such a manner that it can be incorporated by this complete resection procedure. Increasingly, resections are being performed with smaller margins (2 mm to 1 cm) in an attempt to spare function. The addition of postoperative irradiation has been effective in drastically reducing local recurrence, allowing the wide local excision to replace amputation in many instances. Chemotherapy of advanced or metastatic disease continues to have a low response rate of about 15% to 20%, and complete responses are rare. For particular tumor types, preoperative chemotherapy may be administered after a diagnostic biopsy, and the percent necrosis/fibrosis is recorded. Adjuvant chemotherapy is often administered now for some rare but aggressive tumors, such as angiosarcoma and synovial sarcoma, through the use of histology-specific protocols.
METHODS OF DETERMINING LINE OF DIFFERENTIATION
Major advances have been made in the diagnosis of STTs as a result of the application of ancillary techniques (e.g., immunohistochemistry, electron microscopy, cytogenetics) within recent years. A brief discussion of each of these methods is presented with the intent of increasing awareness of their usefulness and limitations.
CYTOGENETICS IN SARCOMAS
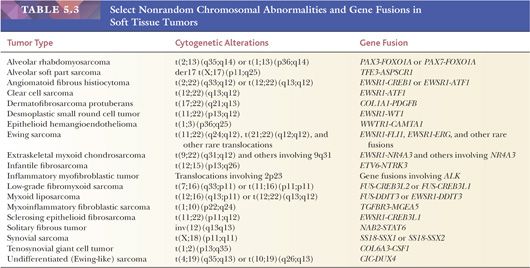
The analysis of chromosomal patterns in sarcomas over two decades has demonstrated that many STTs with recognizable nonrandom chromosomal aberrations occur (Table 5.3) (38,39). The chromosomal translocations are specific for various neoplastic phenotypes and may have diagnostic value. Processing tissue for cytogenetic analysis is encouraged for those pathologists who have access to this technology in their region. Newer fluorescence in situ hybridization (FISH) techniques are being applied in some diagnostic laboratories. With further developments in the molecular analysis of sarcomas, molecular assays have replaced cytogenetic karyotyping in many laboratories (40).
MOLECULAR DIAGNOSIS OF SOFT TISSUE TUMORS
The molecular diagnosis of STTs is a rapidly advancing field, and selected information is supplied in Table 5.3. The findings are clearly useful diagnostically. Specific tumor growth is apparently related to the activation of transcription factors through the formation of new fusion proteins. Some tumors have more than one genetic change, and some genes are involved in multiple tumor types. Recently, multiplex molecular testing of any given tumor for a panel of genetic changes has become available (using massively parallel, or “next generation” sequencing, for example), making assays simpler and the diagnosis easier. The usefulness of molecular analysis in sarcoma prognosis has been documented, particularly in alveolar RMS (41).
OVERVIEW OF IMMUNOHISTOCHEMISTRY
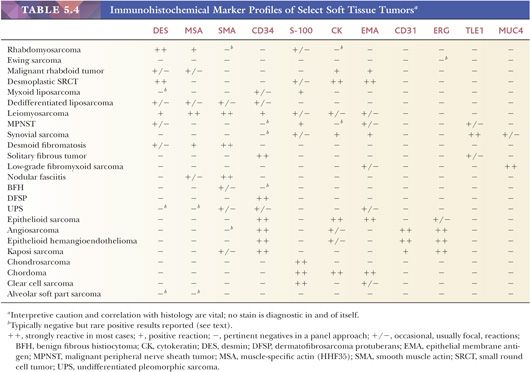
The application of immunohistochemical techniques has had a major impact on the diagnosis of soft tissue lesions. Most laboratories use the avidin-biotin, streptavidin, or newer polymer-based detection techniques because of their increased sensitivity, and frequently, automated equipment is used along with a panel of markers. The usefulness of immunohistochemistry stems from several facts, including the following: (a) some soft tissue phenotypes are associated with an identifiable marker protein; (b) marker proteins are often preserved in paraffin-embedded tissue blocks; (c) the sensitivity and specificity of markers have been reasonably good (although major exceptions do exist, creating problems, which are discussed later); and (d) new markers are forthcoming. With the increased sensitivity resulting from antigen unmasking (“retrieval”) through microwave, pressure cooker, or enzymatic pretreatment, a high percentage of tumors do stain. However, widespread use has led to the identification of nonspecific reactions; therefore, the results of this procedure must be subjected to strict interpretive caution with a foreknowledge of possible aberrant reactions. Furthermore, a panel approach using multiple markers is generally employed. With this technology, approximately 50% to 75% of all STTs contain relatively specific immunoreactivity, enabling the confirmation of a given phenotype or line of differentiation. Emphasis should always be placed on interpretation within the context of the standard histologic features. Typical marker profiles for STTs are listed in Table 5.4.
Problems with Immunohistochemistry
Serious interpretative difficulties have arisen within the past several years as numerous immunohistochemical markers have been applied to the vast array of mesenchymal lesions. The work of many authors has highlighted problems relating to nonspecificity on the one hand and aberrant immunoreactivity on the other.
Marker Negativity. One crucial fact seems to have been forgotten whenever stains are assessed for diagnosis or a contribution to the literature—any given marker does not react with 100% of its proper tumor type. In fact, many studies show that only 50% to 90% of given tumor types show reactivity for the specific marker tested. Even with antigen retrieval, not all cases react. Therefore, the significant lesson that must be relearned is that one may make a correct diagnosis in the face of marker negativity. For example, not all RMS tumors are desmin positive, not all MPNSTs are S-100 positive, and not all monophasic synovial sarcomas are CK positive. In such instances, substantial other support is required, but in certain cases, some blocks are useless. Therefore, perfection in an immunohistochemical marker should not be expected.
Nonspecificity. The initial rush of enthusiasm accompanying the appearance of a new “specific” marker is often replaced with harsh realism once the marker is subjected to a series of scientific studies. The results frequently show that the marker is useful but less than completely specific. For example, as a group, the muscle markers were originally thought to be specific for the smooth and skeletal muscle cell types, but actins react with myofibroblastic and fibrohistiocytic lesions, with SMA expected in myofibroblasts. Hence, marker reactivity alone does not lead to a correct diagnosis; the cell and nuclear size and shape, growth pattern, tumor location, and patient information must be integrated with the results of marker staining.
Aberrant Immunoreactivity. The phenomenon of marker staining occurring when it is theoretically unexpected is referred to as aberrant immunoreactivity. This began with a report of the epithelial marker CK in nonepithelial neoplasms, such as leiomyosarcoma (42), and since then, CK has been identified in nearly every mesenchymal phenotype and tumor. This reactivity is real, it has reflected actual protein expression whenever it has been studied, and it represents the neoplastic expression of markers seen in either the fetal state or in the process of proliferation itself. The problem also involves the other intermediate filaments—neurofilaments can be found in RMS (43), and desmin can be found in endothelial tumors and rare carcinomas (J.S.J.B., personal observation, 1993). Several leukocytic antibodies stain mesenchymal cell types, and skeletal muscle actin antibodies rarely can stain smooth muscle tumors. Pathologists must take this phenomenon into account in the interpretation of immunohistochemistry. Luckily, most instances of aberrant reactivity show only patchy rather than diffuse staining.
Such aberrant immunoreactivity likely is the result of the direct visualization of the generation of neoplastic heterogeneity resulting from gene derepression during tumor progression. In fact, a closer examination of aberrant immunoreactivity may explain why spindle cell carcinomas and carcinosarcomas exist; one hypothesis that has been proposed is that the successive loss of one marker (CK) through the clonal expansion of an epithelial neoplasm is followed by the gain of another marker (e.g., vimentin, desmin), with a consequent shift in morphologic appearance. The fundamental significance of this hypothesis lies in its denial of the need for stem cells or primitive precursors in the development of multiple-lineage neoplasms.
GENERAL MARKERS
Vimentin has been hailed as the intermediate filament for mesenchymal tissues because it is found within essentially all normal mesenchymal tissue elements and most sarcomas. However, vimentin immunoreactivity is not that informative from a diagnostic viewpoint because many tumors, including carcinomas and melanoma, contain vimentin. In mesenchymal tumors, vimentin can be used in three situations. First, as Battifora (44) demonstrated, vimentin staining highlights those areas on a slide, which are likely to contain other forms of reactivity; it acts as a test of a block for marker reactivity in general, and it localizes regions for proper interpretation. Second, vimentin positivity helps confirm a diagnosis in those few tumors, such as epithelioid sarcoma, known to coexpress CK and vimentin. Third, vimentin reactivity provides a better visualization of the cell shape, which may aid in tumor identification. Nonetheless, given its lack of specificity, vimentin has limited utility in the diagnosis of STTs and has been entirely abandoned in diagnostic practice by many experts.
Several STTs may express specific collagen types, but the collagen typing of sarcomas appears to be a complex affair and some time may elapse before specific patterns of collagen immunoreactivity become helpful diagnostically. Immunoreactive laminin is commonly seen in neoplasms demonstrating a basal lamina ultrastructurally (schwannian and smooth muscle lesions) (45). Although laminin was not identified in fibroblastic tumors, fibroblastic and/or fibrohistiocytic processes appear to be capable of laminin production. Antibodies that recognize collagens and laminin are rarely used clinically.
CD34, AN ANCILLARY MARKER
CD34 is a 110-kd transmembrane glycoprotein found on human hematopoietic progenitor cells and vascular endothelial cells; it may play a role in cell adhesion and signal transduction. The first reported antibody to CD34 was MY10, raised against a human myeloid leukemia cell line; this differs somewhat from a more common CD34 antibody, QBEND/10. In 1990, Ramani et al. (46) first used CD34 as a vascular marker. Other studies of vascular lesions soon followed, and they showed its sensitivity for endothelial neoplasms; CD34 decorates 70% to 80% of angiosarcomas, 90% of Kaposi sarcomas, and 90% of epithelioid hemangioendotheliomas.
Clearly, CD34 has a much broader reactivity (Table 5.5) (47,48). Three important CD34-positive lesions are solitary fibrous tumors, gastrointestinal stromal tumors, and dermatofibrosarcoma protuberans (DFSP) (49). Some lipomatous tumors, such as spindle cell lipoma and the fibrous areas of atypical lipomatous tumor, are also positive for CD34.

In normal skin, a select group of fibroblastic cells and dermal dendrocytes display CD34. Cohen et al. (50) demonstrated the usefulness of CD34 in distinguishing DFSP from benign fibrous histiocytoma/dermatofibroma (BFH/DF); more than 95% of DFSPs are CD34 positive, whereas BFH/DF is characteristically CD34 negative. Occasional BFH cases contain CD34, particularly at the periphery; this finding should not deter one from the diagnosis (51).
Finally, CD34 is absent in practically all carcinomas (with the notable exception of NUT midline carcinoma), but it is present in up to 50% of epithelioid sarcomas. Thus, very few tumor types appear to be simultaneously positive for CK and CD34, most notably epithelioid sarcoma and epithelioid angiosarcoma.
BCL-2, AN ANCILLARY MARKER
BCL-2 is actually a family of proteins involved in the apoptosis pathway in cell growth and death. BCL-2 is present in selected STTs (e.g., solitary fibrous tumor, synovial sarcoma, DFSP), but given its lack of specificity, it is not particularly helpful in the diagnosis of STTs (52).
ENDOTHELIAL MARKERS
CD31, AN ENDOTHELIAL MARKER
CD31 is a 130-kd membrane glycoprotein (gp IIa) of the immunoglobulin supergene family expressed in some hematopoietic cells and endothelial cells (53). Originally called platelet endothelial cell adhesion molecule (PECAM-1), CD31 was first reported to have 100% sensitivity and specificity for endothelial lesions; other studies reported reactivity in 78% of 27 angiosarcomas stained (54), in epithelioid angiosarcomas, and in all Kaposi sarcomas (55). CD31 appeared more sensitive than factor VIII–related antigen in detecting tumors of endothelial derivation when these two markers were compared head to head. Work on angiosarcomas has confirmed the excellent sensitivity of CD31 in these malignancies.
Despite the high specificity reported for CD31, weak immunostaining has been noted in occasional carcinomas and mesothelioma (56), and it has been detected by immunoblotting and by polymerase chain reaction in a group of solid tumor cell lines (57). Furthermore, membrane staining is present on histiocytes and macrophages, a possible source of confusion (58). Thus, there is a lack of absolute specificity of CD31 for endothelial cells, and interpretation should take this into account.
FACTOR VIII–RELATED ANTIGEN AND ERG
Factor VIII–related antigen is a large protein produced by endothelial cells that has been identified in numerous benign and malignant endothelial lesions. This antigen may be found to some degree in lymphatic endothelium and lymphangiomas but only focally in Kaposi sarcoma (59). When compared with CD31, factor VIII–related antigen is not as sensitive. Factor VIII–related antigen has largely been supplanted by CD31 and the ETS family endothelial cell transcription factor ERG, which is expressed in nearly all benign, intermediate, and malignant endothelial neoplasms (including poorly differentiated angiosarcomas with spindle cell or epithelioid morphology) (60,61). Nuclear immunoreactivity for ERG is also highly specific for endothelial differentiation among mesenchymal tumors (60).
FIBROHISTIOCYTIC MARKERS
No specific markers for the fibrohistiocytic phenotype are known. Because of the poor specificity of α1-antichymotrypsin, α1-antitrypsin, lysozyme, and ferritin, these markers are no longer used. Muscle markers stain a percentage of fibrohistiocytic lesions (see “Muscle Markers”), so these can be used to advantage in limited situations.
CD68 AND CD163, “HISTIOCYTIC” MARKERS
CD68 is a 110-kd glycoprotein found in the lysosomes of monocytes and macrophages and in the primary granules of neutrophils. KP1 is a commercially available CD68 monoclonal antibody raised against a lysosomal fraction of human lung macrophages; other CD68 antibodies include Y2/131, Y1/82A, Ki-M6, Ki-M7, and PG-M1 (62). The histiocyte-like lineage of the stromal cells in tenosynovial giant cell tumors has been documented with CD68. A similar diffuse reaction is observed in giant cell tumor of soft parts. When one is faced with the differential diagnosis of epithelioid sarcoma versus a reactive granulomatous process, CD68 is useful as a counterpoint to CK. The many tumor-infiltrating histiocytes within a variety of neoplasms—carcinomas, sarcomas, and melanomas—are highlighted with CD68 antibody and should not be confused with tumor reactivity.
