Chapter 48
Shock, Multiple Organ Dysfunction Syndrome, and Burns in Adults
Linda L. Martin, Dennis J. Cheek and Stephen E. Morris

Shock occurs when the cardiovascular system fails to perfuse tissues, cells, and organs adequately, resulting in widespread impairment of cellular metabolism and tissue function. Tissue perfusion is defined as the adequacy of blood flow through the small vessels of the extremities to maintain tissue function.1 Tissue perfusion can be disrupted by any factor that alters delivery of blood to cells and tissues and increases oxygen consumption, such as decreased cardiac output, blood volume, or blood pressure. Shock has many causes and clinical manifestations related to the type of shock. Ultimately, shock from any cause will progress to organ failure and death, unless physiologic mechanisms reverse the process or clinical intervention succeeds in stopping the progression. Untreated, severe shock overwhelms the body’s compensatory mechanisms that may result in multisystem organ failure.
Shock
Cellular Alterations and Impairment of Cellular Metabolism
The body consists of many organs, tissues, and cells that may function or malfunction at different stages of metabolic impairment. The common pathway in all types of shock is impairment of cellular metabolism, attributable to decreased delivery of oxygen and nutrients, frequently coupled with an increased demand and consumption of oxygen and nutrients and a decreased removal of cellular waste products.2 Figure 48-1 illustrates the pathophysiology of shock at the cellular level.
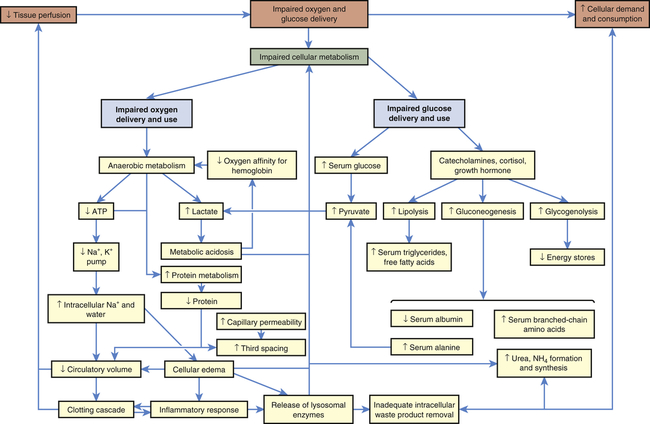
Impairment of Oxygen Delivery and Use
In all types of shock, the cells do not receive adequate amounts of oxygen because of decreased delivery or increased cellular consumption, or both, and are therefore unable to use oxygen normally (see Figure 48-1). In cardiogenic shock cardiac output is too low to deliver adequate oxygen to the cell. In hypovolemic shock, oxygen delivery is impaired by inadequate numbers of red cells that carry oxygen and inadequate blood volume. In neurogenic, anaphylactic, and septic shock, systemic vascular resistance (SVR) becomes very low because of vasodilation, resulting in inadequate perfusion pressure in the capillaries. Thus, the pressure needed to drive oxygen and nutrients across cell membranes is inadequate. In septic shock, hypoxia is worsened by increased inflammatory responses, such as fever, that increase the cell’s oxygen demand and consumption rate, and by direct toxic and inflammatory chemical disruption of cellular metabolism, that impairs the cells’ ability to use oxygen.
Both positive and negative compensatory mechanisms, such as anaerobic metabolism, lysosomal enzyme release, decreased intravascular volume, and activation of the clotting cascade, may further impair oxygen delivery and use. Anaerobic metabolism results in disruption of electrolyte and lysosomal enzymatic processes, thus changing the normal ionic and osmotic levels in cells governed by the physical law of diffusion. Diffusion of nutrients and wastes into and out of the cell takes longer, and cellular metabolism is further altered. Without oxygen, the cell shifts from aerobic to anaerobic metabolism. Anaerobic metabolism is a less efficient method of extracting energy from carbon bonds, and the cell begins to use adenosine triphosphate (ATP) faster than it can be replaced. Without ATP the cell loses its ability to maintain an electrochemical gradient across its selectively permeable membrane. Specifically, the cell cannot operate the sodium-potassium pump. Sodium and chloride accumulate inside the cell and potassium exits the cell. Cells of the nervous system and myocardium are profoundly and immediately affected. The resting potentials of these cells are reduced, and the action potentials decrease in amplitude (see Chapter 1). Myocardial depressant factor also decreases the contractility of the heart. A variety of clinical manifestations of impaired central nervous system and myocardial function result.
In addition to decreasing ATP stores, anaerobic metabolism affects the pH of the cell, and metabolic acidosis develops. A compensatory mechanism is initiated that enables cardiac and skeletal muscles to use lactic acid as a fuel source, but only for a limited time. The decreasing pH of the cell that is functioning under anaerobic conditions has serious consequences. Enzymes necessary for cellular function dissociate under acidic conditions. Enzyme dissociation stops cell function, repair, and division. As lactic acid is released systemically, blood pH drops, reducing the oxygen-carrying capacity of the blood (see Chapter 2), and the heart’s ability to pump. Therefore, less oxygen is delivered to the cells and further hypoxia results in acidosis, which triggers more lysosomal enzyme release caused by disruption of lysosomal membrane integrity. Lysosomal enzymes released during shock injure the cell that released them and injure adjacent cells. By damaging the mechanisms of surrounding cells, lysosomal enzymes extend areas of impaired metabolism and cellular injury.
Intravascular fluid loss into the intracellular and interstitial spaces, described as “third spacing” earlier, is amplified when serum albumin and other plasma proteins are consumed for fuel, which results in decreased intravascular osmotic pressure, shift of fluid to the interstitial or extracellular spaces, and decreased circulatory volume. Decreased circulatory volume magnifies decreased tissue perfusion in all types of shock. Decreased intravascular volume causes decreased cardiac output in septic shock and further reduces cardiac output in cardiogenic shock. In individuals with anaphylactic, neurogenic, or septic shock and an already dilated vasculature, hypotension worsens as a result of decreased circulatory volume. New data on additional mechanisms for hypotension (vasodilation) are illustrated in Figure 48-7 (p. 1678).
Concurrently, diffusion across capillary membranes occurs more slowly, as blood flow in the capillary beds becomes sluggish, caused by decreased fluid volume with increased viscosity of blood. Sluggish capillary flow decreases tissue perfusion further, microvascular clots form, and activation of the clotting cascade may begin (see Chapter 27). Microemboli formation and platelet aggregation account for common complications of shock, such as acute tubular necrosis, acute respiratory distress syndrome (ARDS), and disseminated intravascular coagulation (DIC). It also may activate or be activated by the inflammatory response.3
Impairment of Glucose Delivery and Use
Impaired glucose use can be caused by either impaired glucose delivery or impaired glucose uptake by the cells (see Figure 48-1). The reasons for inadequate glucose delivery are the same as those enumerated for inadequate oxygen delivery. In addition, in septic and anaphylactic shock, glucose metabolism may be increased or disrupted because of fever or bacteria, and glucose uptake can be prevented by the presence of vasoactive toxins, endotoxins, histamine, and kinins.
