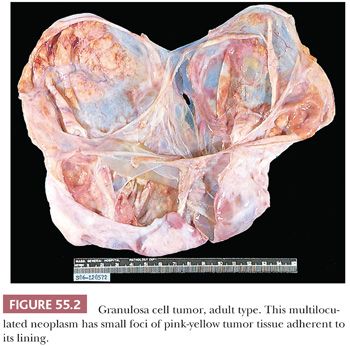
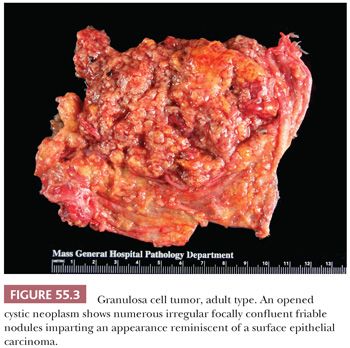
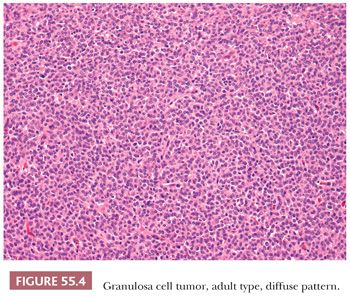
Microscopic Features. At low-power, several patterns may be encountered, and typically two or more are present in the same tumor (Figs. 55.4 to 55.9). The most common is a diffuse (Fig. 55.4) growth of round, oval, or, less often, spindle-shaped cells that almost always is associated with minor foci of obvious epithelial patterns, most often in the form of delicate cords (Fig. 55.6). In these tumors, the stroma is usually minimal to absent. Tumors in which spindled granulosa cells dominate have been referred to as sarcomatoid, but that designation is discouraged as it may cause confusion from the management viewpoint.
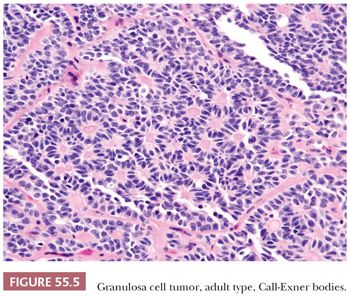
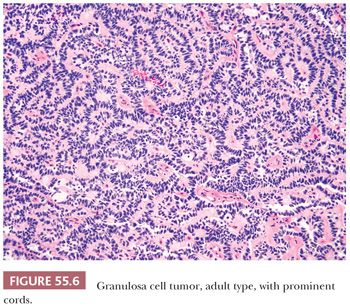
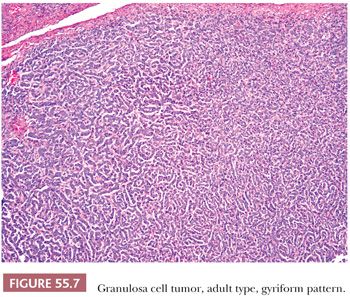
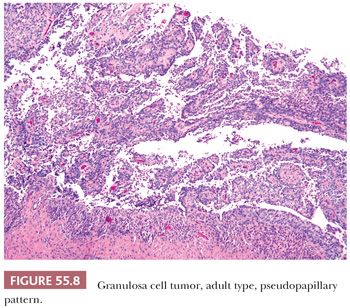
A second common appearance has obvious epithelial arrangements, including insular and trabecular as well as anastomosing cords of granulosa cells, on a background of conspicuous but usually minor fibrothecomatous stroma. Although follicular patterns are often emphasized, they are absent in the majority of tumors and are conspicuous in only a minority. The microfollicular pattern (Fig. 55.5) is characterized by small, generally regular, follicles (Call-Exner bodies) that may contain eosinophilic material with nuclear debris, hyalinized basement membrane–like material, or, rarely, basophilic secretion. The macrofollicular pattern is even less common and displays large, relatively uniform follicles typically containing eosinophilic secretions. Uncommon patterns include “watered-silk” (parallel, thin, winding cords) and gyriform (zigzag arrangement of cords) (Fig. 55.7). Rare tumors have a pseudopapillary pattern (8) (Fig. 55.8). Infrequently, hollow or solid tubules (Sertoli-like) are present, usually in small numbers.
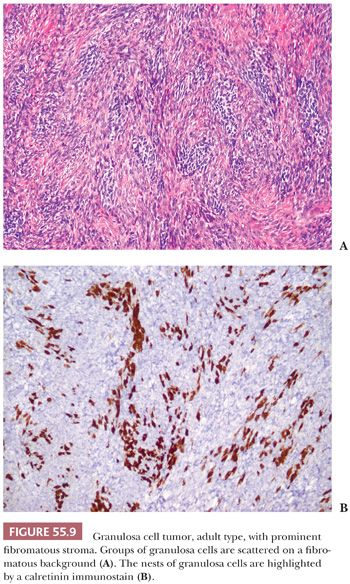
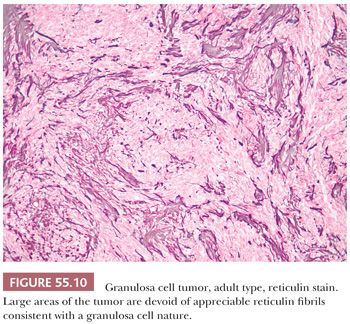
A third low-power appearance may closely resemble either a cellular fibroma or a thecoma due to a prominent stroma, but when a minority (but >10%) component of the tumor is composed of granulosa cells on that background, the tumor is considered a GCT (Fig. 55.9). Not infrequently, granulosa cell elements are best seen at the periphery in these tumors. Yet another low-power pattern is that of cells with pale cytoplasm (similar to that seen in thecoma) sometimes growing in large nodules. Epithelial differentiation indicating granulosa differentiation in the tumor may be subtle, but it is highlighted by a reticulin stain (Fig. 55.10).