Initial reports of prominent CD68 reactivity in UPS raised questions concerning a possible histiocytic histogenesis. However, a high percentage of positive cells clearly is consistent with reactive histiocytes. Nonetheless, UPS tumor cells do exhibit variable CD68 (KP1) reactivity in many cases (63). The significance of this finding has been modified by the realization that any tumor with lysosomal granules may be positive for CD68. Granular cell tumors are typically CD68 positive (64), along with some nerve sheath tumors and many other tumor types, including some carcinomas and melanomas (65). CD68 can be expected in cells that exhibit phagolysosomes or lysosome-like granules, indicating phagocytosis or autophagy. Large studies of CD68 have affirmed its wide spectrum of reactivity and nonspecificity (65). Because of this lack of fidelity to the histiocytic lineage and its reactivity in diverse tumors, CD68 cannot be used as a diagnostic criterion for UPS.
In contrast to CD68, the hemoglobin scavenger receptor CD163 is highly specific for histiocytic differentiation and can be used to support the diagnosis of the exceptionally rare histiocytic sarcoma (as well as benign and reactive histiocytic lesions) (66). CD163 is not expressed by mesenchymal neoplasms.
FACTOR XIIIA, A HISTIOCYTIC MARKER
An intracellular form of the fibrin-stabilizing factor, F13a, is the last enzyme generated in the fibrin coagulation cascade. Normally present in the serum, it may be subject to uptake by tumor cells. F13a is routinely expressed by histiocytes, as well as the so-called dermal dendrocytes in the skin and similar cells in other sites (67). F13a has been used in the differential diagnosis of BFH/DF versus DFSP (68) and in that of juvenile xanthogranuloma versus Langerhans cell histiocytosis. Most BFHs show prominent reactivity. Hyperplasia of the dermal dendrocytes accounts for the reactivity in Kaposi sarcoma (69). With regard to the F13a reactivity commonly found in UPS, most of the positive cells appear to be infiltrating histiocyte-like cells rather than true tumor cells. Although F13a has been said to be of some help in distinguishing UPS from other spindle cell and pleomorphic sarcomas, reactivity can be significant in leiomyosarcomas and MPNSTs. As such, it is not lineage restricted. F13a should be used with extreme caution (if at all), and one should realize that it is only an “ancillary” marker at best. Many pathologists with expertise in STTs do not use F13a in diagnostic practice.
MUSCLE MARKERS
DESMIN
The intermediate filament desmin is a sensitive marker for muscle lesions found in tumors of both smooth and skeletal muscle origin. In skeletal muscle, it links Z-bands of adjacent myofibrils. More than 90% of RMS cases are positive, including those that are very poorly differentiated. In several comparative studies, desmin has been proven to be superior to other muscle markers for the identification of RMS (70,71). In smooth muscle tumors, the immunoreactivity for desmin is variable, depending particularly on tumor site. Esophageal and uterine tumors essentially always appear positive for desmin, whereas a proportion in the soft tissue (72–74) and skin appear to be less immunoreactive. This problem is in part a consequence of fixation; desmin is affected adversely by formalin fixation. However, certain normal smooth muscle phenotypes, particularly those around vessels, are known to be negative for desmin. Desmin staining has been favorably affected by antigen retrieval methods.
Desmin can be found in 17% of nonmyogenic STTs (75). Therefore, desmin positivity indicates a myoid phenotype, not necessarily a muscle phenotype. Myofibroblastic tumors, such as desmoid fibromatosis, may show at least focal desmin reactivity, and sometimes, the reactivity is diffuse. This might cause an erroneous diagnosis of leiomyosarcoma if one depends on the immunoprofile alone. The fact that some cases of UPS also contain spotty desmin (76) may be explained by the presence of myofibroblasts ultrastructurally.
MYOGLOBIN
Myoglobin is found exclusively in skeletal muscle lesions. Although it is specific for RMS, its sensitivity is far less than that of desmin (77,78). It tends to stain cells with relatively abundant cytoplasm. More specific skeletal muscle markers have largely replaced it.
MUSCLE-SPECIFIC ACTIN
In the 1980s, monoclonal antibodies to muscle-specific actin (HHF35 or MSA) and myosin were described. MSA decorates most leiomyomatous tumors and cases of RMS. Like desmin, MSA is not specific for muscle phenotypes because lesions of myofibroblastic and fibrohistiocytic differentiation may be positive for MSA (79). Unlike desmin, MSA stains myoepithelial lesions, which may be mistaken for smooth muscle tumors in several organs. As actins are produced by pericytes, myopericytoma is also positive. The tumor formerly known as hemangiopericytoma (HPC), now known to be synonymous with solitary fibrous tumor, shows fibroblastic (not pericytic) differentiation and is therefore usually negative for MSA.
SMOOTH MUSCLE ACTIN
SMA is restricted in its recognition of actin isoforms; it does not detect other α-actins (skeletal and cardiac) or γ-SMA. SMA is readily identified in smooth muscle neoplasms (80), but it is also found in nonmuscle lesions with a so-called myoid phenotype. With regard to this, understanding that myofibroblastic lesions, such as nodular fasciitis, desmoid fibromatosis, and inflammatory myofibroblastic tumor, are SMA positive is crucial (81); the recognition of this helps prevent their overdiagnosis as leiomyosarcoma, and it can be used to significant advantage in certain differential diagnoses. In reality, SMA expression is the hallmark of the myofibroblastic phenotype (SMA+/MSA+/desmin−). Other tumors showing some SMA-positive myoid reactivity are fibrohistiocytic tumors (82,83), ossifying fibromyxoid tumor (84), some gastrointestinal stromal tumors, endometrial stromal tumors (85), and dedifferentiated liposarcoma (86).
Despite its name, SMA reactivity can be seen in the following: rhabdomyoma (87); some cases of RMS (88), particularly the botryoid type (J.S.J.B., personal observation, 1998); spindle cell carcinomas (10); myoepitheliomas (89); mesothelioma (90); and melanoma (91). Finally, although angiosarcomas are generally SMA negative, reactivity in rare cases (92) has apparently signified the presence or induction of a pericytic cell type.
MYOD1, MYOGENIN, CALPONIN, AND CALDESMON
MyoD1 is a myogenic regulatory gene on the short arm of chromosome 11 that encodes a 45-kd nuclear phosphoprotein expressed only in skeletal muscle (nuclear reactivity). It transactivates other myogenic genes (myogenin and myf5) in the process of development before the other myogenic proteins, such as desmin. In diagnostic pathology, it clearly is a useful marker for RMS (86,93,94). MyoD1 had been used to support the myogenic origin of alveolar soft part sarcoma (95), but reactivity was not nuclear, and other studies have refuted this finding (96).
Myogenin immunoreactivity appears to be specific for the skeletal muscle phenotype. The majority of cases of RMS are positive with nuclear staining (93,94,97). In contrast to MyoD1, myogenin does not show cytoplasmic staining in some nonrhabdomyoblastic tumors.
Calponin is another marker present in smooth muscle; it is found in leiomyosarcoma, myoepithelioma, and myofibroblastic tumors. However, it is not specific, and reactivity can be seen in epithelium and other mesenchymal tumors (98,99). It can complement SMA in putative leiomyosarcomas negative for other myoid markers.
Caldesmon, specifically h-caldesmon, the high–molecular-weight form, is more specific for smooth muscle than is calponin, and it is not found in myofibroblastic lesions, such as fibromatosis or in fibrohistiocytic tumors (99), although reactivity for h-caldesmon is observed in the majority of gastrointestinal stromal tumors (GISTs) (98).
Other muscle markers (e.g., titin, Z-band protein, isoenzymes of creatine kinase) are not used diagnostically.
NEURAL MARKERS
For neural tumors, markers can be roughly divided into those that mark lesions of neuronal origin (e.g., neurofilament) and those that react with nonneuronal elements, including Schwann cell lesions (e.g., S-100 protein and SOX10). Neurofilament, the intermediate filament of neurons, is of value in identifying peripheral tumors, such as neuroblastoma and ganglioneuroblastoma (100–102). Importantly, it does not react with nonneuronal nerve sheath lesions. Ewing sarcoma may also contain neurofilament. Originally, neuron-specific enolase was considered fairly specific for neuronal lesions, as well as selected other tumors. However, this has turned out not to be the case, and a vast array of human tumors are immunoreactive for neuron-specific enolase (103). Thus, the usefulness of this protein as a marker is limited, and the meaning of its immunoreactivity is unclear, despite the availability of monoclonal antibodies to the supposedly more specific γ-isoenzyme. Neuron-specific enolase must never be used alone, the results must be interpreted with extreme caution, and it must be used only when necessary. Synaptophysin and, to a lesser extent, chromogranin are additional proteins to mark neuroblastoma, Ewing sarcoma, and paraganglioma (104,105).
S-100 PROTEIN
Despite all that has been written about it, S-100 antigen is nonetheless quite useful in diagnostic immunohistochemistry, even though it is found in a wide variety of tissues and cell types. In mesenchymal cells, S-100 immunoreactivity is found within chondrocytes, adipocytes, and lesions of schwannian origin. Because the differential diagnosis of a problematic soft tissue lesion rarely involves more than one of these three phenotypes at the same time, S-100 immunoreactivity has real meaning. Around 50% of MPNSTs are positive for S-100 (106–108). Indeed, a positive result of an S-100 stain can be taken as supportive evidence for a tumor of schwannian derivation in the presence of the appropriate histology. Occasional cases of leiomyosarcoma exhibit some S-100 immunoreactivity, usually weak staining that likely is caused by the less specific α-subunit of S-100. Perineurial cells and tumors thereof are S-100 negative but epithelial membrane antigen (EMA) positive (109). Clear cell sarcoma is another S-100–positive tumor, and the cartilaginous areas in chondromatous tumors are immunoreactive. Although S-100 should theoretically stain most myxoid liposarcomas, many are negative or show only focal positivity. Therefore, a negative S-100 result should be considered inconclusive. Interestingly, the dedifferentiated areas of liposarcoma are S-100 negative, as are cases of myxofibrosarcoma. Hashimoto et al. (110) emphasized its usefulness in distinguishing pleomorphic liposarcoma from myxofibrosarcoma. Other S-100–positive tumors include ossifying fibromyxoid tumor (111), myoepithelial tumors, and some cases of synovial sarcoma (112).
SOX10
SOX10 is a neuroectodermal transcription factor that largely parallels S-100 in expression patterns (23,113). Although all benign schwannian neoplasms show diffuse nuclear staining for SOX10, SOX10 is only positive in at most 50% of MPNSTs, and, similar to S-100, usually only a subset of tumor cells expresses this determinant. Nonetheless, the combination of S-100 and SOX10 may be helpful to support a diagnosis of MPNST. Of note, spindle cell and desmoplastic melanomas generally show strong and diffuse staining for SOX10; when such a pattern is encountered, the possibility of melanoma should be considered. Among STTs, SOX10 is also positive in myoepithelioma (23).
CD57 OR LEU-7
CD57 has also been reported in lesions of nerve sheath origin; this may be a nonspecific reaction (114). CD57 has limited (if any) usefulness. Likewise, the specificity of myelin basic protein within selected lesions, particularly those of schwannian origin, is doubtful. Desmin has been detected in rare nerve sheath lesions (115).
NESTIN
This is relatively common in MPNST, but strong reactivity may also be identified in RMS, leiomyosarcoma, melanoma, and some UPS (116). Nestin is therefore not useful for differential diagnosis.
CD99 OR MIC2
MIC2 is a 30- to 32-kd cell surface glycoprotein marker encoded by a pseudoautosomal gene on chromosomes X and Y. With the monoclonal antibodies that are used (O13, 12E7, and HBA71), CD99 is uniformly expressed as membrane staining in Ewing sarcoma, but it is absent in neuroblastoma (117). However, like other surface markers, CD99 is not specific, and it can occasionally be seen in RMS, lymphoma, mesothelioma, UPS, synovial sarcoma, solitary fibrous tumor, nuchal fibroma, and desmoplastic small round cell tumor, among others (117). Nonetheless, in the appropriate context, diffuse membranous staining for CD99 (when other possibilities have been excluded) is helpful to support a diagnosis of Ewing sarcoma.
EPITHELIAL MARKERS
The two sarcomas that characteristically display epithelial markers are synovial sarcoma and epithelioid sarcoma. Biphasic synovial sarcomas are essentially always positive for CK (118) and EMA (119), whereas monophasic tumors exhibit CK and EMA immunoreactivity in approximately 75% to 90% of cases. However, these two markers may not always be present in the same tumor; therefore, performing immunostains for both increases the diagnostic yield. Epithelioid sarcoma is also frequently positive for CK and EMA (120). Other tumors, such as malignant rhabdoid tumor of soft tissue, myoepithelioma, and chordoma, frequently exhibit CK immunoreactivity.
Aberrant CK immunoreactivity can be found in many other sarcomas and in virtually every mesenchymal phenotype. CK has been reported in a significant percentage of smooth muscle tumors (121) and vascular tumors, including angiosarcoma, epithelioid hemangioendothelioma, and pseudomyogenic (epithelioid sarcoma–like) hemangioendothelioma (122,123). Other tumors, such as nerve sheath lesions, occasionally exhibit this phenomenon. This has complicated the interpretation of epithelial markers in mesenchymal tumors and spindle cell lesions in general. When frozen tissues are tested, more tumors and a greater percentage of cells are found to be positive than in paraffin sections. Luckily, aberrant CK in paraffin sections is rarely diffuse; instead, it marks only individual cells widely scattered throughout a given tumor (i.e., a pattern that is different from that of carcinomas and the epithelial-like sarcomas). Nonetheless, the problem of distinguishing the latter from other sarcomas and sarcomas from spindle cell carcinomas is more complicated. To confuse matters further, many spindle cell carcinomas exhibit limited (if any) staining for standard epithelial markers.
EMA does not exhibit complete specificity either. EMA immunoreactivity has been detected in MPNSTs and leiomyosarcomas. Usually, however, the immunoreactivity in these tumor types is focal. EMA reactivity in nerve sheath tumors may reflect a perineurial cell element (109). EMA is also expressed by chordoma, myoepithelial tumors, and the majority of low-grade fibromyxoid sarcomas. When a large series of mesenchymal tumors was tested, promiscuous EMA reactivity was the rule (124), so again, positivity is not definitive for any tumor type.
OTHER MARKERS
MDM2 AND CDK4
These two markers have substantial specificity for liposarcoma including both well-differentiated and dedifferentiated forms (125). Reactivity reflects corresponding molecular changes in liposarcoma. Indeed, use of these markers has demonstrated that many retroperitoneal UPSs are in fact dedifferentiated liposarcomas (126).