Some of the compensatory mechanisms activated by shock contribute to decreased glucose uptake and use by the cells. High serum levels of cortisol, growth hormone, and catecholamines account for hyperglycemia and insulin resistance, tachycardia, increased SVR, and increased cardiac contractility. Cells shift to glycogenolysis, gluconeogenesis, and lipolysis to generate fuel for survival (see Chapter 1). Except in the liver, kidneys, and muscles, the body’s cells have extremely limited stores of glycogen. In fact, total body stores can fuel the metabolism for only about 10 hours. The depletion of fat and glycogen stores is not itself a cause of organ failure, but the energy costs of glycogenolysis and lipolysis are considerable and contribute to the cellular failure.
Types of Shock, Clinical Manifestations, and Treatment
Simultaneous to correcting the underlying cause or condition, treatment is directed at targeting improvement of microcirculatory tissue perfusion.4 Optimizing oxygenation is an absolute necessity in all shock states. The goal has a dual purpose to both optimize oxygen delivery and reduce oxygen consumption. Intravenous fluid is administered to expand intravascular volume and thereby improve tissue perfusion by increasing blood pressure and cardiac output, except in cases of cardiogenic shock, which require diuresis to reduce preload.
Effective glucose level control in various shock states has been shown to improve outcomes. Hyperglycemia, caused by insulin resistance in the liver and muscle, is a common finding in the critically ill. This is most likely an adaptive response providing glucose for the brain, red blood cells, and wound healing. The extent of appropriate glucose level control has been evaluated in recent years, leading to more aggressive treatment with continuous, titrating insulin infusions to maintain blood glucose levels closer to normal, thus promoting normal cellular function and healing.5
Cardiogenic Shock
Cardiogenic shock results from the inability of the heart to pump adequate blood to tissues and end organs from any cause, the most common being the short-term consequences of an acute myocardial infarction or a severe episode of myocardial ischemia. Cardiogenic shock is defined as persistent hypotension and tissue hypoperfusion caused by cardiac dysfunction in the presence of adequate intravascular volume and left ventricular filling pressure.6 Pathologic conditions that reduce contractility, cause pump failure, impair diastolic filling, or cause obstruction can lead to cardiogenic shock. Decreased contractility and pump failure can result from (1) acute myocardial infarction (AMI), cardiomyopathy, sepsis, myocarditis, pericarditis, aneurysm, dysrhythmias, contusion, metabolic abnormalities, papillary muscle rupture; (2) impaired diastolic filling related to dysrhythmias; and (3) obstruction attributable to pulmonary embolism, cardiac tamponade, valvular disorders, tumors, and wall rupture or defects.7
Overall hospital mortality because of cardiogenic shock has decreased from approximately 90% in the 1970s to a recent estimate of 50%.8
As cardiac output decreases, compensatory adaptive responses are activated, such as the renin-angiotensin, neurohormonal, and sympathetic nervous systems, that lead to fluid retention, systemic vasoconstriction, and tachycardia.9 Activation of the inflammatory response (resulting in expression of inducible nitric oxide synthase), activation of inflammatory cytokines, and activation of the complement system appear to play an important role in the pathogenesis and outcome of cardiogenic shock.10 Increases in contractility, heart rate, and blood pressure are maintained in mild shock states through vasoconstriction in response to catecholamine release from the adrenals. Vasoconstriction increases vascular resistance to normalize blood pressure and increases cardiac performance by returning more blood volume and increasing perfusion to the heart; however, this increases myocardial demand and consumption of oxygen and nutrients by the heart. Increasing myocardial requirements burden the already failing heart, which can no longer pump an adequate volume of blood with sufficient force to perfuse the tissues. Thus increased coronary, tissue, and cellular ischemia further deteriorates myocardial function and shock.
Mortality in cardiogenic shock is reduced by use of intra-aortic balloon counterpulsation (IABP); cardiosupportive drug regimens and early coronary interventions have also improved outcomes.10–12 Following myocardial infarction, reperfusion and revascularization can be achieved with treatment modalities that include fibrinolytic therapies (medications that disintegrate the coronary thrombus) and percutaneous interventions (balloon angioplasty, stent placement, and thrombectomies). Surgery (coronary artery bypass, ventriculoplasty, or heart transplant) is also utilized to open the coronary vessels during an acute myocardial infarction or to replace irreparable heart muscle. Cardio-supportive drug and fluid regimens are initiated to maintain adequate blood pressure and essential fluid and electrolyte balance, and to optimize coronary perfusion to the myocardium. Mechanical assist devices, specifically intra-aortic balloon pumps and percutaneous or ventricular assist devices (VADs), are used to support cardiac output temporarily until the individual improves or transplantation is possible.11 Implantable VADs, pacemakers, or internal defibrillator devices are sometimes used as permanent treatment for those who survive cardiogenic shock. Continuous hemodynamic monitoring should be employed to evaluate vascular volume and pressures, optimize fluid levels, and monitor drug administration with the goal of improving cardiac output and tissue perfusion.13
The clinical manifestations of cardiogenic shock are caused by inadequate perfusion to the heart and end organs (Figure 48-2). Subjective complaints of chest pain, dyspnea, and faintness, along with feelings of impending doom, are often revealed. Classic observable signs and symptoms of tachycardia, tachypnea, hypotension, jugular venous distention, dysrhythmia, and low measured cardiac output are hallmarks. Cyanosis; skin mottling; rapid, faint, or irregular pulses; low urine output; and occasional peripheral edema are additional signs and symptoms of end-organ hypoperfusion. Myocardial dysfunction from fluid overload may result in extra heart sounds, pulmonary edema, hypoxemia, and elevated end-organ laboratory values. The amount of brain natriuretic peptide, produced when the heart stretches in relation to increased volume with decreased cardiac output, is increased to help diurese the fluid volume excess.9 Pulmonary edema increases as volume accumulates and expands from the heart into the lungs as evidenced by audible crackles and wheezes as well as abnormal vascular congestion on chest radiography. Metabolic abnormalities involving electrolyte imbalances and elevated levels of inflammatory markers may result from or concur with the cardiac cascade of shock.
