44
Slow Viruses & Prions
CHAPTER CONTENTS
INTRODUCTION
“Slow” infectious diseases are caused by a heterogeneous group of agents containing both conventional viruses and unconventional agents that are not viruses (e.g., prions). Prions are protein-containing particles with no detectable nucleic acid that are highly resistant to inactivation by heat, formaldehyde, and ultraviolet light at doses that will inactivate viruses. Note that prions are resistant to the temperatures usually employed in cooking, a fact that may be important in their suspected ability to be transmitted by food (see variant Creutzfeldt-Jakob disease [CJD] later). Prions are, however, inactivated by protein- and lipid-disrupting agents such as phenol, ether, NaOH, and hypochlorite (see Chapter 28).
The prion protein is encoded by a normal cellular gene and is thought to function in a signal transduction pathway in neurons. The normal prion protein (known as PRPC, or prion protein cellular) has a significant amount of alpha-helical conformation. When the alpha-helical conformation changes to a beta-pleated sheet (known as PrPSC, or prion protein scrapie), these abnormal forms aggregate into filaments, which disrupt neuron function and cause cell death. Prions, therefore, “reproduce” by the abnormal beta-pleated sheet form recruiting normal alpha-helical forms to change their conformation. Note that the normal alpha-helical form and the abnormal beta-pleated sheet form have the same amino acid sequence. It is only their conformation that differs. A specific cellular RNA enhances this conformational change. Prions are described in more detail in Chapter 28.
Pathogenic prion proteins can be thought of conceptually as misfolded proteins. These misfolded proteins not only cause CJD in humans and “mad cow” disease in cattle but are suspected of being involved in the pathogenesis of other important diseases of the central nervous system, such as Alzheimer’s disease and Parkinson’s disease.
In humans, the “slow” agents cause central nervous system diseases characterized by a long incubation period, a gradual onset, and a progressive, invariably fatal course. There is no antimicrobial therapy for these diseases. Note that the term slow refers to the disease, not to the rate of replication of those viruses that cause these slow diseases. The replication rate of these viruses is similar to that of most other viruses.
The human prion-mediated diseases (e.g., kuru and CJD) are called transmissible spongiform encephalopathies (TSE). The term spongiform refers to the spongy, Swiss cheese–like holes seen in the brain parenchyma that are caused by the death of the neurons (Figure 44–1). No virus particles are seen in the brain of people with these diseases.
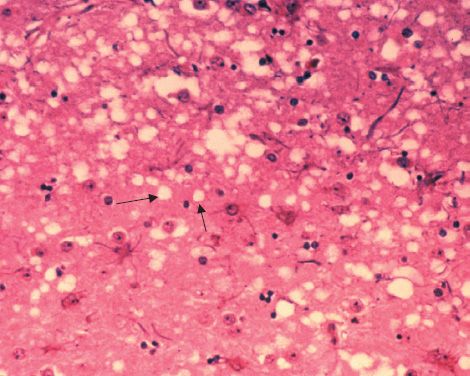
FIGURE 44–1 Prion-mediated spongiform encephalopathy (mad cow disease)—two arrows point to the spongiform appearance (Swiss cheese–like holes) in the brain of a cow with mad cow disease. The brain of a patient with Creutzfeldt-Jakob disease has a similar appearance. (Figure courtesy of Dr. Al Jenny, Public Health Image Library, Centers for Disease Control and Prevention.)
The term encephalopathy refers to a pathologic process in the brain without signs of inflammation. In contrast, encephalitis refers to an inflammatory brain process in which either neutrophils or lymphocytes are present. In TSEs, there are no inflammatory changes in the brain.
The transmissibility of the agent of kuru and CJD (“prions”) was initially established by inoculation of material from the brains of infected patients into the brains of primates followed by serial transfer to the brains of other primates.
Note, however, that both kuru and variant CJD (and bovine spongiform encephalopathy [BSE]—“mad cow” disease) are acquired by ingestion. In this route, the prion protein must survive digestion in the intestinal tract and then penetrate the gut mucosa. The prion protein is then amplified within follicle dendritic cells in lymphatic tissue, such as Peyer’s patches. Prions then spread to the spleen, carried by migrating dendritic cells. From the spleen, prions spread to the central nervous system probably via the sympathetic nerves.
It is also possible that prions reach the brain within lymphocytes, as there is a documented case of CJD that was acquired by transfused blood. In addition, CJD has been transmitted iatrogenically (i.e., in a medical context, via corneal transplants, dura mater grafts, implanted brain electrodes, and growth hormone extracts made from human pituitary glands).
There is evidence that quinacrine and other acridine analogues inhibit the formation of the pathologic PrPSC form in cell culture. These drugs are currently being tested in animal models for their ability to treat or prevent prion diseases.