β-CATENIN
The protein product of the CTNNB1 gene, β-catenin has recently become a useful marker to support the diagnosis of desmoid fibromatosis (127–129). Normally expressed on the cell membrane, β-catenin shows an aberrant nuclear pattern of staining in 70% to 90% of desmoid tumors. This finding is particularly helpful in limited core needle biopsy samples, in which desmoid fibromatosis may be difficult to distinguish from scar tissue and some other mesenchymal neoplasms (129). However, select other tumor types may also show nuclear staining for β-catenin, such as solitary fibrous tumor (in up to 40% of cases) and low-grade myofibroblastic sarcoma (in around 30% of cases) (128).
SMARCB1 (INI1)
Originally implicated in the pathogenesis of malignant rhabdoid tumor, SMARCB1 (also known as INI1) is a ubiquitously expressed nuclear protein involved in transcriptional regulation (130). Loss of SMARCB1 protein expression (which reflects biallelic genetic inactivation, usually secondary to mutations) is a consistent finding in malignant rhabdoid tumor and has become a standard way to confirm the diagnosis (131,132). SMARCB1 deficiency is not specific for malignant rhabdoid tumor; loss of protein expression is also observed in around 90% of cases of epithelioid sarcoma, the majority of epithelioid MPNSTs, and a small subset of myoepithelial carcinomas of soft tissue (133–135).
TFE3, ANAPLASTIC LYMPHOMA KINASE, AND STAT6
The finding of specific chromosomal translocations can also be exploited by immunohistochemistry. Several antibodies that recognize the protein products of gene rearrangements are available for diagnostic immunohistochemistry. Notable examples include TFE3 (rearranged in alveolar soft part sarcoma) (136), anaplastic lymphoma kinase (ALK; involved in translocations in around 50% of inflammatory myofibroblastic tumors) (137), and STAT6 (rearranged in solitary fibrous tumor) (138). Whereas TFE3 and STAT6 are nuclear markers, ALK is usually expressed in the cytoplasm of tumors harboring gene rearrangements. Application of these markers is discussed in more detail in the sections of this chapter devoted to each of these tumor types.
DOG1, TLE1, AND MUC4
Several diagnostic immunohistochemical markers have been identified by gene expression profiling. DOG1 (also known as anoctamin 1, ANO1) is a highly sensitive and specific marker for GIST (139). DOG1 shows a predominantly membranous pattern of staining and is most useful to support the diagnosis of gastric epithelioid tumors that are negative for (or show limited reactivity for) KIT (140). TLE1 is an excellent discriminator of synovial sarcoma from histologic mimics (141). This nuclear protein is expressed at high levels in 80% to 90% of synovial sarcomas, whereas weak nuclear staining is observed in only 10% to 20% of MPNSTs and solitary fibrous tumors (142). TLE1 is also useful to distinguish poorly differentiated (round cell) synovial sarcoma from Ewing sarcoma because the latter tumor type is consistently negative for this marker. MUC4 is a normally epithelial apomucin that is abnormally expressed in two fibroblastic sarcomas, low-grade fibromyxoid sarcoma and sclerosing epithelioid fibrosarcoma (143,144). Because MUC4 is negative in nearly all other STTs, this marker is very useful in differential diagnosis.
WT1
Although this marker was originally identified as present in most desmoplastic small round cell tumors, immunoreactivity and mRNA expression have since been observed in many sarcoma types. Of note, antibodies that recognize the C-terminus of WT1 (as opposed to the standard WT1 antibodies directed against the N-terminus of the protein, which are widely used for mesothelioma and serous carcinoma) must be applied to support the diagnosis of desmoplastic small round cell tumor because the characteristic translocation results in expression of only the C-terminus of the protein (145).
PROGNOSTIC IMMUNOHISTOCHEMISTRY
Relatively few studies have focused on the usefulness of oncoprotein and proliferation markers in assessing patient prognosis in soft tissue sarcomas. Therefore, no conclusive prognostic markers are available, although the following proteins have been studied in selected tumors: p53 (146), Rb protein (147), Ki-67 (148), MDM2 (149), vascular endothelial growth factor (VEGF) (150), and adhesion molecules (151). The nm23 gene has also been evaluated (152). WT1 and p16 may have prognostic value.
ELECTRON MICROSCOPY
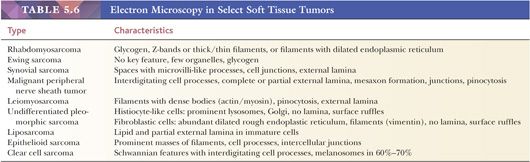
Electron microscopy is a well-studied technology, so it is discussed only briefly here. Review articles on STT ultrastructure are fairly common (153,154), and further references are provided under the relevant individual headings. Table 5.6, a partial summary of electron microscopy findings, is meant to be a starting point for the ultrastructure of STT. Electron microscopy, which is more expensive than immunohistochemistry and is occasionally subject to artifact and sampling error, has largely been replaced by immunohistochemistry for STT diagnosis. Most institutions lack expertise in the soft tissue field, and interpretation can be difficult for the novice.
EVALUATION OF PROGNOSTIC FACTORS
GRADE
The grading of sarcomas (155–159) has always been complex, variable, and somewhat subjective because agreed-on standards are lacking. The area is made all the more confusing when certain lesions are excluded from the grading process and are assigned automatic grades. The rationale for automatic grading is based on the knowledge of the natural history; nonmetastatic lesions, such as DFSP and well-differentiated liposarcoma, have been assigned to grade 1, whereas highly metastatic aggressive tumors, such as RMS, synovial sarcoma, and angiosarcoma, have been assigned to grade 3. Still other lesions are denied the appropriateness of a grade 1 designation (e.g., epithelioid and clear cell sarcomas).
The following four types of grading systems are in use: (a) the original three-tiered system proposed by the American Joint Committee on Cancer (AJCC) (157); (b) another three-tiered system based on necrosis (National Institutes of Health system) (155); (c) a combination of differentiation, mitotic rate, and necrosis (French Federation of Cancer Centers Sarcoma Group system [FNCLCC]) (156,160,161); and (d) the “low/high” two-tiered system used by surgeons (surgical staging system) (162). In the older, widely used AJCC system, the automatic grading already discussed was used, and only the category of spindle cell sarcomas permitted assignment to all three grades. However, the exact histologic distinction between grades was not clarified. Traditionally, the degree of cellularity, pleomorphism, and mitotic activity determined the grade of spindle cell sarcoma. Costa et al. (155) defined the key importance of necrosis in determining the prognosis. Therefore, they assigned cases with no necrosis to grade 1, cases with 15% necrosis to grade 2, and cases with more than 15% necrosis to grade 3. This system had definite predictive value, but they studied very few cases of RMS, and they did not address the question of whether certain histologic types have a poor prognosis in the absence of necrosis. Multivariate analyses have documented the prognostic importance of necrosis, tumor size, mitotic rate, depth, and status of margins (163,164).
The issue is confounded by the fact that no one system has been universally accepted. Some authors have proposed a two-tiered system—a low and a high grade—that is helpful for planning surgery. However, because most sarcomas are not low grade, this produces an inordinate number of high-grade lesions, and thus, it is not a true prognostication scheme. Furthermore, histologic features may affect some automatically graded tumors, such as synovial sarcoma and angiosarcoma, such that automatic grading is made unduly deceptive. A generally acceptable grading system must be simple, and it should be able to be applied without exception. The French system (FNCLCC) is based on key factors such as necrosis and mitotic rate proven to affect prognosis; it is being used increasingly in this country. However, until agreement is reached on a grading system, pathologists may continue to use any one of several systems while alerting clinicians about which grading system is being used. Using multiple systems is also appropriate—for example, “sarcoma, high grade, grade 2 of 3 with French system.” Of note, the most recent (seventh) edition of the AJCC Cancer Staging Manual (165) adopted the FNCLCC grading system, which has also been recommended in the College of American Pathologists Cancer Protocols.
STAGING SYSTEM
The current staging system for sarcomas is critically dependent on the grade assignment, further emphasizing the need for a uniform grading system. In the AJCC staging system (165), stage I is reserved for grade 1 tumors (irrespective of size or depth). Tumors that are grade 2 or 3 and no greater than 5 cm in greatest dimension are assigned stage IIA, whereas larger tumors that are grade 2 are assigned stage IIB. If lymph node involvement is present, or the tumor is grade 3, deep, and greater than 5 cm, the tumor is assigned to stage III. Stage IV is any tumor, regardless of grade, with distant metastasis. When this system is used, the 5-year survival rates for extremity soft tissue sarcomas are as follows: for stage I, 90%; stage II, 81%; and stage III, 56% (165).
FUTURE PROGNOSTICATION
Recently, prognostic nomograms incorporating histologic type, grade, tumor size, patient age, depth, and anatomic site have been developed to help stratify patients with sarcoma for risk of metastasis (166–168). However, most such systems were developed based on the relatively common sarcoma types; it is therefore not clear whether these nomograms are reliable for rare tumor types. Tumor type–specific nomograms have also recently been established (169,170).
OTHER PROGNOSTIC FEATURES, INCLUDING DNA PLOIDY
Factors such as tumor size, depth, and location are accepted prognostic indicators. In general, the prognosis is more favorable if a tumor is small, if it is superficial rather than deep, and if it is distal rather than proximal. The degree of cellular differentiation, a feature that is frequently difficult to quantify, nonetheless appears to have some impact on the prognosis. DNA flow cytometric analysis has shown a rough correlation between ploidy and grade, but the lack of aneuploidy does not imply a low grade; several highly aggressive grade 3 tumors (e.g., angiosarcoma, RMS) are frequently diploid. Several studies have documented the effect of DNA ploidy on sarcoma prognosis. In childhood tumors such as neuroblastoma, aneuploidy is a favorable finding; in RMS, the data are complex, but aneuploidy may again be favorable in some instances. In adult sarcomas, the opposite is true; aneuploidy has been shown, in general, to be unfavorable for sarcomas and specifically for certain histologies, such as uterine leiomyosarcoma, GISTs, synovial sarcoma, epithelioid sarcoma, clear cell sarcoma, and possibly UPS. The S-phase percentage may be important, as two studies have shown (171,172). None of these methodologies are used routinely in clinical practice.
OTHER STUDIES AND FUTURE PROSPECTS
Surprisingly, certain STTs contain hormonal receptors of one kind or another, and they occasionally respond to antihormonal therapy (173–175). In the future, cell proliferation markers and oncogene proteins or messages may assist in the assignment of patients to favorable or unfavorable prognostic categories. With immunotherapeutic approaches gaining promise, the local interaction between immune effector cells and sarcoma cells merits further investigation (176,177). With UPS as an example, a favorable prognostic significance has been demonstrated for human leukocyte antigen (HLA)-DR–positive tumor cells (178) and for heat shock protein (HSP)-27 positivity (179). Finally, mutation analysis with targeted molecule-based therapy, which has already been used in GISTs, will be important in the future.
SARCOMA SYNDROMES
Many genetic and clinical syndromes are associated with mesenchymal tumors, in addition to that which usually comes to mind—namely, neurofibromatosis type 1 (180). The clinical group can be subdivided into those with a constellation of findings, such as Carney complex (181), and those related to laboratory information, such as hypercalcemia (182) and hypoglycemia (183). The Carney triad is the association between gastric GIST, pulmonary chondroma, and paraganglioma (184); pathologists should consider it when gastric GIST is encountered in a young patient. Kasabach-Merritt syndrome is defined as disseminated intravascular coagulation resulting from benign (185) or malignant (186) vascular tumors. Tryptophan ingestion and eosinophilic fasciitis and/or myalgia syndrome (187,188) is another such association. Genetic sarcoma syndromes include the spectrum of cancer families with sarcomas, as well as the following: breast, lung, endometrial, and adrenal cortical carcinomas (189,190); Gardner syndrome (lipomas, mesenteric fibromatosis) (191); tuberous sclerosis (hamartomas, renal angiomyolipomas) (192); and Bannayan-Riley-Ruvalcaba syndrome (lipomas, hemangiomas, macrocephaly) (193), to mention only a few.
SOFT TISSUE LESIONS: LESIONS MIMICKING SARCOMAS (PSEUDOSARCOMAS)
NODULAR FASCIITIS
Nodular fasciitis (NF), which is commonly mistaken for a sarcoma, is a myofibroblastic proliferation (194–199) characterized by extremely rapid growth (more so than the usual sarcoma); it achieves its small size of 2 to 3 cm in a matter of weeks. Lesions are only rarely larger or of longer duration (3 months to 1 year). A few patients report pain and a history of trauma to the area. Most cases occur in persons between the ages of 20 and 50 years, with men and women being equally affected. NF is commonly found on the forearm, arm, face, and shoulder, although it may occur in any exterior location. NF, which is usually well circumscribed, is tan to gray-white with a myxoid appearance, and it is not very distinctive grossly.
In NF, several histologic findings are key, the most important of which is apparent at low power; nearly all cases have a characteristic architecture in the form of a zonation effect (Fig. 5.3). The center is hypocellular, and it may have an eosinophilic fibrinous area; alternatively, some cases show central light hyalinization. At the periphery, the appearance is more hypercellular, with small vessels in a lobular array abutting a collagenized zone. In between, spindly cells populate a loose myxoid area. The lesion typically is located just above the muscle layer, with the lateral thick bands of collagen representing the fascial layer. The predominant cell is a variably sized bipolar spindle cell with long cytoplasmic processes. Scattered cells have the characteristic tripolar or stellate shape of the myofibroblast, the cell type identified ultrastructurally. Immunohistochemically, many of these cells exhibit strong and diffuse staining with SMA (Fig. 5.3D) (81), which can be used to diagnostic advantage. Some cells may react with MSA, but desmin is usually absent. Although the cells may appear worrisome with their active enlarged vesicular nuclei and nucleoli, they are uniform with no pleomorphism. The hallmark is the loose arrangement of the cells in a “tissue culture”–like manner. Although most cases have a random pattern, some “cellular” variants contain storiform areas, interconnecting bundles, or cystic areas focally, even in the same lesion; in these tumors, small microcysts with mucin should be searched for at low power. An inflammatory component of lymphocytes and macrophages is practically always present to some degree; scattered mast cells and histiocytic giant cells can also be seen. Thus, CD68 shows many histiocytes and giant cells, unlike fibromatosis and fibrosarcoma. Plasma cells and neutrophils are rare; large numbers of these should cause alarm (195) because they are a common component of sarcomas. Scattered red blood cells are often seen, and they may be superficially reminiscent of Kaposi sarcoma; however, the pattern is different, the lesion is too deep, and hemosiderin is hardly ever found. Lastly, mitotic activity may be prominent. Most cases contain one to five mitotic figures per 5 hpf; lesions with a higher mitotic rate should be viewed with caution because they may be a malignant process. Nonetheless, NF is the classic example of an important dictum—benign lesions may be quite mitotically active.