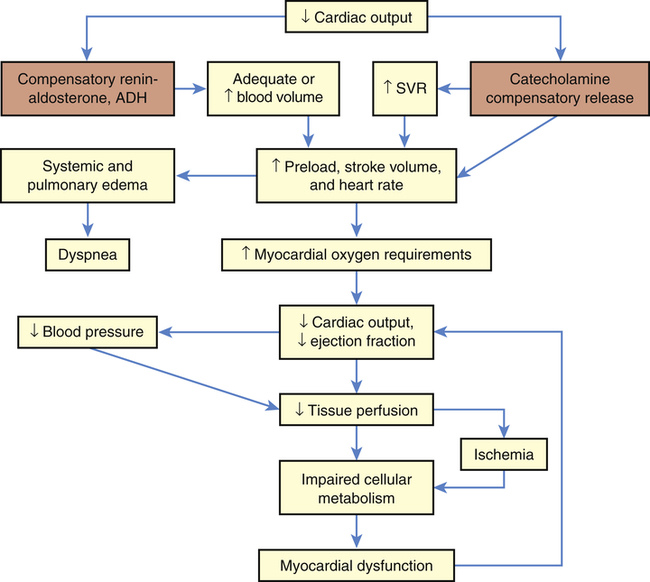
Hypovolemic Shock
Hypovolemia is offset initially by compensatory mechanisms (Figure 48-3). Heart rate and SVR increase as a result of catecholamine release by the adrenals. This boosts cardiac output and tissue perfusion pressures. Compelled by a decrease in capillary hydrostatic pressures, interstitial fluid moves into the vascular compartment. The liver and spleen add to blood volume by disgorging stored red blood cells and plasma. In the kidneys, renin (through several intermediaries) stimulates aldosterone release and the retention of sodium (and hence water), whereas antidiuretic hormone (ADH, or vasopressin) from the posterior pituitary gland increases water retention. Data on the compensation of ADH, however, show that as shock worsens, ADH in plasma decreases. Hypovolemic shock results in compensatory vasoconstriction and increased SVR and afterload in order to improve blood pressure and perfusion to core organs of the body.
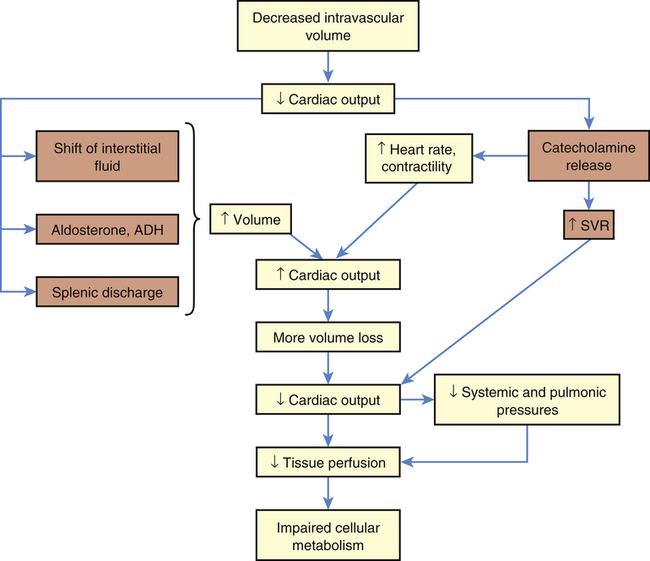
This type of shock becomes life threatening when compensatory mechanisms (orange boxes) are overwhelmed by continued loss of intravascular volume. ADH, Antidiuretic hormone; SVR, systemic vascular resistance.
These compensatory mechanisms are, however, finite. If the initial fluid or blood loss is great or if loss continues, compensation fails, resulting in decreased tissue perfusion. Nutrient delivery to the cells is impaired, and cellular metabolism fails. Mortality from traumatic hemorrhagic shock ranges from 10% to 31%. Prompt control of hemorrhage is the treatment of choice. Fluid replacement is also important, but the type of fluid to be used and the rate of replacement are controversial.14,15 The clinical manifestations of hypovolemic shock include high SVR, poor skin turgor, increased thirst, oliguria, low systemic and pulmonary preloads, and rapid heart rates.
Neurogenic Shock
Neurogenic shock also is called vasogenic shock. Both terms refer to a widespread and massive vasodilation that results from an imbalance between parasympathetic and sympathetic stimulation of vascular smooth muscle (see Chapter 31). Occasionally, parasympathetic overstimulation or sympathetic understimulation persists, causing vasodilation for an extended period. Extreme, persistent vasodilation leads to neurogenic shock (Figure 48-4). Neurogenic shock creates “relative hypovolemia.” Blood volume has not changed, but the amount of space containing the blood has increased, so that SVR decreases drastically; thus pressure in the vessels is inadequate to drive nutrients across capillary membranes, and nutrient delivery to the cells is impaired. As with other types of shock, this leads to impaired cellular metabolism.
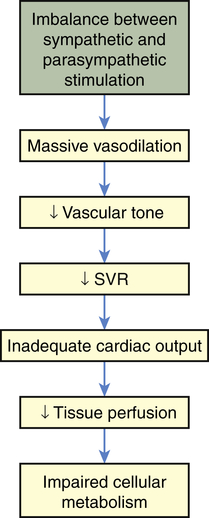
Neurogenic shock can be caused by any factor that stimulates parasympathetic activity or inhibits sympathetic activity of vascular smooth muscle. (Parasympathetic stimulation automatically inhibits sympathetic activity and vice versa; see Chapter 31.) Normally, sympathetic stimulation maintains muscle tone. If sympathetic stimulation is interrupted or inhibited, vasodilation occurs. Therefore, trauma to the spinal cord or medulla, conditions that interrupt the supply of oxygen to the medulla, or conditions that deprive the medulla of glucose (e.g., insulin reactions) can cause neurogenic shock by interrupting sympathetic activity. Depressive drugs, anesthetic agents, and severe emotional stress and pain are other causes of neurogenic shock.
Anaphylactic Shock
Anaphylactic shock is the outcome of a widespread hypersensitivity to an allergen that triggers a reaction known as anaphylaxis. (An allergen is an antigen to which an individual is hypersensitive; see Chapters 7, 8, and 9 for discussions of immunity, inflammation, and hypersensitivity.) Immunologic causes are related to the inflammatory and vasodilatory effects triggered by a pathologic allergic reaction to an antigen. Production of mast cells, IgE, and low-affinity IgE receptor (FceRI) is induced by cellular response to the antigen. Anaphylactoid type reactions, which are nonimmunologic in origin, can be related to cold weather, exercise, and contaminants in medications.16 Physiologic alterations related to the inflammatory and immune response, similar to neurogenic shock, are massive—vasodilation, peripheral pooling, and relative hypovolemia lead to decreased tissue perfusion and impaired cellular metabolism (Figure 48-5). Anaphylactic shock is often severe and has immediate symptoms, including an itchy rash, throat swelling, and low blood pressure, related to the massive vasodilation and systemic inflammation that may progress to death in minutes if emergency treatment is not rendered.
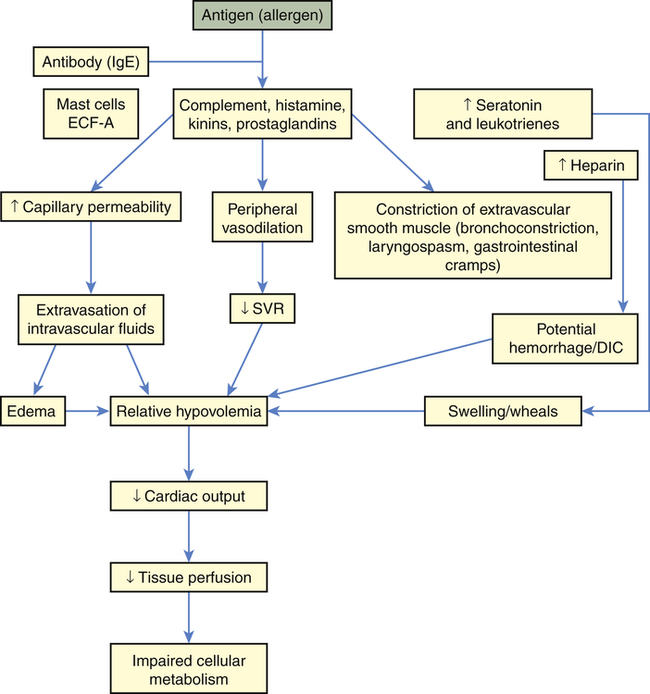
DIC, Disseminated intravascular coagulation; ECF-A, eosinophil chemotactic factor anaphylaxis; IgE, immunoglobulin E; SVR, systemic vascular resistance.