The small group of cystic AGCTs features cysts that are usually lined by many layers of granulosa cells, some of which may show focal follicle formation; granulosa cells may also be present in the cyst walls (7). Denudation of the cyst lining may occasionally be significant and cause confusion with other cystic lesions.
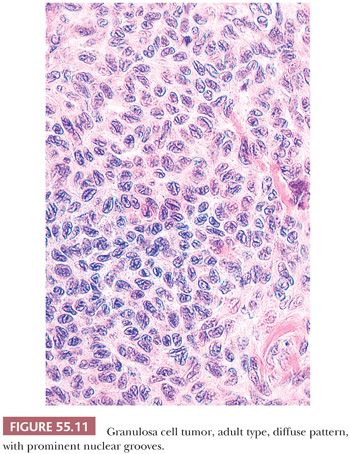
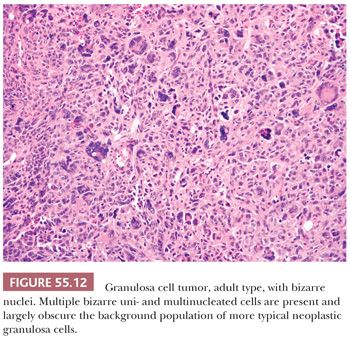
The granulosa cells usually have scant cytoplasm, but rarely, it may be abundant and eosinophilic, resulting in a luteinized appearance (9,10). The nuclei are typically pale and round, oval, or angular and are often haphazardly oriented. Nuclear grooves are common (Fig. 55.11) but may be relatively inconspicuous especially in tumors with a diffuse pattern. Nucleoli are occasionally moderately prominent, particularly in luteinized tumors which often lack nuclear grooves. Significant pleomorphism is usually absent, but approximately 2% contain cells with large, bizarre, hyperchromatic nuclei (11) (Fig. 55.12). The mitotic rate is usually less than or equal to two mitotic figures per 10 high-power fields (hpf), but higher counts do not exclude the diagnosis. A thecomatous or fibromatous stromal component is usually present and may predominate and be richly vascular. Stroma with the appearance of a high-grade sarcoma has been reported (12). Exceptionally, hepatocytes and Leydig cells (13) are seen. Many tumors have conspicuous foci of old or recent hemorrhage as well as nonspecific changes such as fibrosis.
Immunohistochemical Features. As this is the first discussion of immunohistochemistry in this chapter, we consider at this junction findings in AGCT but also other neoplasms. Immunohistochemistry may be helpful in differentiating AGCTs from tumors other than those in the sex cord–stromal category, and their immunohistochemical profile is shared with JGCT. Inhibin is usually positive in GCTs and most other sex cord–stromal and steroid cell tumors, (2,3,14–18) but to a lesser extent in fibromas (15,19) and is typically negative in fibrosarcoma; carcinomas, including small cell carcinoma of hypercalcemic type; and carcinoid (15). It is important to note that up to 20% of AGCT have been reported to be inhibin negative (19). Calretinin is a more sensitive but a less specific marker of sex cord and steroid cell tumors and should be used in conjunction with inhibin (14,20,21) and epithelial membrane antigen (EMA) or CK7 in the differential diagnosis with carcinomas (2,3). It has been shown that recurrent AGCT have a slightly lower percentage of positivity for these two markers (22). SF-1 has been postulated as the most sensitive sex cord–stromal marker among the most common types of sex cord–stromal tumors, showing higher frequency of positivity over inhibin and calretinin (19,23). FOXL2 has been recently described as helpful in the diagnosis of AGCT, but this marker is also positive in other sex cord or stromal tumors (24–27). Other positive markers in AGCT include melan-A, CD99, WT1, CD56, and not infrequently, CD10 and AE1/3 (19,28–32), but they are much less specific for this diagnosis as many of them are positive in other sex cord–stromal tumors as well as in a wide variety of other neoplasms.
Molecular Features. FOXL2 mutations are detected in the vast majority of AGCT but are typically absent in other sex cord tumors (33,34).
Differential Diagnosis. Undifferentiated or poorly differentiated carcinomas are occasionally misinterpreted as a diffuse AGCT, but in contrast to the latter, they are bilateral in more than 25% of cases; often have spread beyond the ovary; and may be extensively necrotic. On microscopic inspection, carcinomas typically have hyperchromatic, often pleomorphic nuclei with many, often atypical, mitotic figures; they may also contain psammoma bodies, surface epithelial–type glands, and intracellular mucin. Stroma, when present, is often desmoplastic, in contrast to the fibrothecomatous stroma of the AGCT. Tumors with pseudopapillary patterns (8) are distinguished from other neoplasms that are typically or sometimes papillary such as serous carcinoma with transitional cell features on the basis of associated morphology and cytology (high cytologic atypia as well as brisk mitotic index).