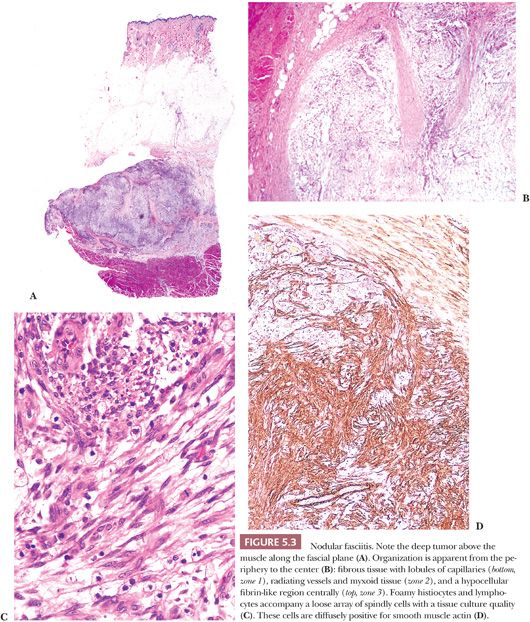
Perhaps the simplest and most useful subclassification of NF is that of Bernstein and Lattes (195). They divide NF into the following five subtypes: (a) the usual type already described; (b) the reactive type, with its radially oriented vessels around a central loose area, which corresponds to the “repair” variant of Allen (194); (c) the cellular type, with microcysts and little or no zonation effect and imperfect storiform regions resembling fibrous histiocytoma; (d) a metaplastic type, with focal osteoid or chondroid metaplasia; and (e) the proliferative type, which is the same as “proliferative fasciitis” described by others and which Bernstein and Lattes (195) and others believe is part of the spectrum of NF. Shimizu et al. (198) analyzed 250 cases and noted that the three subtypes they described were related to the duration of the lesion; the myxoid type had a short history, the cellular type was of intermediate duration, and the fibrous type had the longest duration. The long-standing belief that NF is reactive in nature has recently been proven incorrect; nearly all cases harbor rearrangements of the USP6 locus (17p13), most often resulting in an MYH9-USP6 gene fusion (200,201).
The most common diagnostic difficulty arises with other fibrous tumors, including fibromatosis, fibrosarcoma, BFH, and UPS. However, unlike NF, each of these has a uniform pattern, and none has the microcysts often seen in the cellular NF variant. Neither fibromatosis nor fibrosarcoma has the loose texture of NF; a uniformly collagenized matrix distinguishes the former, whereas the tight “herringbone” pattern separates the latter. The inflammatory “MFH” pattern of dedifferentiated liposarcoma may offer a significant problem, but nuclear atypia, foam cells, plasma cells, and neutrophils are not found in NF. Differentiation between cellular NF and a deep BFH is occasionally extremely difficult; the factors that favor fibrous histiocytoma include a more superficial location, a closely knit pattern of cells, wide storiform areas, a lack of microcysts, and an infiltrating border. Nuclear pleomorphism favors a fibrohistiocytic lesion. Problematic is distinction from the rare low-grade myofibroblastic sarcoma that mimics fasciitis and is often superficial; however, it lacks microcysts and CD68-positive histiocytes and giant cells and has an infiltrative border.
The points to remember in the diagnosis of this lesion are that (a) the nuclei of NF are never hyperchromatic or pleomorphic, (b) NF rarely arises in the dermis, (c) the average mitotic rate in NF is one per hpf, (d) plasma cells and neutrophils are unusual in NF, and (e) NF manifests rapid growth. The diagnosis of NF by aspiration biopsy cytology, although possible, is probably hazardous. Bernstein and Lattes (195) have elegantly demonstrated that NF is clearly a nonrecurrent lesion. Of their 134 cases, all of the recurrent cases were rediagnosed as something else on review, and even partially excised NF did not recur. If all of the reports are surveyed, well-documented NFs are found to recur rarely (1% of cases) (198).
PROLIFERATIVE FASCIITIS
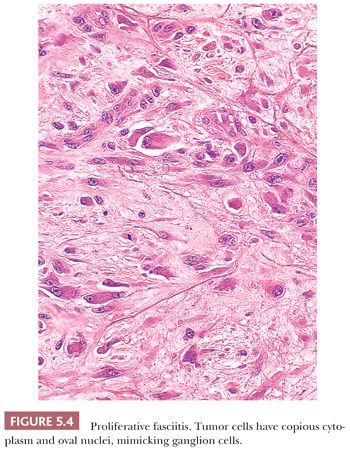
Proliferative fasciitis (202) resembles NF in the following respects: the location within deep tissues, the small size, the presence of a zonation pattern, and the loose quality of cell growth. Strikingly different are the cells themselves. More polygonal in shape, they have abundant amphophilic cytoplasm surrounding very large, but oval to round, vesicular nuclei with prominent nucleoli that highly resemble ganglion cells (Fig. 5.4). They are dispersed singly within a slightly myxoid or collagenized stroma, and they are more common around the central fibrinous area than at the periphery. To those familiar with it, the lesion is unique, and it is not easily mistaken for anything else. Unlike NF, it occurs in an older age group (>50 years), but otherwise, it has a similar presentation and a nonrecurrent natural history.
PROLIFERATIVE MYOSITIS
Proliferative myositis (203) is a related lesion in which ganglion-like cells proliferate between muscle fibers and then separate each of them so that, at low power, a distinctive “checkerboard” appearance is visible. These cells resemble fibroblasts ultrastructurally and immunohistochemically, and they are clearly nonmuscular. Although the checkerboard pattern is occasionally mimicked by an infiltrating lymphoma or fibromatosis, in neither of these lesions are the cells as unusual looking or as dispersed.
POSTOPERATIVE SPINDLE CELL NODULE
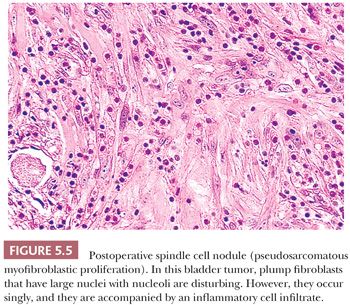
Although postoperative spindle cell nodule does not occur in the soft tissues, it does raise the differential diagnosis of sarcoma. In the original report (204), tumorlike masses of the vagina, prostatic urethra, or bladder developed within 5 weeks to 3 months after a prior surgical procedure. An edematous ulcerating lesion with infiltrating borders emerged. Plump spindle cells with abundant amphophilic cytoplasm are found, accompanied by a prominent chronic inflammatory infiltrate and even extravasated red blood cells (Fig. 5.5). The mitotic rate may vary between 1 and 25 per 10 hpf. Although the postoperative spindle cell nodule simulates sarcoma, the cells are not hyperchromatic, and atypical mitotic figures are not seen; the myxoid quality with cell separation and inflammation, when combined with the history, are characteristic. Immunohistochemically, the proliferating cells have the staining pattern of myofibroblasts (positive for desmin and muscle actin), but they may display CK reactivity, which causes spindle cell carcinoma to be considered in the differential diagnosis. The postoperative spindle cell nodule is not limited to the genitourinary tract, but it may rarely be seen in the endometrium. Similar reactive lesions known as pseudosarcomatous fibromyxoid tumors or pseudosarcomatous myofibroblastic proliferation also occur in the genitourinary tract, but they are not associated with antecedent surgery or trauma (205,206). Around 50% of cases are immunoreactive for ALK (206), although ALK gene rearrangements are rarely detected.
SPINDLE CELL LIPOMA
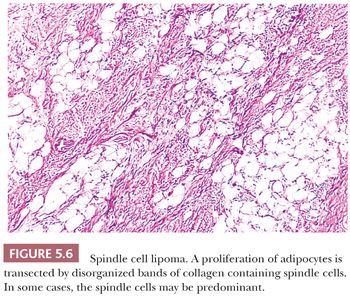
The name spindle cell lipoma is highly descriptive of the pathology—a lipoma with a quite variable content of bland spindle cells (Fig. 5.6). The following two variations are found: the more common myxoid lesion and a nonmyxoid fibrous type. Notably absent from either type are the lipoblasts and the capillary network of myxoid liposarcoma. The spindle cells morphologically remind one of those seen in neurofibroma. The nuclei may be wavy, and they seem to stream along in the same direction. In actuality, distinguishing between a neurofibroma and a fibrous histiocytoma deep within fat can be difficult in those cases in which spindle cells predominate. The spindle cell lipoma (207,208), however, fails to exhibit a storiform pattern; it is frequently more cellular than the usual neurofibroma; and it is S-100 negative and CD34 positive. A recent study has documented consistent loss of expression of the retinoblastoma protein (RB1) by immunohistochemistry in spindle cell lipoma, as well as other STTs characterized by 13q14 chromosomal losses (mammary-type myofibroblastoma and cellular angiofibroma) (209); this marker may be helpful in differential diagnosis. Its clinical presentation is characteristic, with a vast majority of lesions occurring on the back of the neck or on the shoulder in elderly men; however, odd locations have been reported. In the fibrous variant, thin bands of collagen accompany the proliferation in a manner that is similar to that seen in solitary fibrous tumor. For the tumor to recur is unusual.
PLEOMORPHIC LIPOMA
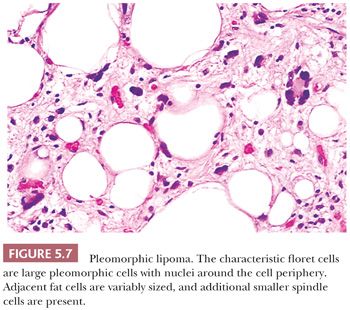
As the name implies, this unusual but benign tumor (210,211) is characterized by very large cells against a background of fibroadipose tissue; practically, all cases are superficial—in the subcutaneous tissue. The following four histologic features should be noticed: (a) circumscription with encapsulation (unlike liposarcoma), (b) a wreathlike arrangement of the nuclear lobes in the characteristic floret cells (Fig. 5.7), (c) a lack of prominent vascularity, and (d) a rarity of lipoblasts. Smaller spindly cells with elongated nuclei in a lightly collagenized background are also seen, and these are reminiscent of spindle cell lipoma. The clinical setting of this CD34-positive tumor is similar to that of spindle cell lipoma; it is a superficial tumor found almost exclusively on the back of the neck in older men. Cases in women or unusual locations are infrequent. At one time, the floret cells were considered pathognomonic for this tumor, but they can occasionally be identified in liposarcoma. Extremely rare mitotic figures and a large size do not detract from the diagnosis. No aggressive behavior has been reported; however, one or two instances of local recurrence have been documented. Pleomorphic lipoma is rare, being only one-tenth as common as the spindle cell lipoma, which in turn accounts for only 1.5% of all adipocyte tumors. The cytogenetic findings show a relationship to spindle cell lipoma and clearly differ from those of atypical lipomatous tumor (212).
LIPOBLASTOMA
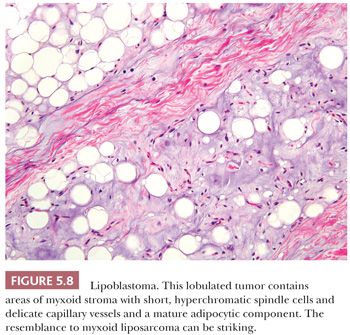
Vellios et al. (213) initially described the benign fatty tumor called lipoblastoma. It characteristically occurs on the extremities of young children, particularly boys, before the age of 3 years (213), but rare cases can occur in adulthood. In this lesion, prominent fibrous septa divide the adipose tissue into lobules, and a light myxoid quality is visible at low power. A range of differentiation, from short spindle cells within the myxoid matrix to increasingly larger and more vacuolated cells to the mature adipocyte (Fig. 5.8), can be noted at high power. Despite the presence of these scattered developing lipoblasts, the lesion usually lacks the capillary network of myxoid liposarcoma. Furthermore, although myxoid liposarcoma harbors DDIT3 rearrangements (with either FUS or EWSR1, which can be detected by FISH), lipoblastoma is characterized by PLAG1 rearrangements (214,215), leading to nuclear expression of the PLAG1 protein, which can be detected by immunohistochemistry (216). If these lesions are followed for some time, they gradually mature, with the disappearance of the spindle and myxoid elements. Such lesions are virtually indistinguishable from conventional lipomas, other than the presence of a lobulated architecture with thin fibrous septa. The clinician should have a natural reluctance to diagnose liposarcoma in children because such lesions are exceedingly rare at that age.
CELLULAR ANGIOLIPOMA
Most angiolipomas are easily recognized as the mainly fatty subcutaneous tumors that they are. Unusual examples include an overgrowth of the vascular component mimicking Kaposi sarcoma (217) or even other solid lesions, such as smooth muscle tumors. Because the vessels are probably small venules, spindle cells representing pericytes dominate the appearance, and these are intermingled with endothelial cells (Fig. 5.9). The cellular angiolipoma is set apart from Kaposi sarcoma by the following features: (a) it is deeper because it is a subcutaneous tumor; (b) it is circumscribed; (c) it nearly always contains small vessel thrombi; and (d) it incorporates mature fat (however little) within its confines. When angiolipomas arise in the subcutaneous tissue of the breast, they may be mistaken for mammary angiosarcoma; however, the latter tumor type arises in the breast parenchyma and entraps breast ducts and lobules.
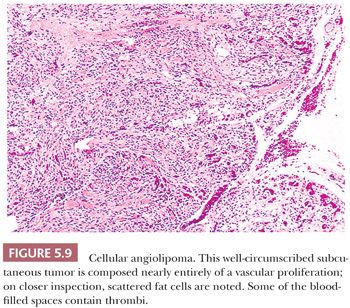
BIZARRE LEIOMYOMA
That pleomorphism is not a sine qua non for sarcoma is best exemplified by this highly pleomorphic but benign smooth muscle tumor, which is most frequently found in the uterus. Under various designations (e.g., atypical, symplastic, apoplectic), these tumors are characterized by quite large, atypical-appearing nuclei in an otherwise classic smooth muscle tumor (Fig. 5.10). They tend to be small and to have either very few or no mitotic figures. Whenever nuclear atypia is encountered in a putative soft tissue or retroperitoneal leiomyoma, mitoses should be sought very carefully, and a malignant diagnosis should be seriously considered.