Some allergens known to cause hypersensitivity reactions are foods, insect and snake venoms, pollens, medications, and latex. Once in the body, the allergen causes the extensive immune and inflammatory response. The vascular effects of this response include vasodilation and increased vascular permeability, resulting in peripheral pooling and tissue edema. The extravascular effects include constriction of extravascular smooth muscle. Constriction often causes respiratory difficulty because it tends to affect smooth muscle layers in the airway walls (e.g., the larynx and bronchioles; see Chapter 34).
Clinical manifestations of anaphylactic and anaphylactoid shock may be anxiety, difficulty in breathing, gastrointestinal cramps, edema, hives (urticaria), sensations of burning or itching of the skin, fever, and hemolysis. A precipitous decrease in blood pressure occurs and is followed by impaired mentation. Other signs include decreased SVR (with high or normal cardiac output) and oliguria. Treatment begins with removal of the antigen (if possible). Epinephrine is administered to decrease mast cell and basophil degranulation, cause vasoconstriction, and reverse airway constriction.17 Volume expanders (e.g., lactated Ringer solution) are given intravenously to reverse the relative hypovolemia, and antihistamines and corticosteroids are given to stop the inflammatory reaction.
In response to the need for improved anaphylaxis diagnosis and treatment, the World Allergy Organization (WAO) formulated global guidelines in 2011 for assessment and management of anaphylaxis.17 These guidelines were further updated in 2012 to include recommendations for validation of the clinical criteria for diagnosis, use of epinephrine, development of in vitro tests to distinguish clinical risk from asymptomatic sensitization, and immune modulation to prevent anaphylaxis16 (see What’s New? Anaphylactic Shock).
Septic Shock
Septic shock is the endpoint of a continuum of progressive dysfunction.18 The syndrome begins with systemic inflammatory response syndrome (SIRS), then sepsis, then severe sepsis, and then septic shock. Consensus on definitions of each component was updated at an international sepsis conference in 2003 (Table 48-1).18 The International Sepsis Forum reviewed research for sepsis to identify and define the six most common infection sites (pneumonia, bloodstream, intravascular catheter, intra-abdominal, urosepsis, and surgical wound infection) associated with sepsis in the intensive care setting.19
TABLE 48-1
DEFINITIONS OF SEPTIC SHOCK COMPONENTS
| TERM | BASIC DEFINITION | COMMENTS |
| Infection | A pathologic process that results from an invasion of a normal part of the body by pathogenic or potentially pathogenic microorganisms | Still viewed as an imperfect definition Infection can be strongly suspected without microbiologic confirmation |
| Bacteremia | Presence of viable bacteria in the blood | Neither necessary nor sufficient to make a diagnosis of sepsis |
| SIRS | Systemic inflammatory response syndrome is manifested by two or more of general variables:1a through 1d or 2a through 2c | Diagnostic criteria: a. Fever—core temperature >38.3° C (100.9° F) b. Hypothermia—core temperature <36° C (96.8° F) c. Heart rate >90 beats/minute f. Significant edema or positive fluid balance (>20 ml/kg over 24 hr) g. Hyperglycemia (blood sugar >140 mg/dl) in the absence of diabetes a. Leukocytosis (WBC count >12,000) b. Leukopenia (WBC count <4000) c. Normal WBC with >10% immature forms d. Plasma C-reactive protein >2 SD above the normal value 4. Organ dysfunction variables a. Arterial hypoxemia (PaO2/FiO2 <300) b. Acute oliguria (urine output <0.5 ml/kg/hr or 45 ml for at least 2 hr) c. Creatinine increase >0.5 mg/dl d. Coagulation abnormalities (INR >1.5 or a PTT >60) f. Thrombocytopenia (platelet count <100,000) g. Hyperbilirubinemia (plasma total bilirubin >4 mg/dl or 70 mmol/L) |
| Sepsis | Systemic response to infection, which is manifested by two or more of the SIRS criteria | |
| Severe sepsis | Sepsis complicated with one or more organ system dysfunctions | May be difficult to differentiate underlying organ dysfunction from sepsis-related organ dysfunction |
| Septic shock | Severe sepsis complicated by persistent hypotension refractory to early fluid therapy | Persistent systolic blood pressure <90 mmHg |
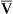
Data from Dellinger RP et al: Crit Care Med 41(2):580–637, 2013; Levy MM et al: Crit Care Med 312:1250, 2003; Opal SM, Scand J Infect Dis 35:529, 2003.
Severe sepsis is the tenth most common cause of death in the United States.20 Mortality ranges from 28% to 60%.21 Septic shock is caused by gram-negative bacteria, gram-positive bacteria, and fungi. Advances in antibiotic therapy for gram-negative sepsis have made gram-positive bacteria the leading cause of sepsis.22 Even when properly treated with available therapies, it carries a high mortality rate. Prognosis is significantly affected by the source and virulence of the infectious microorganism.
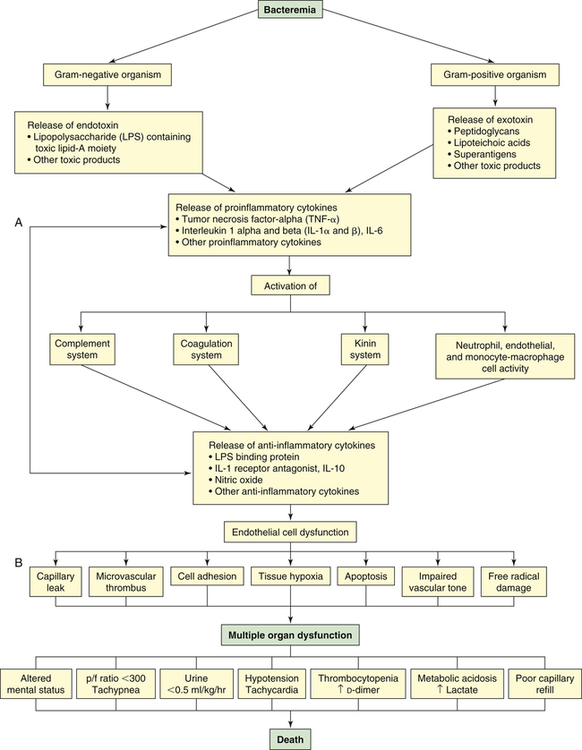
Septic shock begins with a nidus of infection that may be readily discernible or extremely difficult to locate (Figure 48-6). Bacteria and fungi enter the bloodstream to produce bacteremia in one of two ways: (1) directly from the site of infection, or (2) indirectly from toxic substances released by the bacteria directly into the bloodstream. These toxic substances, which act as triggering molecules in the septic syndrome, include endotoxins released by gram-negative microorganisms, lipoteichoic acids and peptidoglycan released by gram-positive microorganisms, and superantigens.22
p/f (PaO2/FiO2), Oxygenation ratio. (A from Lazaron V, Barke RA: Urol Clin North Am 26[4]:687, 1999. B © 2003, Eli Lilly and Company. All rights reserved. Reprinted with permission from Eli Lilly and Company.)