Other tumors in the differential diagnosis are pure stromal tumors, particularly thecoma and cellular fibroma, and exceptionally fibrosarcoma. The differential with thecoma is challenging in cases in which the granulosa cells have pale gray cytoplasm more typical of thecomas. A reticulin stain typically shows abundant fibrils coursing around individual cells in these tumors, unlike AGCTs where groups of tumor cells are surrounded by fibrils. Although stromal tumors may have sex cord elements resembling granulosa cells, they are by definition minor (<10%). Thecomas typically have cells with relatively abundant pale gray cytoplasm, and both thecomas and fibromas are often associated with hyaline plaques, a rare feature of AGCT. Fibromas in contrast to AGCT are typically inhibin negative or show minimal and weak positivity. Furthermore, fibromas, even when cellular, as well as thecomas typically lack FOXL2 mutations (with rare exceptions), although they do show FOXL2 nuclear positivity (24,25,35). Extensively luteinized AGCTs may be difficult to distinguish from steroid cell tumors, but thorough sampling usually discloses foci with the typical microscopic features of AGCT. Some immunohistochemical stains may aid in this differential diagnosis as steroid cell tumors are frequently negative for WT1 and FOXL2 (19,24), but the final diagnosis should rely on both histologic and immunohistochemical findings. Distinction of AGCT from endometrioid carcinoma, carcinoid tumor, endometrioid stromal sarcoma, and small cell carcinoma of the hypercalcemic type is discussed in other chapters. Distinguishing a unilocular or multilocular AGCT from follicular cysts may rarely be troublesome. In nonpregnant patients, the mere size of most cystic GCTs (≥8 cm) almost always excludes a follicle cyst. Typical follicle cysts of nonpregnant patients lack neoplastic granulosa cells within their walls. If a patient is pregnant or in the puerperium, the large, solitary follicle cyst (LSFC) may be indistinguishable on gross inspection from a unilocular AGCT. However, in the LSFC, the cells are typically uniformly luteinized, whereas in AGCT, luteinization is rarely extensive and typically focal. Bizarre nuclei, seen in only up to 2% of AGCT, are almost a constant finding in LSFC. The latter lacks follicles in the cyst lining, whereas they are present in some cystic AGCTs (36). Nonneoplastic granulosa cell proliferations of the type sometimes seen in the ovary of a pregnant patient are microscopic and often multifocal (37).
Juvenile Granulosa Cell Tumor
Gross Features. This tumor is also typically both solid and cystic, with the cysts often filled with blood. However, uniformly solid or cystic tumors are also seen (38–40). The cut surface may be gray, cream-colored, brown, or yellow; necrosis or hemorrhage may be present but are overall infrequent.
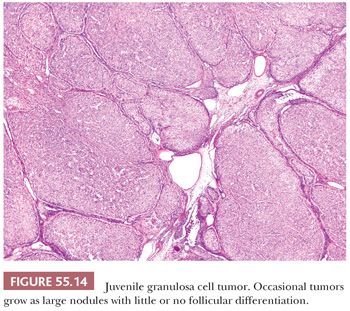
Microscopic Features. Sheets or nodular aggregates of neoplastic granulosa cells are usually punctured by varying numbers of follicles that vary in size and shape but are typically medium-sized (Fig. 55.13) and often contain eosinophilic or basophilic mucicarminophilic secretions. Rare tumors have scant or no follicles (Fig. 55.14) and Call-Exner bodies are rare. In the diffuse areas, a basophilic background can be noted. In some cases, the nodules of granulosa cells may undergo extensive sclerosis. Theca cells are often present between the nodules of granulosa cells; occasionally, both cell types are arranged in a disorderly fashion and are difficult to distinguish. Tumors from pregnant patients may exhibit prominent edema (41). JGCT may also show a pseudopapillary pattern as seen in AGCT (8).

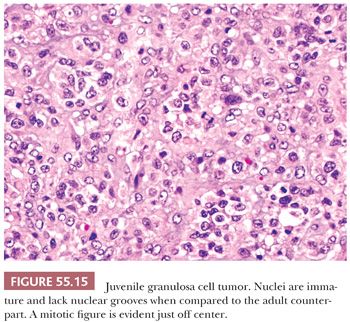
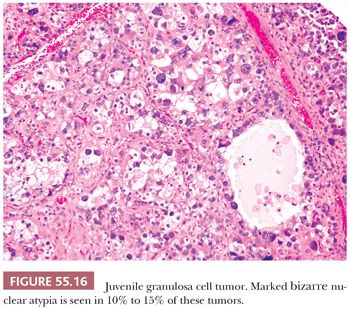
Both granulosa and theca cells usually have abundant eosinophilic cytoplasm (Fig. 55.8); occasionally, it is vacuolated. The neoplastic cells have hyperchromatic, round, and typically nongrooved nuclei (Fig. 55.15). Apical nuclei (hobnail cells) may be seen lining the follicles. Mitoses, which may be atypical, are often numerous (Fig. 55.15). In 10% to 15% of tumors, bizarre-type nuclear atypia may be present (Fig. 55.16), but the orderly follicular and solid patterns of the tumor are usually retained (39). Rarely, an organized growth is not retained, and a diffuse growth of highly atypical, extremely mitotically active cells with necrosis is seen. Our anecdotal experience with these so-called anaplastic tumors suggests that they account for the majority of clinically malignant JGCTs. Occasionally, minor components of AGCT or Sertoli-Leydig cell tumor of intermediate differentiation may be noted in association with JGCT.