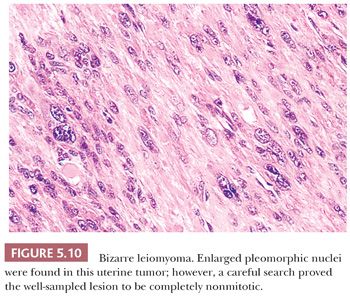
FETAL AND GENITAL RHABDOMYOMA
The fetal rhabdomyoma, which is occasionally confused with the better differentiated myotubular forms of RMS, occurs mainly in the head and neck of toddlers (218). A similar lesion is found in the adult female genital tract (219). Small parallel bundles of easily recognized rhabdomyocytes are separated by thin strands of collagen and vascular tissue or by less differentiated cells. The rhabdomyocyte groups intersect in a random fashion, although the entire lesion retains an architectural structure of separated bundles of streaming tumor cells (Fig. 5.11). The nuclei are bland in appearance, and only occasional nucleoli can be seen. Notably, the tumor lacks mitotic activity, a cambium layer, and the jaggedly infiltrating pattern of RMS at its periphery. Thus, whereas the rhabdomyoma is not really circumscribed, most tumor cells tend to stop at roughly the same location in the lateral and deep aspects of the tumor. When the fetal type is highly cellular and compact, it becomes more difficult to distinguish from RMS (209). A recent study has identified PTCH1 mutations and deletions in fetal rhabdomyoma (220). In the female genital tract, the genital type occurs at a rather advanced mean age (36 years) in comparison with genital RMS, and it is much less cellular than the fetal type already described.

PAPILLARY ENDOTHELIAL HYPERPLASIA
Although papillary endothelial hyperplasia (221–223) may commonly be mistaken for an angiosarcoma, these can be easily differentiated. First, most of these reactive lesions occur within a vessel or a vessel-like structure, and therefore, unlike the malignant tumor, they are circumscribed. Second, the nuclei are quite bland. Third, rather than ramifying the vascular channels, the papillary formations tend to occupy a contiguous large space that can be traced around much of the proliferation (Fig. 5.12). In foci, a fibrin-like matrix probably representing a stage of an organizing thrombus is observed in this lesion. Although the lesion may occur almost anywhere, it is frequently found on the fingers as a painful nodule. Pathologists should be aware that it can proliferate in a hematoma or blood-filled space within another tumor, causing further confusion, or that it can result from procedures such as fine-needle aspiration of organs, where it is noncircumscribed, simulating angiosarcoma even further.
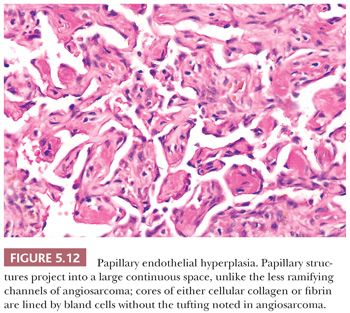
ATYPICAL FIBROUS POLYPS
Atypical fibrous polyps are unusual but benign lesions that occur most often around orifices such as the nose, vagina, and anus, although they also form elsewhere. Within what otherwise is bland fibrous tissue, highly atypical cells are seen superficially resembling those in a sarcoma. However, unlike the latter, atypical fibrous polyps exhibit multiple but uniform oval nuclear lobes that all are the same size (Fig. 5.13). These lobes are often clustered, and they overlap (similar to the floret cell). Another characteristic is that the atypical cells are scattered singly throughout the lesions, never appearing to group. Therefore, another rule of STT pathology is that neoplasms tend to exhibit crowding and adjacent cell growth, in contrast to the single-cell growth of reactive fibroblastic lesions. Furthermore, cells with multiple nuclear lobes are not truly atypical or pleomorphic and should be ignored when considering malignancy.
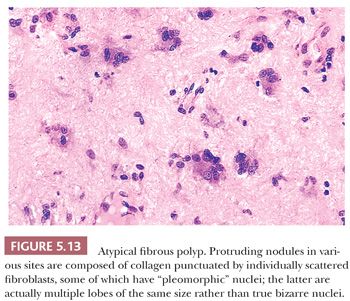
GIANT CELL FIBROBLASTOMA
Giant cell fibroblastomas are discussed in “Dermatofibrosarcoma Protuberans.”
MYXOMAS
Intramuscular myxoma is an extremely gelatinous lesion that commonly appears as a circumscribed mass deep within the muscle, usually in an extremity (224,225). Strangely enough for a nearly avascular lesion (Table 5.7, Fig. 5.14), an occasional myxoma may reach a considerable size (10 to 13 cm). The bulk of the lesion consists of a slightly basophilic proteoglycan matrix, and at high power, only a few stellate or bipolar cells with oval nuclei are seen. Lesions showing higher cellularity are often referred to as cellular myxoma. Unlike myxoid liposarcoma, the lesion is circumscribed and very hypocellular, and it lacks any significant vascularity. An inapparent architecture is present; toward the periphery, an increased number of spindle cells abut on a lightly collagenized capsule. Centrally, a mucinous cyst or focal eosinophilic fibrinous quality can often be appreciated. A GNAS mutation can be found in most examples of intramuscular myxoma (226,227). Intramuscular myxoma rarely, if ever, recurs. The cardiac myxoma (228) is different because it contains endothelial cells in a fibrinous or myxoid matrix; cardiac myxoma is a frequent component of the Carney complex (229). Most cardiac myxomas in Carney complex exhibit mutations in PRKAR1A (230), which result in loss of expression of the PRKAR1A protein by immunohistochemistry (231). Nonsyndromic tumors lack such mutations but may also show loss of protein expression in some cases (231).
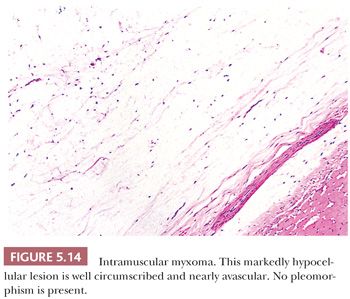

MYOSITIS OSSIFICANS
Massive calcification and other findings appear quickly in myositis ossificans (232). Within the span of 4 to 6 weeks after the commonly associated traumatic injury, pain and tenderness develop, followed by a mass lesion within the extremity musculature, typically in a young male patient (Fig. 5.15). The massive calcification of the lesion is apparent on radiographic study, and histologically, it has the configuration of lamellar bone. It is a solitary and well-circumscribed lesion with organization into the following three zones: a periphery of well-formed lamellar bone gradually maturing from poorly formed trabeculae of osteoid in the middle zone and, in the center, a fibroblastic proliferation with remarkable similarity to NF. Older, mature lesions resemble soft tissue osteomas. Before the lesion is well developed, it may resemble osteosarcoma, but, in extraosseous osteosarcomas, the osteoid is usually located more centrally, which is the reverse of myositis. Putative cases of “malignant transformation” in myositis ossificans have been reported; it seems likely that such cases were instead examples of extraskeletal osteosarcoma with areas showing histologic overlap with myositis ossificans. Like other reactive lesions, myositis ossificans shows zonation at low power.
OTHERS
Many other pseudosarcomatous lesions (233–235) have been recognized, and considering them in the differential diagnosis and keeping the possibility of a sarcoma mimic in mind at all times are important. To reiterate, the possibility of spindle cell carcinoma should always be entertained whenever a lesion is in or near an organ or the skin; likewise, melanoma is always part of the differential diagnosis.
FIBROUS LESIONS
NF, proliferative myositis, and atypical fibrous polyps have already been described in “Soft Tissue Lesions: Lesions Mimicking Sarcomas (Pseudosarcomas).”
EOSINOPHILIC FASCIITIS AND EOSINOPHILIA-MYALGIA SYNDROME
In 1975, Shulman described a diffuse fasciitis with eosinophilia, a fibrosing inflammatory condition of skin, subcutaneous tissue, and fascia that has come to be known as eosinophilic fasciitis (236). Patients had painful symmetric thickenings, particularly on the thighs. The infiltrates of eosinophils were typically accompanied by clusters of lymphocytes, mast cells, and histiocytes, and the resultant lesions were either fibrotic or fibromyxoid (Fig. 5.16). Myopathy was observed only occasionally. In 1990, the ingestion of l-tryptophan became associated with a similar disease, the eosinophilia-myalgia syndrome, which is characterized by myositis and neuritis. After a flurry of articles and investigations, a toxin contaminating the drug during manufacturing was identified as a likely cause of eosinophilia-myalgia syndrome (187,237,238). The pathologic findings of eosinophilia-myalgia syndrome overlap with those of eosinophilic fasciitis, and only a history of l-tryptophan can distinguish between the two.
TUMEFACTIVE FIBROINFLAMMATORY LESIONS
Cases in which the surgeon observes a tumor but the biopsy specimen shows only fibrosis and mild inflammation are referred to as tumefactive fibroinflammatory lesions. Many such cases present as head and neck tumors, although they can also be seen on the extremity (239). The fibrosis is characteristically dense, and occasionally, obliterated vascular structures are identified on elastic stain. The etiology is unknown.
Focal myositis is another histologically bland entity with a mass-forming presentation (240).
RETROPERITONEAL FIBROSIS
Although true fibromatosis of the retroperitoneum may occur in Gardner syndrome, most cases of retroperitoneal fibrosis do not appear to be a proliferation of fibroblasts as much as they do a scarring process (241). This lesion typically shows bilateral impingement on the ureters, resulting in hydronephrosis. Fibrosis with clusters of lymphocytes and plasma cells is found. Occasionally, large presacral masses can be seen (242). The pathogenesis may be related to autoimmunity or to certain drugs, such as methysergide.
KELOID
The keloid, which is easily recognized as a hypertrophic scar, is typified by the presence of very thick, highly eosinophilic collagen bands. The reason for mentioning this entity is that the presence of such “keloidal collagen” in another lesion, such as fibromatosis, may provide a clue to its fibroblastic nature.
FIBROMAS
Several distinct tumor types are referred to as fibromas. Hypocellularity accompanied by dense collagen is the main feature in the sclerotic fibroma (243), but pleomorphic skin tumors have been described (244). If a pleomorphic fibroma is a consideration, excluding the desmoplastic melanoma with an S-100 stain is wise.
Better known are the examples of fibroma of tendon sheath (245). In the usual clinical setting, a male patient between 30 and 50 years of age presents with a nodule on the fingers, hands, or wrist. These well-circumscribed, generally small tumors may be lobulated. The dense collagen is variably stained, and small vascular slits in the tumor separate the widely scattered spindled fibroblasts (Fig. 5.17). Focally, the lesions may be cellular, particularly at the periphery, but mitotic figures are scarce. Some authors have suggested a relationship to giant cell tumor of tendon sheath; however, giant cells are rarely seen, and this assertion has been criticized. A subtype with pleomorphism in which giant nuclei are dispersed singly, as in atypical fibrous polyps, has been described (246).
The nuchal fibroma often occurs on the back of the neck and presents as fibrofatty tissue with very dense collagen and occasional cartilaginous metaplasia (247).
Associated with familial adenomatous polyposis syndrome, the Gardner fibroma is a highly collagenized and hypocellular tumor (Fig. 5.18) similar to nuchal fibroma occurring predominately in the back, chest wall, and flank of children (248). Similar to desmoid fibromatosis, Gardner fibroma usually shows nuclear immunoreactivity for β-catenin (248).
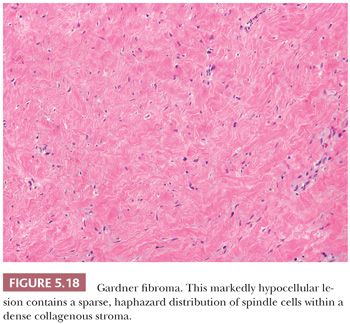
COLLAGENOUS FIBROMA
Collagenous fibroma, which is also called desmoplastic fibroblastoma (249), is a subcutaneous lesion without primitive elements (250). It most closely resembles a very hypocellular fibromatosis with scattered stellate spindle cells (Fig. 5.19), some of which show SMA positivity. Although some lesions are infiltrative, most are circumscribed. No recurrences have been reported.
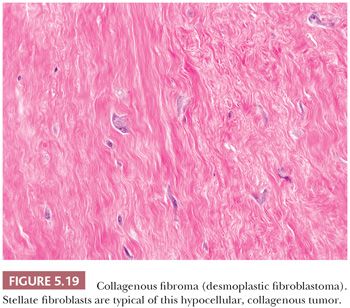
NASOPHARYNGEAL ANGIOFIBROMA, CELLULAR ANGIOFIBROMA, AND SOFT TISSUE ANGIOFIBROMA
Nasopharyngeal angiofibroma is a tumor that occurs in young male patients, particularly in the nasopharynx (251) (see Chapter 21). It consists of bland fibroblastic cells proliferating among irregular, thin-walled blood vessels. Cellular angiofibroma has a predilection for the vulva and inguinal region. This usually cellular tumor is composed of short fascicles of bland spindle cells with prominent thick-walled, hyalinized blood vessels and variably fat (Fig. 5.20) (252). These lesions show considerable histologic overlap with and have similar cytogenetic features as spindle cell lipoma (253). Angiofibroma of soft tissue is composed of small hyperchromatic spindle cells in a collagenous or myxoid stroma with numerous, branching blood vessels superficially resembling those in myxoid liposarcoma but with thicker walls (Fig. 5.21) (254). Soft tissue angiofibroma has a t(5;8)(p15;q13) translocation, resulting in an AHRR-NCOA2 fusion (255).
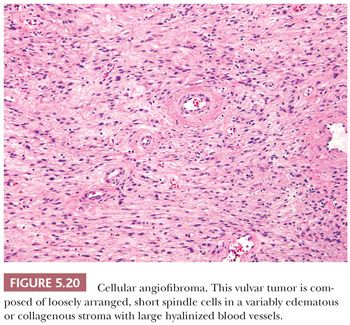
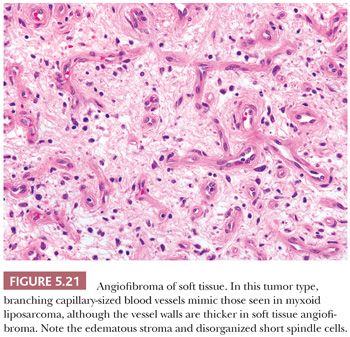
ELASTOFIBROMA
Nearly every example of elastofibroma has occurred in the mid back and scapular region (256,257). On low power, it appears to be a fibroblastic or fibrofatty proliferation that is densely collagenized, but at higher power, circular and snakelike eosinophilic bands of elastic tissue are noted (Fig. 5.22). Usually, these elastic fibers have a beaded appearance and constitute most of the stroma. The underlying cause of this pseudotumor produced by abnormal elastogenesis is unknown, but the surgically excised lesion has no tendency to recur.