The triggering molecules cause the host to initiate a proinflammatory response. Proinflammatory cells released include polymorphonuclear leukocytes, macrophages, monocytes, and platelets. Proinflammatory mediators released include cytokines (interleukins [IL]-1, IL-2, IL-6, IL-8, and IL-15; tumor necrosis factor-alpha [TNF-α]; and granulocyte cell–stimulating factor), complement and complement cascade activation, kinins, arachidonic acid metabolites (prostaglandins, prostacyclin, leukotrienes, and thromboxane), soluble adhesion molecules, platelet-activating factor, endorphins, vasoactive neuropeptides, histamine, serotonin, monocyte chemoattractant proteins 1 and 2, proteolytic enzymes (e.g., elastase and lysosomal enzymes), protein kinase, tyrosine kinase, CD14, toxic oxygen metabolites (e.g., superoxide, hydroxyl radical, hydrogen peroxide, peroxynitrite), neopterin, and clotting cascade activation.3,23,24 Proinflammatory cytokines enhance tissue factors, which initiates coagulation. Diminished thrombomodulin (cell surface glycoprotein of endothelial cells) inhibits the conversion of protein C and activated protein C. A compensatory anti-inflammatory response syndrome is presumed to follow this response.3,23,24 Anti-inflammatory mediators released include lipopolysaccharide-binding protein; IL-1 receptor antagonist; soluble CD-14; type 2 IL-1 receptor; leukotriene B4 receptor antagonist; IL-4, IL-10, and IL-13; soluble tumor necrosis factor receptor; transforming growth factor-beta (TGF-β); epinephrine; and nitric oxide.3,23,24 Presumably the end result is a mixed antagonistic response syndrome as proinflammatory and anti-inflammatory mediators respond, intensify, and lead the host into MODS.
Clinical manifestations of septic shock are persistent low arterial pressure, low SVR from vasodilation, and an alteration in oxygen extraction by all cells. Septic shock and states of prolonged shock causing tissue hypoxia with lactic acidosis increase nitric oxide synthesis, activate ATP-sensitive and calcium-regulated potassium channels (KATP and KCa, respectively) in vascular smooth muscle (see Chapter 31), and lead to depletion of ADH (vasopressin) (Figure 48-7). Tachycardia causes cardiac output to remain normal or become elevated, although myocardial contractility is reduced. Temperature instability is present, ranging from hyperthermia to hypothermia. Effects on other organ systems may result in deranged renal function, gastrointestinal mucosa changes that result in release of bacteria from the gut, jaundice, clotting abnormalities, deterioration of mental status, and tachypnea that often progresses to ARDS.
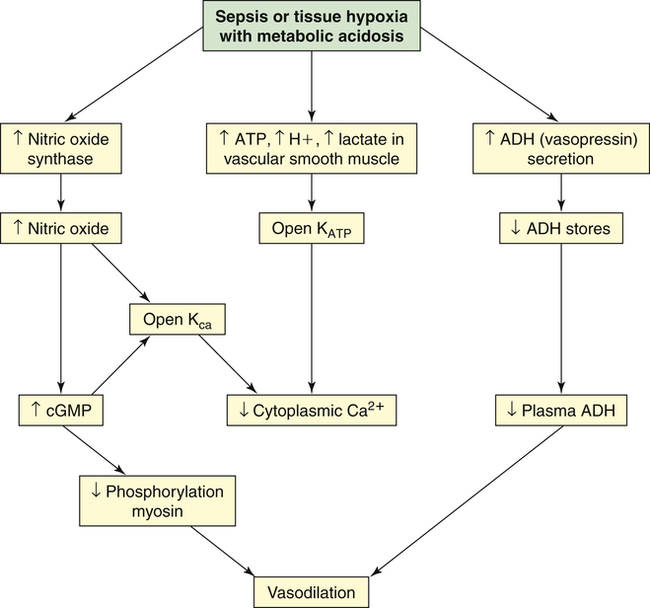
Vasodilatory shock is caused by the inappropriate activation of vasodilatory mechanisms and the failure of constrictor mechanisms. Unregulated nitric oxide, by regulating guanylate cyclase and generating cyclic guanosine monophosphate (cGMP), causes dephosphorylation of myosin and, thus, vasodilation. Nitric oxide synthesis and metabolic acidosis activate the potassium channels (KATP and Kca) in the plasma membrane of vascular smooth muscle. The resulting hyperpolarization (see Chapter 3) of the membrane presents the calcium that mediates norepinephrine and angiotensin II–induced vasoconstriction from entering the cell. Therefore, hypotension and vasodilation stubbornly persist despite high plasma levels of these hormones. In contrast, and unexpectedly, the plasma level of antidiuretic hormone (ADH) (vasopressin) is low despite the presence of hypotension. The early, massive release of ADH may result in future depletion.