Differential Diagnosis. The follicles of AGCT are more regular in size and shape than those of JGCT, and its cells usually show scant cytoplasm, with nuclei that have a “coffee bean” appearance due to grooves. The eosinophilic basement membrane material, often accompanied by degenerating nuclei, that is usually present in the microfollicles of AGCTs also differs from the basophilic follicular contents in JGCT. From the immunohistochemical point of view, both AGCT and JGCT show a similar immunohistochemical profile, expressing inhibin, calretinin, SF-1, WT1, CD99, CD56, and FOXL2 (2,3,19,20,24,25,35). However, in contrast to AGCT, JGCT do not show FOXL2 mutations (42–44). JGCTs may be misdiagnosed as primitive germ cell tumors. Perhaps the most realistic differential diagnosis is with yolk sac tumor (YST). The follicular pattern of JGCTs is different from the reticular or microcystic anastomosing spaces seen in YST, the only overlap being with the cysts of the polyvesicular YST. However, in the latter, the cysts may show eccentric constrictions simulating subdivision of primary yolk sac vesicle, and they lack an associated thecomatous component. By immunohistochemistry, YSTs including the polyvesicular variant are typically α-fetoprotein, glypican-3, or SALL-4 positive and inhibin, calretinin, and SF-1 negative in contrast to JGCT, which shows the opposite profile (19,20,23,45–48). Cytokeratins and EMA are not useful in this differential diagnosis (49).
JGCT is sometimes misinterpreted as thecoma because of occasional absence or rarity of follicles, abundant cytoplasm of the neoplastic cells, and occasional predominance of theca cells. Thorough sampling to demonstrate follicles, as well as reticulin stains to establish the granulosa nature of at least some of the tumor cells, is helpful as the immunohistochemical profile of both tumors is very similar. Furthermore, thecomas rarely exhibit significant mitotic activity, infrequently occur before 30 years of age, and are rare in children. A predominantly diffuse pattern in a JGCT may suggest the diagnosis of steroid cell tumor, but the uniform architecture, typical absence of follicles, and cytologic features including the presence of multiple small intracytoplasmic vacuoles in the latter would be unusual for JGCT. Finally, steroid cell tumors rarely show FOXL2 nuclear positivity in contrast to JGCT (24) and are typically WT1 negative. Pregnancy luteomas contain rounded, follicle-like spaces in three-fourths of the cases and may resemble a JGCT but only contain colloid-like and not basophilic material. Furthermore, they are multiple and bilateral in one-half and one-third of the cases, respectively, and their cells are cytologically uniform. The pregnant status of the patient is obviously helpful (50).
Surface epithelial tumors that may enter in the differential diagnosis of JGCTs are primarily clear cell, serous carcinoma with transitional features, and undifferentiated carcinomas. The tubulocystic variant of clear cell carcinoma is suggested when follicles in JGCTs are lined by hobnail cells. A pseudopapillary architecture in a JGCT at low power may suggest a serous carcinoma with transitional features, but often, the latter is associated with a component of recognizable serous carcinoma (8). JGCTs with high-grade nuclear atypia may suggest undifferentiated carcinoma. The usual young age of the patient and the presence of follicles provide evidence for JGCT, whereas carcinomas generally occur in older patients and also have other distinctive features. Inhibin, EMA, and PAX8 can help to establish the diagnosis in difficult cases. The vast majority of JGCTs are typically EMA negative with rare exceptions (51) as well as PAX8 negative (52), whereas müllerian carcinomas have the opposite profile. The differential diagnosis of JGCT with small cell carcinoma of the hypercalcemic type is discussed in Chapter 56.
Metastatic malignant melanoma can simulate a JGCT when it contains follicle-like spaces and has cells with abundant eosinophilic cytoplasm. Metastatic melanoma, however, is rare in the first two decades of life, when most JGCTs are encountered. Knowledge of the concurrent or prior existence of melanoma is helpful. However, as the previous history of melanoma may be remote or the tumor may have regressed, the possibility of metastatic melanoma should be considered in the differential diagnosis of a JGCT, especially when the patient is older than 20 years. It is more likely that the neoplasm represents metastatic melanoma if there is bilateral ovarian involvement. Differential staining for inhibin, SF-1, FOXL2, and HMB45 should establish the diagnosis in challenging cases. Calretinin has to be used with caution as melanoma may be positive for this marker (53).
PURE STROMAL TUMORS
The majority of tumors in this group are fibromas. Most of the remainder are thecomas or sclerosing stromal tumor, but other rare entities discussed in the following text also fall in this group of tumors including microcystic stromal tumor and signet ring cell stromal tumor.
Fibroma
Fibromas occur at an average age of 48 years (54). They are rare in children, with the exception of those patients with the basal cell nevus (Gorlin) syndrome, which is an autosomal dominant disorder, characterized by congenital malformations and predisposition to basal cell carcinomas, ovarian fibromas, medulloblastomas, and other tumors. The main presenting symptom is abdominal pain, reported in approximately 40% of patients. Fibromas are rarely associated with clinical or pathologic evidence of hormone production, but they have been reported to be associated with increased CA-125, raising clinical suspicion for ovarian carcinoma. Fibromas more than 10 cm in diameter are associated with ascites and Meigs syndrome (ascites and pleural effusion) in up to 40% and approximately 1% of patients, respectively.