Prion-caused diseases can be classified into three categories: some are clearly transmissible (infectious), such as kuru; some are clearly hereditary (genetic), such as fatal familial insomnia; and others are sporadic (neither infectious nor hereditary), such as most cases of CJD. The sporadic cases seem likely to be due to spontaneous somatic mutations in the affected individual.
SLOW DISEASES CAUSED BY CONVENTIONAL VIRUSES
Progressive Multifocal Leukoencephalopathy
Progressive multifocal leukoencephalopathy (PML) is a fatal demyelinating disease of the white matter (i.e., leukoencephalopathy) and involves multiple areas of the brain (i.e., multifocal). Note that it is not an encephalitis because there is no inflammation in the brain.
The clinical picture includes visual field defects, mental status changes, and weakness. The disease rapidly progresses to blindness, dementia, and coma, and most patients die within 6 months. It occurs primarily in individuals with compromised cell-mediated immunity, especially patients with acquired immunodeficiency syndrome (AIDS) and those who are receiving cancer chemotherapy and immunosuppressive drugs following organ transplantation. Some patients undergoing treatment for multiple sclerosis with the monoclonal antibody natalizumab develop PML, and others receiving mycophenolate to prevent transplant rejection have also developed PML. Table 44–1 describes some important features of slow viral diseases in humans caused by conventional viruses.
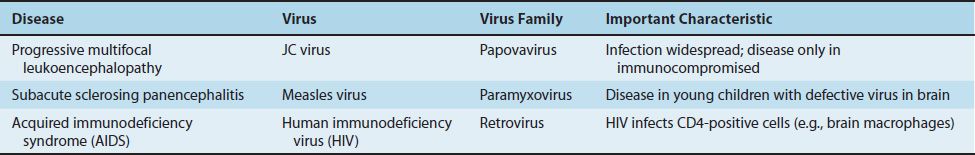
PML is caused by JC virus, a member of the polyomavirus family. Polyomaviruses are nonenveloped viruses with a circular, double-stranded DNA genome. JC virus infects and kills oligodendroglia, causing demyelination. Neurons are unaffected. Antibodies to JC virus are found in approximately 75% of normal human sera, indicating that infection is widespread. Disease occurs when latent JC virus is activated in an immunocompromised patient. The virus persists in the kidney and is excreted in the urine. The diagnosis is typically made by polymerase chain reaction assay of a brain biopsy specimen or spinal fluid. There is no effective antiviral treatment, but cidofovir may be beneficial.
Subacute Sclerosing Panencephalitis
Subacute sclerosing panencephalitis (SSPE) is a slowly progressive disease characterized by inflammatory lesions in many areas of the brain. It is a rare disease of children who were infected by measles virus several years earlier. Unlike PML, immunosuppression is not a predisposing factor. SSPE begins with mild changes in personality and ends with dementia and death.
SSPE is a persistent infection by a variant of measles virus that cannot complete its replication. The evidence for this is as follows:
(1) Inclusion bodies containing helical nucleocapsids, which react with antibody to measles virus, are seen in the affected neurons.
(2) A virus very similar to measles virus can be induced from these cells by cocultivation with permissive cells in culture. The induced virus has a different matrix protein; this protein is important in viral assembly.
(3) Patients have high titers of measles antibody in the blood and spinal fluid.
(4) SSPE has virtually disappeared in the United States since the onset of widespread immunization with measles vaccine.
A progressive panencephalitis can also occur in patients with congenital rubella.
Acquired Immunodeficiency Syndrome
AIDS is caused by human immunodeficiency virus (HIV), a member of the lentivirus group of retroviruses. AIDS is a disease with a long latent period and a progressive course and can involve the central nervous system. See Chapter 45 for more information.
SLOW DISEASES CAUSED BY PRIONS
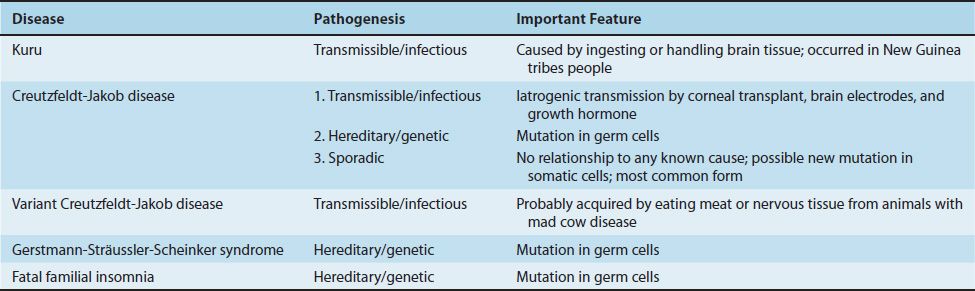
There are five human TSEs caused by prions: kuru, CJD, variant CJD, Gerstmann-Sträussler-Scheinker (GSS) syndrome, and fatal familial insomnia. Table 44–2 describes some important features of slow viral diseases in humans caused by prions.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