Early detection of high-risk illness and rapid implementation of the following standard protocols are increasingly being used by hospitals worldwide: decreasing the time before lactate levels are determined; obtaining blood cultures; starting administration of antibiotics and antifungals; implementing a fluid challenge; achieving goals for blood pressure, central venous pressure (CVP), and central venous oxygen saturation.25,26 Promptly initiated treatment helps reduce mortality and morbidity related to multiple organ dysfunction. Infection and sepsis identified in the early stages and treated with antibiotics and fluid resuscitation may prevent evolution to severe sepsis and shock; however, once these are identified, more stringent treatment is recommended. The current guidelines for severe sepsis and septic shock27 recommend immediate resuscitation in patients with lactate levels >4 mmol/L. Elevated lactate levels indicate tissue hypoperfusion and patients should begin early goal-directed therapy or fluid resuscitation within 6 hours, regardless of blood pressure level.25,26 Specifically, fluid resuscitation with either crystalloid or colloid fluids should be goal-directed to maintain the following values: central venous pressure, 8 to 12 mmHg (or 12 to 15 mmHg when the patient is being mechanically ventilated); mean arterial pressure, 65 mmHg; urine output, 0.5 ml/kg/hr; mixed venous saturation (S o2), 65%. In addition, specific anatomic sites of infection with source identification should be determined so that appropriate antibiotics can be prescribed and control measures can be implemented that eliminate or reduce the spread of infection.27,28 Fungal assays, including 1,3-β-d-glucan assay and mannan and anti-mannan antibody assays, are recommended to identify suspected fungal infection.29 Normal saline, at least 1 L or 30 ml/kg body weight, is the initial fluid of choice and hetastarches with molecular weights >200 daltons are not recommended. Adequate and incremental fluid resuscitation is a prerequisite and should be continued to achieve perfusion; supplementation with vasopressors is recommended if the endpoints of tissue perfusion (CVP and normalized lactate levels) are not met.29
o2), 65%. In addition, specific anatomic sites of infection with source identification should be determined so that appropriate antibiotics can be prescribed and control measures can be implemented that eliminate or reduce the spread of infection.27,28 Fungal assays, including 1,3-β-d-glucan assay and mannan and anti-mannan antibody assays, are recommended to identify suspected fungal infection.29 Normal saline, at least 1 L or 30 ml/kg body weight, is the initial fluid of choice and hetastarches with molecular weights >200 daltons are not recommended. Adequate and incremental fluid resuscitation is a prerequisite and should be continued to achieve perfusion; supplementation with vasopressors is recommended if the endpoints of tissue perfusion (CVP and normalized lactate levels) are not met.29
Pharmacologic hemodynamic support and adjunctive therapy to maintain tissue perfusion include vasoactive agents. Norepinephrine is now considered the initial vasopressor of choice.29 Dopamine, epinephrine, phenylephrine, and vasopressin are alternatives; however, these are less preferred because of potential tachycardia, dysrhythmias, or reduction in splanchnic blood flow. Dobutamine, an inotropic agent, should be used if myocardial dysfunction is present or hypotension remains after intravascular volume and blood pressure are achieved. Similarly, intravenous corticosteroids are suggested in septic shock to help replace intrinsic loss of cortisol, but only when blood pressure remains unresponsive to volume.27 Other supportive therapies in the treatment of severe sepsis include blood product administration to maintain hemoglobin levels between 7 and 9 g/dl, targeted tidal volume (6 ml/kg) when mechanically ventilated with plateau pressure less than 30 cm H2O, utilization of sedation and analgesic protocols when needed, and avoidance of neuromuscular blockers if possible. Glucose concentration is to be maintained at less than 180 mg/dl. If renal dialysis is required, intermittent hemodialysis and continuous venovenous hemofiltration are considered equivalent. Bicarbonate therapy is contraindicated for the purpose of treating hypoperfusion lactic acidemia, unless the pH is <7.15. Use of deep vein thrombosis prophylaxis, such as low-dose unfractionated heparin or low-molecular-weight heparin, or mechanical compression prophylaxis of heparin, is contraindicated. Stress ulcer prophylaxis with H2-receptor antagonists or proton pump inhibitors is indicated. Digestive tract decontamination and selective oropharyngeal decontamination are recommended in the prevention of ventilator-acquired pneumonia.29 It is important to discuss advance directives with the individual and family.27 Seeking to decrease use of antibiotics in the critically ill and, thus, prevent resistance to antibiotics is an important strategy in treating infection (see What’s New? Procalcitonin Levels). Recent research suggests that monitoring serial procalcitonin (PCT) levels, a precursor hormone to calcitonin, may be used to shorten antibiotic use in the treatment of respiratory tract infections.30 PCT, normally not discernible on assay, when elevated may indicate specific proinflammatory response during bacterial infection. PCT levels should not be used as an indicator to start antibiotics, but if monitored sequentially at the start of empiric antibiotics and then dropping to low levels, discontinuation may be clinically indicated.30,31
Multiple Organ Dysfunction Syndrome
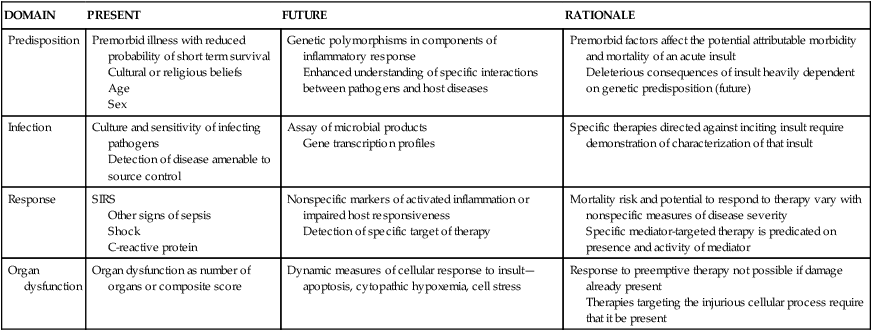
Multiple organ dysfunction syndrome (MODS) is the progressive dysfunction of two or more organ systems resulting from an uncontrolled inflammatory response to a severe illness or injury. The organ dysfunction can progress to organ failure and death. MODS occurs during severe sepsis. In 2001, an international consensus conference developed a set of definitions for sepsis and related disorders (see Table 48-1), and a predisposition-infection-response-organ (PIRO) dysfunction staging system was proposed as a template for staging sepsis (Table 48-2). MODS is the end stage of a variety of injuries that terminate in severe, generalized inflammatory response.
TABLE 48-2
PREDISPOSITION-INFECTION-RESPONSE-ORGAN (PIRO) DYSFUNCTION SYSTEM FOR STAGING SEPSIS
| DOMAIN | PRESENT | FUTURE | RATIONALE |
| Predisposition | Premorbid illness with reduced probability of short term survival Cultural or religious beliefs Age Sex | Genetic polymorphisms in components of inflammatory response Enhanced understanding of specific interactions between pathogens and host diseases | Premorbid factors affect the potential attributable morbidity and mortality of an acute insult Deleterious consequences of insult heavily dependent on genetic predisposition (future) |
| Infection | Culture and sensitivity of infecting pathogens Detection of disease amenable to source control | Assay of microbial products Gene transcription profiles | Specific therapies directed against inciting insult require demonstration of characterization of that insult |
| Response | SIRS Other signs of sepsis Shock C-reactive protein | Nonspecific markers of activated inflammation or impaired host responsiveness Detection of specific target of therapy | Mortality risk and potential to respond to therapy vary with nonspecific measures of disease severity Specific mediator-targeted therapy is predicated on presence and activity of mediator |
| Organ dysfunction | Organ dysfunction as number of organs or composite score | Dynamic measures of cellular response to insult—apoptosis, cytopathic hypoxemia, cell stress | Response to preemptive therapy not possible if damage already present Therapies targeting the injurious cellular process require that it be present |
SIRS, Systemic inflammatory response syndrome.
From Levy MM et al: 2001 Intensive Care Med 29:530, 2003. Reproduced with kind permission of Springer Science + Business Media.