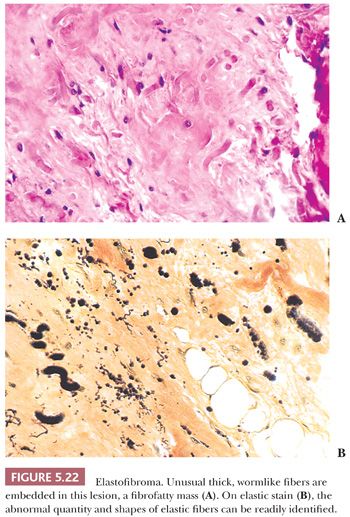
FIBROUS PROLIFERATIONS OF CHILDHOOD
A delineation of every fibrous proliferation that may occur in children would be voluminous (258). Practically, all such lesions are associated with a characteristic age and location, and they may be solitary or multiple; recurrence or regression may or may not occur. When such lesions are encountered, some of which may simulate fibrosarcoma or other sarcomas, consulting soft tissue or pediatric pathology texts or review articles is best. Only selected features of certain lesions are discussed here. The fibrous hamartoma of infancy is a solitary, poorly circumscribed proliferation in which organoid nodules of spindle cells with a myxoid matrix are interspersed within fatty and fibrous tissue (Fig. 5.23A) (259). Infantile digital fibroma/fibromatosis is characterized by fascicles of fibroblasts with prominent stromal collagen (260). The pathognomonic feature is the presence of small, round intracytoplasmic inclusions approximately the size of a lymphocyte nucleus; they appear red on a trichrome stain and are periodic acid-Schiff (PAS) negative; they consist of actin filaments (260). In infantile myofibromatosis, small bundles of spindle-shaped myofibroblasts alternate with more cellular areas containing ovoid cells and thin-walled, branching blood vessels (261). Cutaneous nodules, gingival hypertrophy, and flexure contractures characterize the mesenchymal dysplasia known as juvenile hyaline fibromatosis (262), which is inherited as an autosomal recessive trait caused by mutations in the ANTXR2 gene (263). The amorphous hyaline substance found within it has areas resembling keloidal collagen. Fibromatosis colli presents as a rapidly growing mass in the second to fourth week of life, and microscopically, it appears as a diffuse scar within skeletal muscle. In calcifying aponeurotic fibroma (264), ill-defined and painless masses appear on the hands and feet of children between the ages of 10 and 15 years; in the classic lesion, primitive mesenchymal cells resembling fibromatosis or fibrosarcoma occur in nodules surrounding central calcification (Fig. 5.23B).
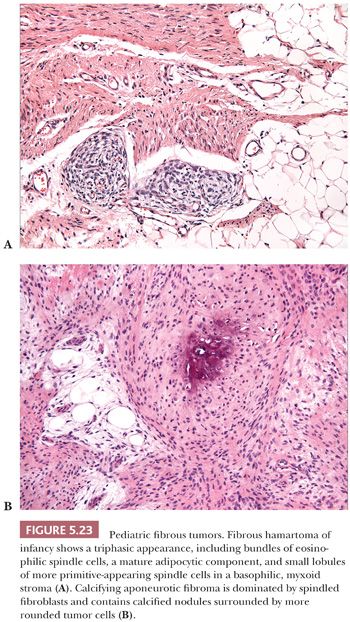
ANGIOMYXOID LESIONS
ANGIOMYOFIBROBLASTOMA
Angiomyofibroblastoma arises in the vulvovaginal region, shows variable cellularity, and is composed of small round to spindle cells accompanied by small but widely arced vessels (265,266). Pleomorphism is absent, and the tumor is circumscribed (Fig. 5.24; see Table 5.7 for the differential diagnosis). Unlike aggressive angiomyxoma, with which angiomyofibroblastoma may be confused, it is more vascular and does not have vessels that are open and round. It can also be mistaken for liposarcoma, but no lipoblasts are found. The immunoprofile, which was originally reported to be distinct, is the same as that of aggressive angiomyxoma—it is positive for desmin but negative for MSA and SMA; thus, the distinction between the two entities must be based on the hematoxylin and eosin appearance (267).
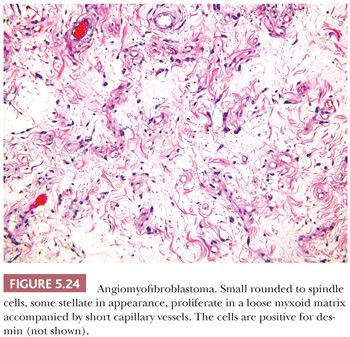
SUPERFICIAL ANGIOMYXOMA
Described in detail by Allen et al. (268), the superficial angiomyxoma is a cutaneous and subcutaneous multinodular hypocellular myxoma-like tumor that may contain epithelial elements in the form of squamous inclusions. Thin-walled, curved blood vessels and stromal neutrophils are typical features (Fig. 5.25). The recurrence rate is 30% (269). Some cases are related to Carney complex (see Table 5.7 and Fig. 5.50 for the differential diagnosis).
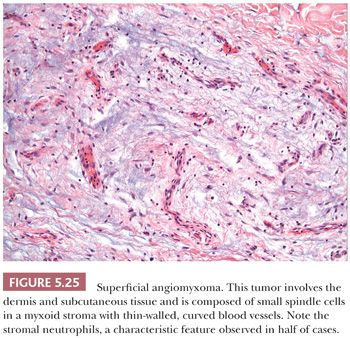
DEEP (“AGGRESSIVE”) ANGIOMYXOMA
Deep (“aggressive”) angiomyxoma (see Chapter 51) has a predilection for the perineum and pelvis of women but also occurs in male patients, and it is rarely found on nongenital regions of the body (270,271); it can be distinguished from angiomyofibroblastoma by its large size, infiltrating border, uniformly hypocellular appearance, and thick rounded vessels (see Table 5.7 and Fig. 5.50 for the differential diagnosis). With only a 30% recurrence rate, it is not as aggressive as originally thought.
FIBROBLASTIC/MYOFIBROBLASTIC NEOPLASMS OF INTERMEDIATE MALIGNANCY
INFLAMMATORY MYOFIBROBLASTIC TUMOR
This myofibroblastic proliferation (272–274) may be seen in a wide variety of locations in both children and adults but has a predilection for the lung, retroperitoneum, and abdominal cavity. Typically, inflammatory myofibroblastic tumor is a circumscribed but nonencapsulated lesion; it contains fascicles of spindle cells proliferating in a background of fibrosis with lymphocytes, plasma cells, histiocytes, foamy macrophages, and, occasionally, eosinophils and neutrophils (Fig. 5.26A). In some cases, the stroma is sclerotic and hypocellular, whereas in other cases, numerous SMA-positive plump spindle cells with pale eosinophilic cytoplasm are noted. Nuclear pleomorphism and atypical mitoses are absent. A rare, usually intra-abdominal variant is dominated by epithelioid cells with amphophilic cytoplasm, often with myxoid stroma and stromal neutrophils (275) (Fig. 5.26B). This aggressive variant is known as epithelioid inflammatory myofibroblastic sarcoma (275). The inflammatory myofibroblastic tumor, which was thought to be reactive in nature at one time, is a true neoplasm with the potential for recurrence (35%) and multifocality; clonal cytogenetic findings support this view. Distant metastases develop in fewer than 5% of patients (276). The cases originally described as inflammatory fibrosarcoma are now considered to belong to this group of lesions (274). Molecular findings show that the inflammatory myofibroblastic tumor demonstrates a fusion of the ALK gene with a wide range of partners in around 50% of cases (137,277); these cases are immunoreactive with ALK antibodies such as ALK1 or p80 (278). Epithelioid inflammatory myofibroblastic sarcoma usually harbors an ALK-RANBP2 fusion and shows a nuclear membrane pattern of ALK staining (275). The small subset of patients whose tumors pursue an aggressive clinical course may be treated with ALK tyrosine kinase inhibitors such as crizotinib (279).
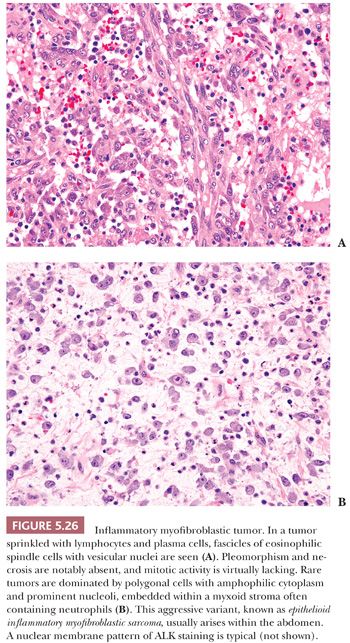
FIBROMATOSIS
Several fibromatoses occur in adults (280–283), mainly in two distinctly different forms. First, lesions occurring on the hands and feet (palmar and plantar, respectively) appear as contractures with small nodules; cellular fibrotic lesions with variable amounts of collagen are seen among the tendinous tissue. Although they are frequently multinodular, a single dominant nodule may be present. The lesions are rarely misdiagnosed. Unlike the proliferation in scars, proliferation in fibromatosis is uniform and relatively hypovascular, and it lacks hemosiderin. Although the palmar and plantar fibromatoses may recur locally after excision, they are localized lesions without the same potential for growth and infiltration that is seen in the desmoid tumors.
The second, or desmoid, type of fibromatosis is noted clinically as a large mass, often on the abdomen or trunk. Here, a uniformly cellular appearance is typical (Fig. 5.27). The fibroblasts in one plane are bipolar with attenuated to invisible cytoplasm and a thin, oval, pointed nucleus. In another plane, the cells are polygonal or stellate, and the periphery of the cytoplasm appears to merge imperceptibly with the surrounding collagen, indicating an intimate relationship with it. This feature is a general aid in identifying cells as myofibroblastic. Infiltration of the surrounding tissues is typical. As a rule, the cells in the fibromatoses are quite evenly scattered, and the overall cellularity is low to intermediate. In other words, cellularity is never marked, and the cells rarely touch one another. Mitoses are usually not identified, or they are rare. The immunoprofile with its SMA-positive myoid reaction can be used to advantage in the differential diagnosis. β-Catenin nuclear positivity can be supportive of the diagnosis (Fig. 5.27B), but it is neither absolutely necessary nor specific (127,128). The abdominal and pelvic fibromatoses occur preferentially in female patients, and they can be more myxoid; some of these are hormonally responsive. In Gardner syndrome (284), the fibromatoses commonly are mesenteric and postoperative, and the rate of recurrence is higher (191). Sporadic desmoid fibromatosis harbors CTNNB1 (β-catenin) gene mutations (285).
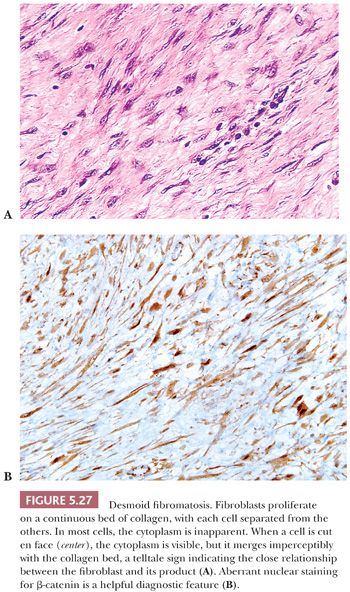
Fibromatosis often recurs, but if it is left alone, it may solidify and stop growing. The particular CTNNB1 mutation may predict recurrence (286,287). Disagreement exists about the ultimate treatment; some recommend only an initial wide excision, followed by observation in the hope that growth may cease, whereas others recommend radiotherapy, which may have long-term untoward consequences. There is a recent trend toward a “watchful waiting” approach, reserving surgery for symptomatic patients and those whose tumors continue to grow (286). Congenital and infantile forms of fibromatosis are highly cellular and mitotically active, and these mimic fibrosarcoma (288).
SOLITARY FIBROUS TUMOR
Tumors described originally as localized fibrous mesothelioma have come to be known as solitary fibrous tumors (SFTs) because they clearly are not of mesothelial origin (289–291). SFTs develop not only from the pleura but also near other serosal surfaces, such as the pericardium, peritoneum, and the surface of the liver. Significantly, they may develop without an association to a serosal surface, such as in the mediastinum, orbit, thyroid, or nasal cavity; and now, they are also commonly reported in the soft tissues.
SFTs belong to the category of fibroblastic tumors. The lipomatous HPC is now regarded to belong to the SFT group of tumors (292,293), and indeed, essentially all HPCs are now considered (uniformly cellular and/or malignant) SFTs (24). SFTs rarely express actin (and desmin), and they have (myo)fibroblastic ultrastructural features. Regardless of location, they are histologically identical to those in the pleura, which are composed of nondescript bland and uniform spindle cells dispersed among elongated, thin, parallel collagen bands in a “patternless” pattern (Fig. 5.28). The nuclei are small, and mitoses are difficult to find in the average case. Foci of storiform or hemangiopericytomatous growth are typical. Actually, the parallel arrangement of the collagen bundles is a characteristic feature that is not seen in fibromatosis; the consistent CD34 positivity is another feature distinguishing SFT from fibromatosis (294). Therefore, two markers distinguish between these two collagenized tumors—fibromatosis (SMA+/CD34−) and SFT (SMA−/CD34+). SFTs are also positive for BCL-2 and CD99. Recent studies have identified a NAB2-STAT6 gene fusion in SFT (295–297). This specific genetic signature of SFT results in nuclear expression of the STAT6 protein (Fig. 5.28C), which can be detected by immunohistochemistry (138). Most benign-appearing SFTs behave in a benign fashion (298). If an uncommon cellular tumor is encountered, the following criteria developed for malignancy in pleural tumors (291) are applicable in soft tissue (299,300): increased cellularity, necrosis, pleomorphism, and an increased mitotic rate (>4 per 10 hpf). In rare cases, malignant SFTs appear to occur as fibrosarcomatous (or pleomorphic) progression next to benign-appearing SFTs (301). Recently, a risk assessment model for SFT has been developed, and pathologists may refer to it in pathology reports (302).