Gross Features. Fibromas average 6 cm in diameter with typically a uniform solid, firm, white appearance, but they may be soft and edematous. Hemorrhage, necrosis, calcification, and cyst formation may be seen. Occasionally, tumors are predominantly cystic. Cellular fibromas are apt to be larger, softer, and yellow, with a much higher frequency of hemorrhage and necrosis than typical fibromas (55). Fibromas associated with Gorlin syndrome, in contrast to typical fibromas, are almost always bilateral, multinodular, and calcified.
Microscopic Features. The classic appearance is that of spindle cells resembling fibroblasts associated with collagen, sometimes arranged in a storiform pattern. Cellularity varies among tumors and within different areas of a tumor but is usually not marked; if striking, the tumor should be placed in the category of cellular fibroma (see “Cellular Mitotically Active Fibroma, and Fibrosarcoma”). Hyaline plaques that vary in thickness may be seen. Some tumors are diffusely edematous, and some have conspicuous vascularity. The cells typically have oval to elongated, wavy and pointed nuclei and scant cytoplasm; however, the latter may rarely display a peculiar granular eosinophilic nature due to the presence of multiple small hyaline globules (periodic acid-Schiff positive but mucicarmine and Alcian blue negative) (56). Cytologic atypia and mitotic activity are usually absent or minimal. Fibromas may contain lutein cells and have been placed in the past in the category of luteinized thecoma, but the current approach is to eschew that designation but mention lutein cells in a notation if indicated (e.g., should it help explain associated endocrine function). Fibromas may have minor sex cord elements seen as granulosa-like elements or Sertoli tubules, but these should represent less than 10% of the neoplasm (57). Other unusual features include bizarre nuclei and Verocay-like areas (55).
Immunohistochemical Features. These tumors are more often positive for calretinin and SF-1 than inhibin; they express CD56 and WT1 but are typically CD99 and CD10 negative (19,30,31,58). They can be positive for smooth muscle actin (58) and they are rarely CD34 and desmin positive. They display FOXL2 nuclear positivity but lack FOXL2 mutations (26).
Molecular Features. Patients with the basal cell nevus syndrome have mutations in the human homologue of Drosophila patched gene (PTCH), localized in chromosome 9q22.3. Some sporadic typical and cellular fibromas have shown loss of heterozygosity (LOH) at 19p13.3, with the highest frequency at microsatellite marker D9S15, which localizes proximal to the PTCH gene. In one study, LOH at 19p13.3 was seen in 50% of cellular fibromas and 25% of conventional fibromas but not in fibrosarcomas. These results indicate that sporadic cellular fibromas may arise through similar genetic pathways as cases associated with the basal cell nevus syndrome (59).
Differential Diagnosis. As fibromas and thecomas are both derived from ovarian stromal cells and because a spectrum exists between the two tumor types, any distinction is arbitrary. We place tumors in the fibroma category unless an appreciable component of a stromal neoplasm is composed of thecoma cells (see the following discussion). Some use the term fibrothecoma for tumors with hybrid features between fibroma and thecoma. Occasional fibromas are quite vascular but lack the other distinctive features of a sclerosing stromal tumor (see the following discussion). Tumors with cystic degeneration may be misdiagnosed as cystadenofibromas if the lack of an epithelial lining of the cystic spaces is not appreciated. Krukenberg tumor, Brenner tumor, and carcinoid tumor may resemble a fibroma on gross examination, but their epithelial components facilitate their identification on microscopic inspection. Fibromatosis of the ovary may resemble a fibroma, but it envelops follicles and their derivatives, in contrast to fibroma, which typically displaces them. Similar differences are seen between massive edema of the ovary and edematous fibroma (60). Fibromas can be distinguished from the uncommon smooth muscle and nerve sheath tumors of the ovary based on standard criteria used at other sites. Of note, when using immunohistochemistry, it is important to be aware that fibromas can be positive for smooth muscle actin as well as S100. Furthermore, fibromas and smooth muscle tumors share expression of estrogen receptor (ER), progesterone receptor (PR), WT1, and CD56 (58,61).
Cellular, Mitotically Active Fibroma, and Fibrosarcoma
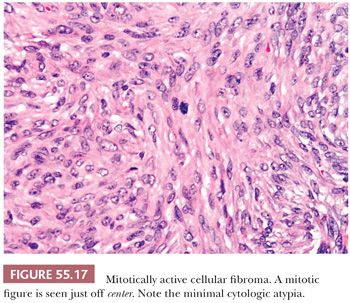
Cellular fibromas are densely cellular with closely packed nuclei and with an increased mitotic rate, with an average of less than or equal to three mitoses per 10 hpf (but a mitotic index higher that of a typical fibroma) (Fig. 55.17). Higher mitotic indices are still compatible with a diagnosis of mitotically active cellular fibroma provided there is no appreciable cytologic atypia (55). In fact, some tumors have been shown to display up to 19 mitoses per 10 hpf in a recent study (55). This subset of fibromas should be descriptively designated as mitotically active cellular fibromas. Cellular fibromas are much less frequently associated with edema or hyaline bands when compared to typical fibroma but frequently show areas of conventional fibroma. Nuclear palisading may be seen. Cellular and/or mitotically active fibromas are almost always benign unless they are adherent to surrounding tissues or ruptured, but even in their absence they may rarely recur.