MODS was first recognized as a distinct clinical syndrome in the mid-1970s32,33 when advances in resuscitation and support technologies allowed many individuals to survive life-threatening illness or trauma only to die of complications of their disease. Today MODS is a leading cause of mortality in surgical intensive care units (ICUs). Mortality for individuals with MODS increases progressively from 54% with two failing organ systems to 100% with five failing organ systems. Moreover, mortality has not improved much over the past 15 to 20 years.34,35
Although sepsis and septic shock are the most common causes, MODS can be initiated by any severe injury or disease process that activates a massive systemic inflammatory response by the host. Documented clinical infection is not necessary for its development. Other common triggers are severe trauma, major surgery, burns, circulatory shock, acute pancreatitis, acute renal failure, ARDS, blood transfusion, heat stroke, liver failure, mesenteric ischemia, propofol infusion syndrome, persistent inflammatory foci, DIC, and necrotic tissue (Box 48-1). MODS is the major cause of death following septic shock, trauma, burn injuries, and ARDS. People at greatest risk for developing MODS are older adults and persons with significant tissue injury or pre-existing disease.18
The PIRO system (see Table 48-2), a clinically useful sepsis staging system, stratifies individuals with disease by baseline risk of adverse outcomes and potential to respond to therapy.18
Pathophysiology
In primary MODS the organ injury is directly associated with a specific insult, most often ischemia or impaired perfusion from an episode of shock or trauma, thermal injury, soft tissue necrosis, or invasive infection.36 This decreased perfusion is local (in the injured organs themselves) and generalized. The generalized hypoperfusion in primary MODS usually cannot be detected clinically. As a result of the insult, a stress response is initiated and stress hormones—in particular, catecholamines—are released. The inflammatory and stress responses are not as evident as they are in secondary MODS. In primary MODS during the inflammatory response, presumably neutrophils and macrophages are “primed” by cytokines.36 Any second insult, such as additional tissue injury, infection, or organ ischemia, may then activate the primed cells to produce an exaggerated response of secondary MODS36,37 (Figure 48-8).
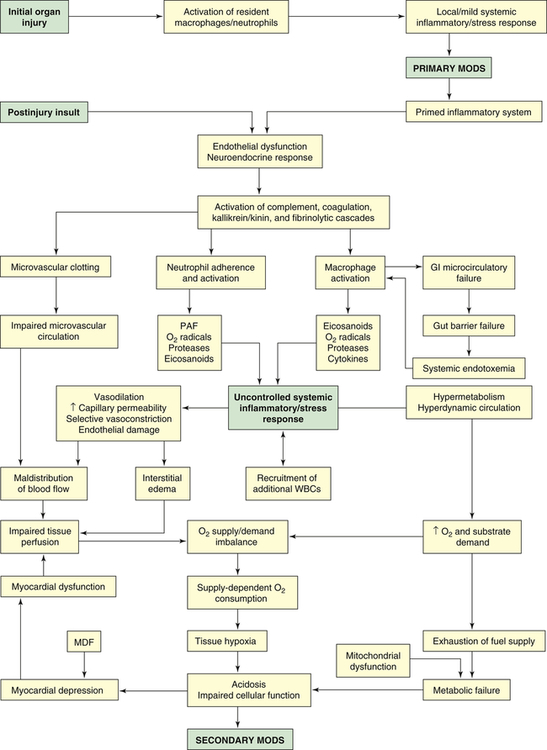
GI, Gastrointestinal; MDF, myocardial depressant factor; MODS, multiple organ dysfunction syndrome; PAF, platelet-activating factor; WBCs, white blood cells.
Secondary MODS is initiated by the delayed postinjury insult as primed macrophages release a barrage of mediators, particularly the cytokines TNF and IL-1. These mediators damage the endothelium throughout the body. If a gram-negative bacterial infection is present, endotoxin released from the bacteria also causes severe damage to endothelial cells. Normal endothelial cells have little interaction with leukocytes, but when stimulated by TNF, IL-1, IL-6, or endotoxin, they change to a proinflammatory state and express adhesion molecules that mediate adhesion of neutrophils. The adhered neutrophils then migrate through the endothelium, aggregate in the area of damaged tissue, and amplify the inflammation.3 The activated endothelial cells increase production and release of nitric oxide (endothelium), a potent vasodilator that is considered an important factor in the blood flow changes and loss of vascular tone noted in systemic inflammation.38 The injured endothelium also becomes much more permeable, allowing fluid and protein to leak into the interstitial spaces. An important function of normal endothelium is anticoagulation. When damaged, the endothelium loses much of its ability to prevent blood clotting, allowing microvascular thrombi to develop.
The postinjury insult also activates the neuroendocrine system, resulting in a second, more extensive stress response. The normal function of the stress response is to maintain basal and stress-related homeostasis39; however, homeostasis cannot be maintained in MODS. In fact, the endocrine response becomes excessive and injurious. There is an early increase in the levels of circulating catecholamines that contributes to many of the clinical manifestations of MODS, such as tachycardia, hypermetabolism, and increased oxygen consumption. Cortisol, glucagon, insulin, human growth hormone, ADH (which may become depleted), and endorphin levels also are increased. Many of these hormones contribute to the extreme catabolic state of MODS, and endorphins, which are vasodilators, decrease SVR. The sympathetic nervous system, to compensate for complications resulting from the injury (e.g., fluid loss, hypotension), also is stimulated. The stimulation persists throughout the period of critical illness.39 The stress response can be amplified by a number of factors, including pain, anxiety, psychosis, and hyperthermia. (The stress response is discussed in detail in Chapter 11.)
Because of endothelial cell dysfunction and the release of mediators, four major plasma cascades are activated: complement, kallikrein-kinin, coagulation, and fibrinolytic.40 Complement components, particularly the anaphylatoxins C3a and C5a, cause vasodilation by stimulating release of histamine from mast cells. They also have strong chemotactic properties. C5a, especially, causes adhesion and the activation and degranulation of neutrophils. Complement is thought to be a powerful trigger for the exaggerated inflammatory response. Activation of the kinin system results in the production of bradykinin, a very potent vasodilator known to decrease SVR. Coagulation mechanisms also are activated, and because tissue injury and endothelial dysfunction are extensive, microvascular thrombosis occurs throughout the body, resulting in impaired microvascular circulation and organ ischemia. Concurrently, fibrinolytic mechanisms are activated. The tendency toward clotting, however, is greater, resulting in a net procoagulant state that can lead to the development of DIC. The overall effect of the activation of the plasma cascades is a hyperinflammatory and hypercoagulant state that contributes to vasodilation, vasopermeability, cardiovascular instability, endothelial dysfunction, and clotting abnormalities.41
Once cytokines and other mediators have been released and the plasma enzyme cascades have been activated, a massive systemic inflammatory response develops. It involves several types of inflammatory cells, particularly neutrophils, macrophages, and mast cells. These cells, having been primed by their response to the initial organ injury, now pour large amounts of chemical mediators into tissues and into the systemic circulation. Neutrophils have tremendous inflammatory potential. The accumulation of activated neutrophils in organs is thought to play a key role in the pathogenesis of MODS.42 When neutrophils adhere to the endothelium, they undergo a “respiratory burst” (oxidative burst) and release oxygen-free radicals. The respiratory burst occurs as the activated neutrophil experiences a sudden increase in oxidative metabolism, producing large quantities of highly toxic oxygen-free radicals. These reactive oxygen species (ROS) cause oxidative stress. The primary ROS produced are superoxide, hydrogen peroxide (H2O2), hydroxyl radical (OH−), and singlet oxygen (O). Oxygen-free radicals are extremely damaging to vascular endothelium and tissue cells, attacking deoxyribonucleic acid (DNA), cross-linking membrane structures, and inducing membrane peroxidation—reactions that disorganize cell membranes and lead to tissue necrosis3,24 (also see Chapter 2).