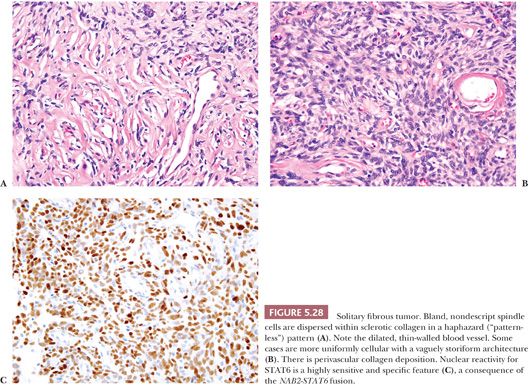
MYOFIBROBLASTOMAS
PALISADED MYOFIBROBLASTOMA
Also known as intranodal hemorrhagic spindle tumor with amianthoid fibers, this tumor was simultaneously described by two groups as a lesion that is found exclusively in inguinal lymph nodes (303,304). It is composed of a spindle cell proliferation replacing the substance of a lymph node and is accompanied by hemorrhage and irregular knots of collagen bundles (the amianthoid fibers with a crystalline appearance). The spindle cells have elongated nuclei that often show nuclear palisading similar to that of a nerve sheath tumor (Fig. 5.29). Their cytoplasm is eosinophilic and tapered, giving a myoid quality and raising the possibility of smooth muscle origin. With actin immunoreactivity, the cell type is likely either a myofibroblast or a specialized smooth muscle cell. The lesion mimics Kaposi sarcoma and a node-based hemorrhagic schwannoma, but neither of these has the characteristic fibers, and the tumor is negative for S-100. This unusual lesion has been reported in other locations (305,306). No evidence of aggressive behavior or even local recurrence has been found.
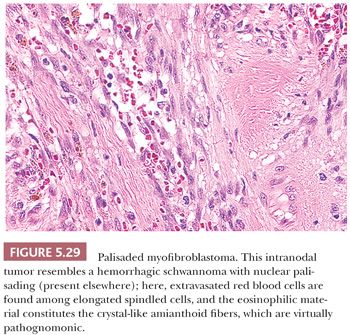
MAMMARY-TYPE MYOFIBROBLASTOMA
Mammary myofibroblastoma is composed of fascicles of bland, short spindle cells with prominent stromal collagen and a variable adipocytic component (Fig. 5.30) (307). Expression of both desmin and CD34 is typical. This tumor shares genetic features (loss of 13q14) with spindle cell lipoma and cellular angiofibroma (253), and loss of RB1 (retinoblastoma) protein expression was recently reported (209). Mammary-type myofibroblastoma may also occur in the inguinal region and other soft tissue sites (308).
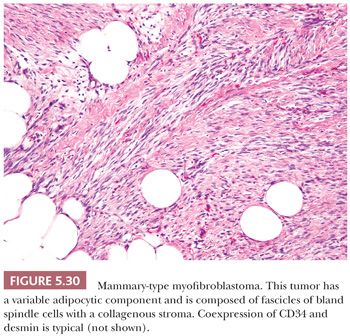
OSSIFYING FIBROMYXOID TUMOR
Ossifying fibromyxoid tumor (OFMT) is a distinctly unusual tumor (84,309). The presence of both calcification and ossification in irregular trabecula-like formations throughout the tumor or concentrated at the periphery raises the clinical diagnosis of a calcified soft tissue mass, such as a synovial sarcoma or osteosarcoma. Usually, a well-circumscribed mass approximately 4 cm in size is found in the subcutaneous tissue or muscle of the upper and lower extremities. More than 80% of cases have an incomplete shell of mature bone in the capsular region. Small rounded cells may exist either as sheets of poorly differentiated cells or as unusual cords and strings suspended in a loose stroma (Fig. 5.31). Two-thirds of the cases show S-100 positivity; approximately one-third have desmin. OFMT harbors PHF1 gene rearrangements in most cases (310–312). Local recurrence has been noted in 25% of cases, and metastatic disease has been reported in 1 of 41 initial patients (309). The lineage of this tumor is unresolved. Evidence for a Schwann cell origin includes the presence of a basal lamina and an absence of type II collagen. Atypical and malignant variants have been reported (the latter with hypercellularity, high nuclear grade, and greater than two mitoses per 50 hpf) (111,313), but there is a nearly negligible rate of metastasis in conventional OFMT.
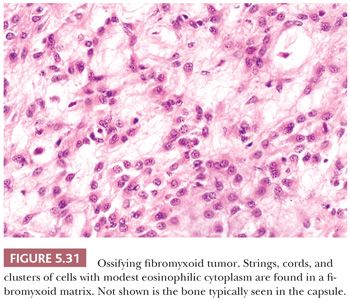
FIBROSARCOMAS
INFLAMMATORY FIBROSARCOMA
Inflammatory fibrosarcoma is considered a variant of inflammatory myofibroblastic tumor (see Inflammatory Myofibroblastic Tumor”).
MYOFIBROSARCOMA
Rare tumors of malignant myofibroblasts have been reported (314). Most of these are low-grade tumors (315), also known as low-grade myofibroblastic sarcoma according to the new World Health Organization classification (24). Composed of SMA-positive myofibroblasts, the tumors mimic NF (although they are infiltrative, have a deeper intramuscular location, and lack histiocytic giant cells), desmoid fibromatosis (although they have notable nuclear atypia), and leiomyosarcoma (although they lack the alternating fascicular pattern of a smooth muscle neoplasm) (Fig. 5.32). Desmin positivity may be seen alone or in combination with SMA. High-grade pleomorphic myofibrosarcomas resemble UPS, but they exhibit prominent rather than spotty SMA reactivity (316).
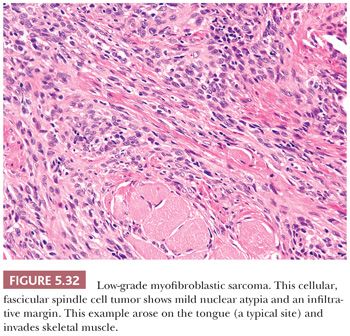
MYXOINFLAMMATORY FIBROBLASTIC SARCOMA
This unusual tumor type occurring mainly on the hands and feet was originally reported in two series (317,318); a recent large study provided additional outcome data (319). These tumors are found in both men and women of all ages, and the morphology is most reminiscent of either inflammatory pseudotumor or myxofibrosarcoma with extensive chronic inflammation. No other tumor has the following constellation of morphologic findings: (a) an infiltrative subcutaneous tumor with a multinodular appearance, (b) areas of dense fibrosis with prominent chronic inflammation, (c) myxoid areas with both spindled and epithelioid cells, (d) scattered pleomorphic cells resembling Reed-Sternberg cells with prominent eosinophilic nucleoli (“virocytes”), and (e) mucin-filled cells in the myxoid areas with a basophilic wispy cytoplasm similar to the extracellular matrix (Fig. 5.33). Positive markers include vimentin, focal weak CK in some, and focal CD68. A characteristic translocation t(1;10)(p22;q24) has been identified (see Table 5.3) (320,321). Recurrences are seen in 25% to 67% of patients, but metastatic disease is rare.
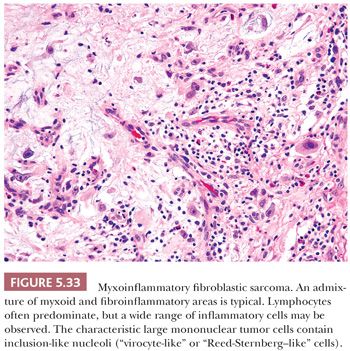
MYXOFIBROSARCOMA
The term myxofibrosarcoma was originally applied to low-grade lesions (322,323), but it has come to encompass all cases previously termed myxoid MFH (see Table 5.7 and Fig. 5.34) (24); therefore, it is discussed here rather than in the section on UPS.
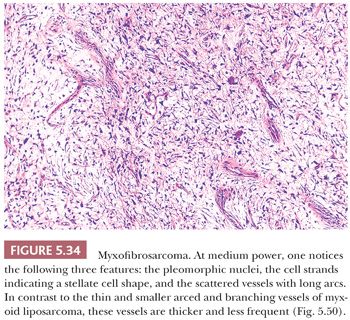
The tumor previously known as the myxoid variant of MFH is more common but less aggressive than the “storiform-pleomorphic” type (UPS), although it is more recurrent locally (324). Unlike myxoid liposarcoma, it often affects elderly adults, is often found in the subcutaneous location, and exhibits several distinctive histologic features (325). First, pleomorphism is prominent, in contrast to the remarkably uniform nature of the much smaller nuclei in most myxoid liposarcomas; furthermore, it is characteristically an S-100–negative tumor, unlike some liposarcomas. Second, the cells have a stellate or tripolar shape, like myofibroblasts (see Fig. 5.34). Third, the vessels are recognizably different, being thick walled and often curved with a wide arc. They are not isolated fine capillaries; instead, they are associated with an eosinophilic substance, and the adherent tumor cells add to their thick appearance at low power (see Fig. 5.50 for comparison with other myxoid tumors). Fourth, another differentiating feature is the presence of nonmyxoid UPS-like elements in many cases. Originally, the definition required more than 50% myxoid histology (324); however, there is no longer a specific requirement for the extent of the myxoid component, so long as the typical low-grade component is identified (24,325). Low-grade myxofibrosarcoma (lacking solid, cellular areas) rarely metastasizes, whereas intermediate- or high-grade tumors have a significant metastatic risk (20% to 35%) (24). Of note, local recurrences often progress to higher histologic grade.
Recently, an epithelioid variant of myxofibrosarcoma has been described with epithelioid cells surrounded by varying amounts of myxoid matrix, growing in sheets, with pleomorphic cells; this variant recalls a differential diagnosis of melanoma, carcinoma, and other epithelioid tumors and has a worse prognosis than other high-grade myxofibrosarcomas (326).
FIBROSARCOMA
The following two highly cellular variations of fibrosarcoma exist: infantile fibrosarcoma and adult fibrosarcoma. In infants and children (327,328), fibrosarcoma is a mitotically active, locally aggressive tumor with a specific gene fusion (ETV6-NTRK3) (329) and a relationship to congenital mesoblastic nephroma. In the past, tumors in adults labeled as fibrosarcomas constituted a high percentage of soft issue sarcomas. More recently, the total number of cases with this designation has decreased precipitously for several reasons (330,331). First, pleomorphic fibroblastic tumors in adults are now conventionally called undifferentiated pleomorphic sarcoma. Second, with the use of immunohistochemical techniques, it has become apparent that several lesions may mimic fibrosarcoma in adults, particularly MPNST and synovial sarcoma. Clear examples of fibrosarcoma exist in adults (331), but this designation should be limited to lesions with the following characteristics: (a) an overall highly cellular spindle cell pattern, (b) a herringbone pattern in which sheets of cells intersect at acute angles (Fig. 5.35), (c) an absence of pleomorphism, and (d) an absence of immunohistochemical staining for S-100 or CK. High-grade fibrosarcoma in adults is exceedingly rare (331).
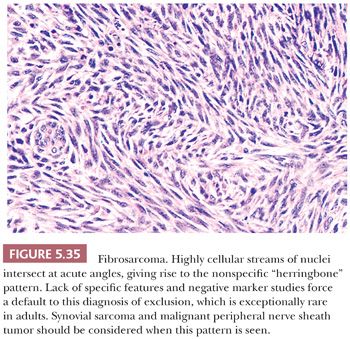
LOW-GRADE FIBROMYXOID SARCOMA
This deceptively bland sclerosing lesion, which is similar in appearance to both fibromatosis and a nerve sheath tumor, does metastasize (332,333). Many of the cases reported thus far have been in female patients, and most of these sarcomas have been identified after the metastases appeared. The metastases develop years or decades after the removal of a primary tumor (334). A slight whorling, partially storiform appearance is noted, and areas may be extremely hypocellular with myxoid nodules and small, thin-walled blood vessels. The cells are small, thin, and spindly, and occasionally, the nuclei are bent and wavy. However, the lesion is negative for S-100, and mitotic activity in the primary lesion can be extremely low.
Some cases may clearly have giant rosettes, and the tumor originally described as hyalinizing spindle cell tumor with giant rosettes (335) is one end of the low-grade fibromyxoid sarcoma spectrum (336). At low power, the tumor resembles a nerve sheath tumor because the hyalinizing rosettes are reminiscent of palisading Verocay bodies. However, these rosettes are large, and they have a collagenized core, and the remainder of the lesion is composed of small spindle cells in a storiform or random distribution (Fig. 5.36). The rate of metastases is less than 5% for all variants in the first 5 years following excision; with long-term follow-up, up to 30% of patients will eventually develop lung or pleural metastases (334). Low-grade fibromyxoid sarcoma has a translocation t(7;16)(q34;p11) or t(11;16)(p11;p11), resulting in FUS-CREB3L2 or FUS-CREB3L1 gene fusions; the former is by far more common (337,338). MUC4 is a highly specific immunohistochemical marker for this tumor type (143). Reactivity for MUC4 is helpful to distinguish low-grade fibromyxoid sarcoma from soft tissue perineurioma, with which it can easily be confused.
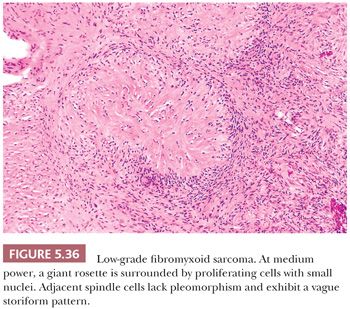
SCLEROSING EPITHELIOID FIBROSARCOMA
In the first series of 25 cases of this unusual tumor, young persons from 20 to 50 years of age had deep tumors on the lower extremity (339). Uniform cellular proliferation was found in distinct nests or cords of epithelioid cells with abundant cytoplasm. Prominent hyaline sclerosis, which was noted in all cases, surrounded the nests (Fig. 5.37), and foci of conventional fibrosarcoma were occasionally seen. The mitotic rate averaged four per 10 hpf, and a few cases showed chondro-osseous differentiation. The tumors were positive for vimentin, and they occasionally stained with EMA, S-100, and CK. Some cases of sclerosing epithelioid fibrosarcoma show areas of low-grade fibromyxoid sarcoma (144,337). The frequent recurrence in at least 50% of cases and metastases in 43% to 86% of cases attest to its aggressiveness, but the course may be long (5 to 15 years) (339,340). Similar to low-grade fibromyxoid sarcoma, most cases of sclerosing epithelioid fibrosarcomas are positive for MUC4 (144). A recent study has identified EWSR1-CREB3L1 gene fusions in sclerosing epithelioid fibrosarcomas (341). Sclerosing epithelioid fibrosarcomas containing a component of low-grade fibromyxoid sarcoma often harbor FUS-CREB3L2 fusions and may represent a form of tumor progression (144,337).