Fibrosarcomas are typically large, fleshy tumors that exhibit moderate to marked nuclear atypia and brisk mitotic activity. An occasional fibrosarcoma may show only mild to moderate atypia and, in individual cases, determining whether tumors should be characterized as mitotically active cellular fibroma (55) or low-grade fibrosarcoma may be difficult. Fibrosarcomas are typically inhibin and CD10 negative but can express calretinin (15,20,31).
Differential Diagnosis. Cellular and/or mitotically active fibromas and rarely fibrosarcomas should be distinguished from smooth muscle tumors using standard criteria. The cellularity of endometrial/endometrioid stromal tumors equates with that of cellular fibromas and may show fibroblastic or fibromatous differentiation (62–65). Extensive sampling will show typical areas of endometrial/endometrioid stromal differentiation. Furthermore, primary ovarian endometrial/endometrioid stromal tumors are frequently associated with endometriosis. Metastatic (or less commonly primary endometrial/endometrioid stromal tumors) frequently involve both ovaries. Immunohistochemistry may be of help as endometrial/endometrioid stromal tumors are typically CD10 positive in contrast to fibromas and fibrosarcomas, which are typically CD10 negative (31). Gastrointestinal stromal tumors (GIST) may secondarily involve the ovary and grow in fascicles of fairly uniform spindle-shaped cells, but these tumors typically lack the storiform growth of some fibromas. Moreover, spindle areas are often associated with sheets of epithelioid cells in GIST (61). Other characteristic histologic features of GIST (lacking in fibromas) include skeinoid fibers and paranuclear cytoplasmic vacuoles. GISTs are typically positive for c-kit and DOG1 and, frequently, CD34 (66). Although fibromas may be positive for smooth muscle markers, they are negative for c-kit and DOG1, and only some tumors show focal CD34 positivity.
Thecoma
Thecomas are approximately one-third as common as GCTs. Many are associated with estrogenic manifestations, often only evidenced by pathologic findings such as endometrial hyperplasia. Rarely, androgenic manifestations may occur (67). In one large study (68), 84% of the patients were postmenopausal, with a mean age of 59 years; only 10% were younger than 30 years old.
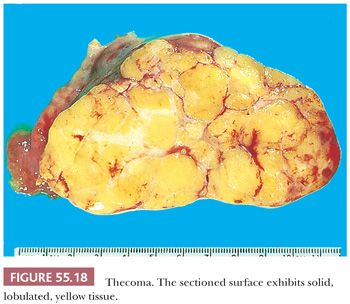
Gross Features. Thecomas are typically solid with a yellow to tan cut surface (Fig. 55.18), although they may be predominantly white and only focally yellow and may show lobulation. Thecomas are usually small (average 5 cm) and are rarely bilateral. Cystic degeneration and gritty areas (due to calcification) may occur but are rarely conspicuous. Necrosis and hemorrhage are uncommon (67).
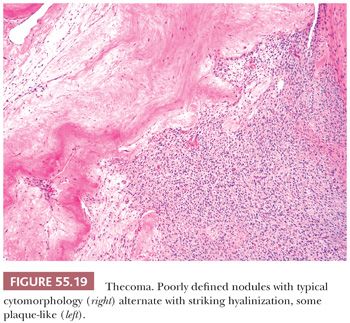
Microscopic Features. The most characteristic appearance is that of medium-sized to large, rounded cells with ill-defined cell membranes and pale, gray cytoplasm, typically growing in sheets or alternating diffuse and nodular growths (Figs. 55.19 and 20). A fibromatous component often separates sheets and nests of theca cells, and it is most typically in the form of brightly eosinophilic hyaline plaques/bands (Figs. 55.19 and 55.21). In some cases, there are large confluent zones of hyalinization, and occasionally, there is a myxoid background that may even impart a vaguely chondroid appearance. Foci of calcification (more common in young women) can be seen in up to 20% of tumors (69), and rarely, adipocytic metaplasia has been reported (67,69). The nuclei are round to oval with visible but inconspicuous nucleoli. The cells show absent to mild cytologic atypia and minimal to absent mitotic activity. Nuclear grooves and bizarre nuclei are rarely seen in otherwise typical tumors (10,67). Significant atypia and conspicuous mitotic activity accompanied by clinically malignant behavior are exceptional. Some tumors are more cellular than usual and are descriptively designated “cellular thecoma.”

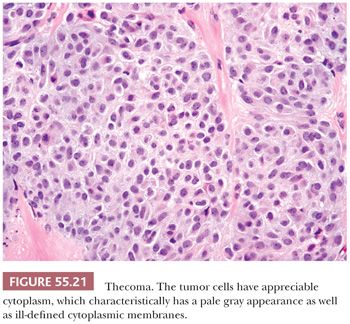
Immunohistochemical Features. Thecomas are typically positive for SF-1 and WT1, inhibin, and calretinin (15,19,20) and may be CD10 positive (31). Thecomas are often FOXL2 positive but do not demonstrate associated mutations (33).