Other important mediators released by neutrophils are proteases, particularly collagenase and elastase. Proteases directly damage endothelium and neighboring cells, resulting in increased capillary permeability and organ damage. When activated, neutrophils also release platelet-activating factor (PAF), a mediator that damages endothelium, stimulates clot formation, and activates increasing numbers of phagocytes. Finally, neutrophils release arachidonic acid metabolites (eicosanoids) as a result of lipid peroxidation of their cell membranes. Of the arachidonic acid metabolites (prostaglandins, thromboxanes, leukotrienes), two are particularly important in the pathogenesis of organ hypoperfusion: prostacyclin (PGI2) and thromboxane A2 (TXA2). TXA2 is a powerful vasoconstrictor, and PGI2 is a potent vasodilator. When released in varying amounts in different organ beds, they are largely responsible for the maldistribution of blood flow characteristic of MODS. In total, neutrophils produce at least 50 to 60 toxins.43,44 Collectively, products released by neutrophils cause endothelial dysfunction, systemic vasodilation, selective vasoconstriction (vasoconstriction of certain organ beds or parts of organ beds), increased vascular permeability, and microvascular coagulation.
Macrophages, present in most tissues, are activated by endotoxin, complement, and monocyte chemotactic substances.45 Macrophages share a key role in the development of the unregulated inflammation of secondary MODS with the neutrophils. Like neutrophils, they produce oxygen-free radicals, proteases, cytokines, nitric oxide, and arachidonic acid metabolites. It has been reported that excessive or prolonged stimulation of macrophages leads to the overproduction of cytokines and nitric oxide that initiate the cycle of harmful effects in MODS.3,24,45 TNF and IL-1, which share many of the same functions and act synergistically, are the major cytokines that mediate inflammation.45,46 TNF has potent metabolic effects, including fever, anorexia, hyperglycemia, hypermetabolism, and weight loss. It activates neutrophils, damages endothelial cells, and potentiates hypotension and shock. IL-1 also has metabolic effects, inducing fever, hypermetabolism, and muscle wasting. Normally, TNF activates cytokines, the coagulation system, fibrinolysis, and neutrophils. With the exception of neutrophil activation, IL-1 causes similar activation in individuals with cancer.47 In the pathogenesis of MODS, the cytokines are linked to all cellular, hemodynamic, and metabolic alterations.
The gastrointestinal mucosa is particularly vulnerable to inflammatory mediators released by macrophages and neutrophils. Under normal circumstances the gut mucosa serves as a barrier to prevent bacteria from the gastrointestinal tract from entering the systemic circulation. Damage to the mucosa results in microcirculatory failure of the gut and consequent loss of the gut barrier function. The loss of intestinal barrier function leads to the systemic spread of bacteria and/or endotoxin from the gut (systemic endotoxemia). This phenomenon is called translocation of bacteria. The idea that the gut acts as a reservoir of bacteria and endotoxin that can initiate or perpetuate the development of MODS is known as the gut hypothesis. The gut hypothesis provides a possible explanation for the fact that an infectious focus is not always found in individuals with MODS. Although this hypothesis has been substantiated by animal studies and has much support, the evidence from human studies is inconclusive.48,49
The numerous inflammatory processes operating in MODS cause maldistribution of blood flow and hypermetabolism. Maldistribution of blood flow refers to the uneven distribution of flow to various organs and between the large vessels and capillary beds of the body. It is caused by generalized vasodilation, increased capillary permeability, selective vasoconstriction, endothelial dysfunction, and impaired microvascular circulation. It is a major factor in the pathophysiology of MODS.40 The alterations in blood flow—which can occur at the cellular, organ, or regional level—lead to impaired tissue perfusion and a decreased supply of oxygen to the cells. The organs most severely affected by hypoperfusion are the lungs, splanchnic bed, liver, and kidneys. Despite supernormal systemic blood flow, oxygen delivery to the tissues decreases. Several factors contribute to the problem. First, blood is shunted past selected regional capillary beds. Shunting, caused by loss of autoregulation in some organs, may be an early indicator of progression of sepsis into MODS.50 This occurs because inflammatory mediators, particularly TXA2, override the normal vascular control mechanisms to cause selective vasoconstriction and because injured endothelial cells are unable to respond to normal vasodilator mediators. Second, interstitial edema, resulting from microvascular permeability, contributes to decreased oxygen delivery to cells by increasing the distance oxygen must travel to reach the cells. Third, capillary obstruction occurs because of the formation of microvascular thrombi and the aggregation of leukocytes.3,24,45
Hypermetabolism, with accompanying alterations in carbohydrate, fat, and lipid metabolism, is initially a compensatory measure to meet the body’s increased demands for energy. Eventually, however, hypermetabolism becomes detrimental, placing enormous demands on the heart. Hypermetabolism is the result of: (1) the neuroendocrine response to stress with the release of catecholamines and cortisol, and (2) the action of TNF and IL-1. With increased metabolism the calorie requirements are markedly increased,34 and the cardiac output increases 1.5 to 2 times normal.51 The alterations in metabolism affect all aspects of substrate use. Most important is the catabolism of protein, primarily of skeletal muscle and visceral organs. The extreme catabolism of protein can rapidly deplete lean body mass. Hyperglycemia occurs as gluconeogenesis by the liver increases and glucose use by the cells decreases. Fatty acids are mobilized from adipose tissue. The net result of the hypermetabolism is depletion of oxygen and fuel supplies.
Myocardial depression also accompanies MODS. The cause remains unclear, but possible explanations are the effects of myocardial depressant factor (MDF), TNF, and IL-1 on cardiac contractility; alterations in α-adrenergic receptors in the heart; and hypoxia of the myocardium.52,53
The decreased oxygen delivery to the cells (resulting from the maldistribution of blood flow) and the increased oxygen needs of the cells (resulting from hypermetabolism) combine to create an imbalance in oxygen supply and demand. This imbalance is critical in the pathogenesis of MODS because it results in a pathologic condition known as supply-dependent oxygen consumption. Ordinarily the amount of oxygen consumed by the cells depends only on the needs of the cells because there is an adequate reserve of oxygen that can be delivered if required. In MODS, however, the reserve has been exhausted and the amount of oxygen consumed becomes dependent on the amount the circulation is able to deliver. Because the amount is inadequate in MODS, the tissues become hypoxic. Compounding the hypoxic damage to cells is a phenomenon called reperfusion injury (see Chapter 2). Much of the organ damage in MODS occurs with the reestablishment of blood flow after a period of ischemia. During the ischemic episode, energy stores and ATP are depleted and the enzyme xanthine dehydrogenase is converted to xanthine oxidase. With reperfusion of the ischemic tissue, oxygen-free radicals are formed from oxygen by the action of xanthine oxidase, and they attack the already damaged tissues. Consequently, although reperfusion is necessary to restore oxygen supply to ischemic organs, it can increase the extent of injury. Therefore, because of supply-dependent oxygen consumption and reperfusion injury, tissues become increasingly hypoxic. The result is cellular acidosis, impaired cellular function, and ultimately multiple organ failure.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