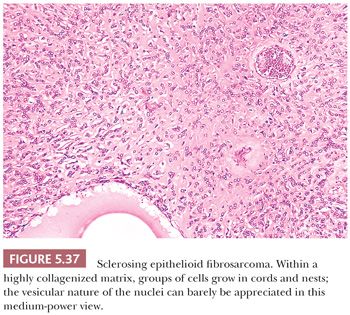
FIBROHISTIOCYTIC (“FIBROCYTIC”) LESIONS
It is now accepted that tumors in this category are not related to true histiocytic lineage (monocyte/macrophage bone marrow–derived cell) but rather are probably derived from a peculiar type of fibroblastic mesenchymal cell. Therefore, the term fibrocytic is gaining acceptance for this group of lesions.
BENIGN FIBROUS HISTIOCYTOMA
BFH usually presents as a small dermal nodule at almost any age. When the tumor is highly sclerotic and hypocellular, it may be termed dermatofibroma. When lesions are larger and more cellular, the hallmark of fibrohistiocytic lesions, the storiform pattern, is seen. In these lesions, the small spindly cells are tightly arranged, and they have very thin, elongated nuclei with scanty cytoplasm. The nuclei have pointed ends, and they practically touch one another, unlike those seen in most smooth muscle lesions. The storiform arrangement is best envisioned as a cartwheel configuration of cells emanating from a common point. Within each portion of the cartwheel, the cells line up in a parallel array, although they lack the nuclear palisading of a schwannoma. An infiltrating border is typical (lateral entrapment of hyaline dermal collagen is a helpful diagnostic feature), and recurrence is more likely if the lesion is incompletely excised or it extends into the subcutaneous fat (82,342). BFHs of deeper soft tissue are rarely encountered (343), and they may raise the possibility of a sarcoma; the benign tumor lacks pleomorphism, and mitoses are uncommon.
Related lesions include atypical fibrous histiocytoma (DF with monster cells) and the epithelioid variant (344,345). Occasional aneurysmal or “angiomatoid” variants also occur (Fig. 5.38) (346). A recent study identified gene fusion involving protein kinase C (PRKCB and PRKCD) in BFH (347).
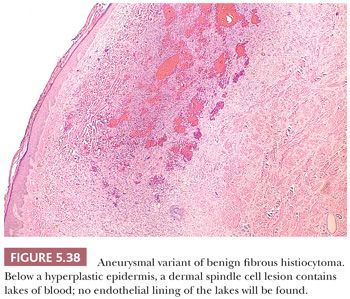
ATYPICAL FIBROUS HISTIOCYTOMA
In reality, a spectrum of lesions, ranging from the completely bland BFH to the lesion with marked nuclear pleomorphism called atypical fibrous histiocytoma (AFH), exists (344). In contrast to an atypical fibroxanthoma (AFX) (see “Atypical Fibroxanthoma”), the AFH commonly presents on the extremities of young to middle-aged adults, rather than on sun-exposed areas, including the head and neck, of older patients.
PLEXIFORM FIBROHISTIOCYTIC TUMOR
In this tumor of children and young adults (348,349), separated nodules surrounded by a rim of fibrous tissue proliferate in the deep dermis and subcutaneous fat. Histiocytoid cells and multinucleated giant cells populate the nodules (Fig. 5.39), accompanied by a chronic inflammatory infiltrate. The combination of giant cells and stromal cells is reminiscent of the giant cell tumor of tendon sheath to some degree, but the superficial location is incorrect. As a rule, mitotic figures are rare (zero to two per 10 hpf), but exceptions do occur. Some nodules appear predominantly fibroblastic. Most of these patients are female. In one study, local recurrence developed in more than one-third of the patients, and 2 of 32 tumors metastasized to the local lymph nodes; distant metastases were not observed (348). The differential diagnosis includes ordinary fibrous histiocytoma, other plexiform tumors (e.g., neurofibroma), fibromatosis, giant cell tumor of soft tissue, and epithelioid sarcoma. The results of studies including SMA positivity suggest that this may be another myofibroblastic proliferation. Atypical and widely metastatic variants have been described (350,351).
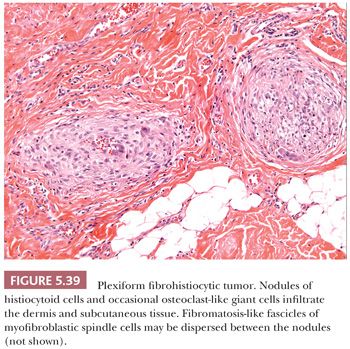
DERMATOFIBROSARCOMA PROTUBERANS
DFSP is discussed elsewhere in this text (see Chapter 2). It is the storiform tumor par excellence, with each and every field showing this characteristic pattern (Fig. 5.40). Other tumors may also show this pattern (Table 5.8). Although the tumor is highly cellular, the nuclei are once again quite thin, with an innocuous appearance. Mitoses are difficult to find, and the lesion is concerning only because of its propensity for local recurrence. In larger, bosselated tumors bulging from the skin surface, the diagnosis is evident (352); however, in smaller lesions, in which DFSP may be confused with fibrous histiocytoma, the immunoprofile is distinctly different because DFSP (CD34+/F13a−) stain reactions differ from those in BFH (CD34−/F13a+).
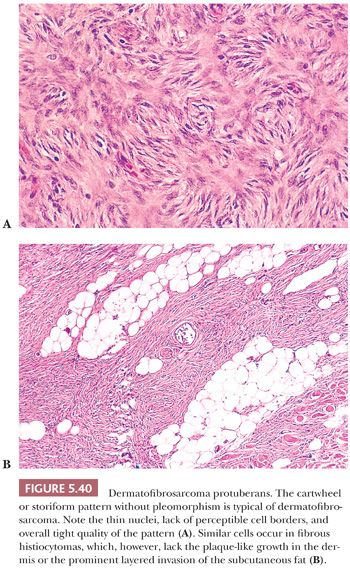
The myxoid variant may be confused with liposarcoma, but it differs in its superficial location either directly under the epidermis or in the subcutaneous tissues (353). DFSP may also be pigmented (“Bednar tumor”) (354), it may acquire a granular cell cytoplasm (355), and it may dedifferentiate with fibrosarcoma-like or UPS-like areas (356).
Some primary DFSPs or recurrences exhibit fibrosarcomatous transformation (DFSP-FS) (357,358). The fibrosarcomatous component is highly cellular, may lose the storiform pattern, frequently retains its CD34 positivity, and exhibits a much higher mitotic rate (>7 per 10 hpf). When such transformation occurs, it is often visible as a dominant gross nodule, and the tumor acquires metastatic potential (10% to 15%) (359). Because of the pathogenetic gene fusion COL1A1-PDGFB (see Table 5.3), patients with recurrences and metastases of DFSP may be treated with imatinib mesylate (360).
GIANT CELL FIBROBLASTOMA
Giant cell fibroblastoma, which is frequently mistaken for a sarcoma, is a rare fibroblastic tumor that occurs mainly in boys younger than 10 years of age (361,362). The tumors are superficial, and they are found in a variety of locations. The lesions may be highly sclerotic and hypocellular, with widely scattered atypical cells; alternatively, the regions may be quite cellular with prominent thick-walled vessels. Adipose tissue is often seen. One telltale characteristic is the formation of wide, vessel-like spaces (Fig. 5.41) that are incompletely lined by fibroblastic (nonendothelial) cells. The lesion may resemble (a) a neurofibroma with ancient change, (b) a liposarcoma lacking capillary vascularity, or (c) a peculiar angiosarcoma in which the huge spaces are unusual and only incompletely lined. Approximately half of patients experience local recurrence, but no metastases have occurred. This odd lesion has acquired the designation childhood DFSP because occasional cases show zones consistent with the storiform adult tumor (363), adult DFSPs may contain giant cell fibroblastoma–like areas (364), and the same gene fusion is detected (365).
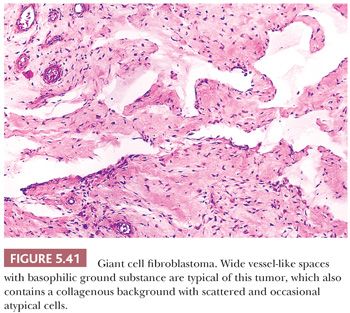
UNDIFFERENTIATED PLEOMORPHIC SARCOMA (PREVIOUSLY CALLED MALIGNANT FIBROUS HISTIOCYTOMA)
More than two decades ago, Fletcher first argued that MFH does not exist as a discrete entity (366); the term pleomorphic fibrosarcoma has also been proposed for otherwise unclassifiable tumors in this former category (367). As mentioned earlier, lesions in this group of tumors bear no relationship to the true histiocyte, and therefore, MFH is a misnomer.
The term malignant fibrous histiocytoma was used to encompass a group of tumor subtypes, and now most of them have been renamed or indeed expanded into a spectrum of lesions. Aside from myxofibrosarcoma (discussed earlier in this chapter), each of these will be described here. The MFH designation is no longer used in the new World Health Organization classification (24).
In most older tallies, the UPS/MFH complex had been the most common soft tissue sarcoma (368–370), accounting for approximately 20% to 25% of cases. Some now suggest that UPS with storiform/pleomorphic features accounts for as little as 5% of adult sarcomas, with myxofibrosarcoma constituting much of the remainder of the tumors formerly included in the MFH category. It may occur in virtually any site of the body. The lower extremity is the most frequent site, and UPS is likely the most common tumor of the thigh, with liposarcoma being a close second. Morphologically, the different “variants” of the complex resemble a wide variety of other tumors and makes the differential diagnosis lengthy. When all of the tumors formerly categorized as MFH are considered, several generalizations emerge. First, the prognosis varies with size, depth, location (distal is better than proximal), and grade. Second, many deep extremity cases may be cured by wide local excision. Third, the vast majority of retroperitoneal and abdominopelvic lesions are in fact dedifferentiated liposarcomas. In presentation, they are similar to most sarcomas, producing mass lesions.
The essence of UPS is pleomorphism. In daily practice, pathologists have a tendency to misuse the words pleomorphism and pleomorphic to characterize peculiar-looking cells. A strict definition of the terms is a mean variation in cytoplasmic and nuclear size throughout a tumor. To be a useful classifier in daily practice, evaluation of this characteristic should occur at low to medium power, thus dividing STTs into “uniform” or “pleomorphic” categories. At that power, pleomorphism is an outstanding feature in UPS. Whereas other tumors, such as liposarcoma and RMS, do exhibit pleomorphism, they tend to do so within the constraints of their phenotype (i.e., they still look somewhat like the cell lineage). In contrast, the bizarre UPS cell has certain characteristics. In its largest form, it has voluminous cytoplasm that may be pale to highly eosinophilic and that occasionally is vacuolated. The cell may range in size from very large to huge; indeed, it may be the most expansive tumor cell in existence. Notably, the cytoplasm does not have the granular or fibrillar appearance of rhabdomyoblasts; however, it may be multivacuolated, simulating a lipoblast. The vacuoles in myxofibrosarcoma are composed of proteoglycan material (Table 5.9). Unlike the cells in leiomyosarcoma, which have elongated, tapering cytoplasm, the UPS cells may be oval or spindled, but they are shorter and do not show long stretches of streaming cells or fascicular growth. Unlike smooth muscle cells, the spindled cells often lack defined cell borders. The nuclear characteristics are also quite typical, with highly bizarre shapes and multiple nuclear lobes of differing sizes. The nucleoli may be not only prominent but also quite large. The nucleus tends to have a thick membrane and, more often than not, exhibits a jagged outline that may be termed “irregularly irregular.” Occasionally, the nuclei contain eosinophilic inclusions similar to those of melanoma or display clear nuclear vacuoles, such as those that may be seen in liposarcoma.
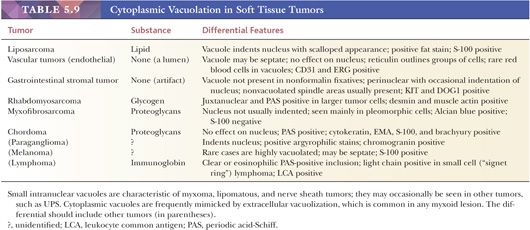
With strict adherence to the histologic features mentioned earlier, coupled with the skeptical approach and a search for more specific morphologic patterns (e.g., alternating fascicles in leiomyosarcoma), many of the mimics of UPS can and should be eliminated.
The UPS cell appears to be a peculiar form of mesenchymal cell—possibly fibroblastic—with a propensity to develop such unusual features. Although it may exhibit immunoreactivity with markers such as CD68, surface marker analysis by several investigators has shown that it does not resemble cells of the bone marrow–derived monocyte, macrophage, and histiocyte series (371). As a mesenchymal cell, it retains the ability to produce laminin, and “histiocyte-like” cells emerge from fibroblast-like cells (372). Compared with most other soft tissue phenotypes, this cell is probably more primitive developmentally.
UPS/MFH tends to be overdiagnosed, a point that Fletcher (366) properly emphasized. Hopefully, careful attention to a strict definition of a storiform pattern, when combined with the realization that UPS-like areas may accompany phenotypically different sarcomas (373) or carcinomas, will alleviate the stress on the use of this term.
ATYPICAL FIBROXANTHOMA (AND “SUPERFICIAL UPS/MFH”)
AFX is covered in detail in Chapter 2. If a tumor occurs in older patients (>50 years) and occurs on sun-exposed skin, the AFX designation is appropriate. Histologically similar tumors in other settings may represent examples of AFH, as discussed earlier, or unclassified dermal sarcomas. Both AFX and AFH bear histologic similarity to UPS, and as such, they may erroneously be diagnosed as sarcomas, although they are small and generally innocuous. Tumors that are large (>3 cm) and involve the subcutaneous tissues may be called (superficial) undifferentiated pleomorphic sarcoma or pleomorphic dermal sarcoma (374). As in deep UPS, pleomorphic cells are apparent at low power, but a great variability in the accompanying smaller cells is also noted. Cells with angulated and curved nuclei are present, as well as an inflammatory component (Fig. 5.42). The (nondistinctive) immunoprofile is similar to that of UPS. The variants include a nonpleomorphic type and cases with benign giant cells. Unlike AFH, AFX lacks a grenz zone or space below the epidermis and the classic features of BFH (344).
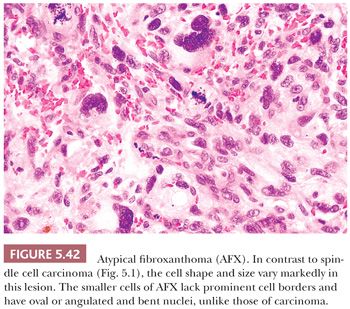
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