Differential Diagnosis. Occasionally, a thecoma may be difficult to distinguish from a GCT when the latter has a prominent thecomatous component; however, when definite epithelial formations are present and account for greater than 10% of the neoplasm, the diagnosis should be that of GCT. Reticulin stain may aid in this distinction because there is a much greater reticulin framework in thecomas, often encircling individual cells while it outlines aggregates of epithelial cells in GCTs. However, it must be noted that this stain is by no means always discriminatory, and occasional thecomas show reticulin surrounding small aggregates of cells. Care should be taken to perform the stain in areas suspicious for granulosa cell neoplasia, as thecomatous areas in the latter will show a pattern similar to a pure thecoma. FOXL2 mutation analysis may be helpful in this distinction, but the same practical points just mentioned for the reticulin should be applied (33). When lutein cells in a thecoma are extensive, it may simulate a steroid cell tumor, not otherwise specified; however, the fibromatous component in steroid cell tumors is minor. Steroid cell tumors, in contrast to thecomas, are typically FOXL2 negative (24).
Diffuse luteinization of thecomas that may be seen in pregnant patients can lead to diagnostic confusion with pregnancy luteomas. The latter, however, are often bilateral, multiple in half of the cases, contain little or no lipid, and have much more abundant eosinophilic cytoplasm than seen in thecomas (50).
Luteinized Thecoma Associated with Sclerosing Peritonitis
This unusual ovarian lesion typically associated with sclerosing peritonitis is considered by some a variant in the thecoma family (70,71) and by others a nonneoplastic proliferation due to its distribution expanding the ovarian cortex (72). It often occurs in young females with an average age of 27 years who typically present with abdominal distension and pain with ascites and sometimes bowel obstruction. These patients may die secondary to complications of their peritoneal pathology.
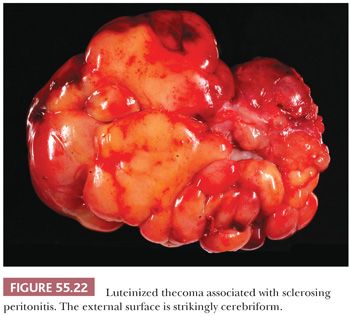
Gross Features. It is nearly always bilateral and often shows irregular enlargement of the ovaries instead of forming a discrete mass, suggesting a hyperplastic rather than neoplastic process. The ovaries frequently have a striking cerebriform, smooth, glistening, often hemorrhagic surface (Fig. 55.22). On sectioning, the lesion is mostly centered within the ovarian cortex, which becomes variably expanded often associated with areas of edema and/or hemorrhage, whereas the medullary region is often preserved.
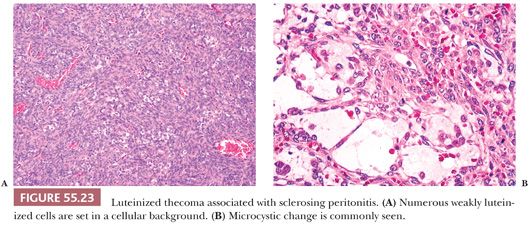
Microscopic Features. The lesion is typically cellular, and spindled cells grow in sheets or a vaguely storiform pattern (Fig. 55.23A). They have bland cytologic features with ovoid, pale nuclei and small nucleoli but brisk mitotic activity. Areas of edema often create a microcystic pattern (Fig. 55.23B), and entrapped follicles are often found. Small lutein cells, singly or in small clusters, are dispersed among the spindle cells (Fig. 55.23A). They are often inconspicuous but always present.
The associated sclerosing peritonitis is characterized by proliferation of bland spindled cells with fibroblastic appearance present within a myxoid, edematous, or densely collagenized background. In the omentum, involvement of the septa between the fat lobules often produces a distinctive lobulated pattern at low power. The pathogenesis of the accompanying sclerosing peritonitis is not known (71).
Immunohistochemical Features. The spindled cells are typically inhibin negative and mostly calretinin and CD56 negative. In contrast, the luteinized cells are in most cases diffusely positive for inhibin, calretinin, and CD56. α-Smooth muscle actin, desmin, and AE1/3 focal positivity can be seen in the spindled cells which can also show focal or diffuse nuclear ER and PR immunoreactivity (71). The spindle cells have recently been reported to be positive for SF-1 and FOXL2 without associated mutations (26).
Differential Diagnosis. Thecomas associated with sclerosing peritonitis should be distinguished from various nonneoplastic and neoplastic conditions, but their unique features should enable their recognition. The distinctive morphology of these unusual luteinized thecomas, including cortical expansile growth, microcystic change, and striking mitotic activity, distinguish them from massive edema or fibromatosis, although some consider the luteinized thecoma with peritonitis related to these benign entities (72).
STROMAL TUMORS WITH MINOR SEX CORD ELEMENTS
Predominantly fibromatous or thecomatous tumors rarely contain scattered sex cord aggregates occupying less than (usually much less) 10% of the tumor (57). Nests composed of cells resembling granulosa cells or indifferent sex cord–type cells, or tubules lined by cells resembling Sertoli cells, may be seen. The overall appearance and behavior of these tumors is akin to those of a fibroma or thecoma rather than those of a sex cord tumor. The diagnosis should be carefully restricted to tumors with limited epithelial elements because, as noted earlier, occasional GCTs are composed predominantly of fibrothecomatous elements, thus sampling is very important.
SCLEROSING STROMAL TUMOR
This distinctive tumor occurs at a younger average age than typical thecoma or fibroma, with more than 80% of patients being younger than 30 years (73). Most patients are first seen for nonspecific symptoms related to an ovarian mass; estrogenic manifestations have been present only occasionally, whereas virilization during pregnancy has been noted (74). All reported tumors were benign.
Gross Features
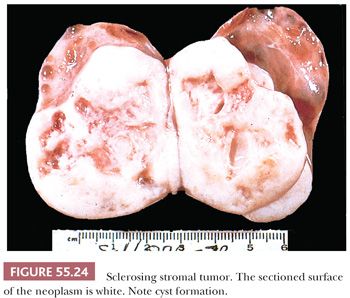
The tumors are typically unilateral and average 10 cm in diameter. They are well demarcated, with a predominantly solid white cut surface with yellow zones; areas of edema and cyst formation are common (Fig. 55.24). Rarely, the tumors may be predominantly cystic.
Microscopic Features
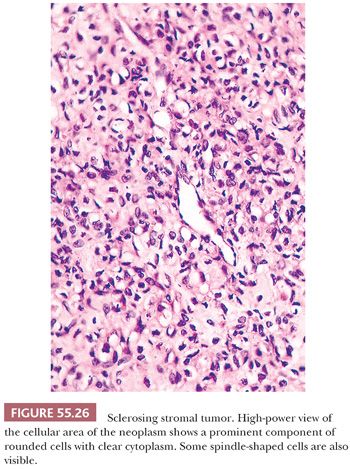
Low-power examination usually shows ill-defined cellular pseudolobules (Fig. 55.25) separated by stroma that varies from densely hyalinized to markedly edematous and even occasionally focally myxoid (75). Two cell types are intermingled within the nodules: spindle cells producing collagen and round to oval cells with small, dark nuclei. The latter have vacuolated cytoplasm containing lipid and represent degenerated lutein cells (Fig. 55.26). In an occasional tumor, the round or oval cells have dense eosinophilic cytoplasm and large nuclei containing prominent nucleoli, characteristic of typical lutein cells. Such cells may become particularly conspicuous if the patient is pregnant. Mitotic figures are absent or present in only small numbers, with rare exceptions (also more often seen during pregnancy). Another distinctive feature is a network of thin-walled, often ectatic blood vessels within and in between the nodules.

Immunohistochemical Features
These tumors are more often positive for calretinin than inhibin (staining typically not strong); they also express ER, PR, CD10, CD56, and FOXL2 (15,20,24,30,76) and may be positive for smooth muscle actin and CD99. No FOXL2 mutations have been detected (24).
Differential Diagnosis
The heterogeneous appearance of SST contrasts with the relative homogeneity of fibromas and thecomas. Although fibromas may be edematous and vascular, edema is generally diffuse rather than focal, and pseudolobules and lutein cells are absent or rare. Hyaline plaques, a conspicuous feature of many fibromas and thecomas, are rare in SSTs. The lutein cells in SSTs typically appear degenerated, unlike their typical robust appearance in thecomas. It has been speculated that the rare ovarian myxoma may be in fact an entirely myxoid SST. This is impossible to disprove, but myxomas in our experience have a different morphologic appearance (77). Rarely, the vacuolated cells in SST have eccentric, compressed nuclei and simulate signet ring cells of a Krukenberg tumor, but vacuoles in SSTs contain lipid rather than mucin and these tumors are typically keratin and EMA negative. The prominent vascularity in SSTs may suggest the diagnosis of hemangiopericytoma, although no well-documented ovarian example of the latter has been reported to date. Finally, a very luteinized SST from a pregnant patient may suggest the possibility of a pregnancy luteoma, but the latter is typically bilateral and multifocal (50).
SIGNET RING STROMAL TUMOR
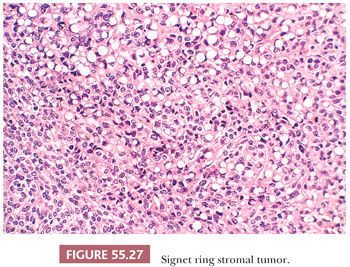
This rare neoplasm occurs in adults and is clinically nonfunctioning (78,79). It has a solid or solid and cystic cut surface. On microscopic examination, spindle cells are diffusely distributed and merge almost imperceptibly with rounded cells containing eccentric nuclei and single, large vacuoles resembling signet ring cells (Fig. 55.27). Stains for lipid and mucin are negative. The tumor cells may be focally positive for keratin and α-smooth muscle actin but are negative for EMA, desmin, inhibin, calretinin, SF-1, and S100 (79,80). Electron microscopic examination has shown that the vacuoles may result from generalized edema of the cytoplasm, from swelling of mitochondria, or as a result of formation of cytoplasmic pseudoinclusions composed of edematous extracellular matrix (78). An important consideration in the differential diagnosis is a Krukenberg tumor, but negative mucin stain as well as lack of expression of EMA exclude that diagnosis. Signet ring stromal tumor lacks the pseudolobulation, lipid-rich cells, and prominent vascularity of a sclerosing stromal tumor. AGCTs or, less commonly, thecomas may contain signet ring cells but also will display typical areas of those tumors. Thus, the designation “signet ring stromal tumor” should be reserved for tumors exclusively composed of signet ring–like cells.
MICROCYSTIC STROMAL TUMOR
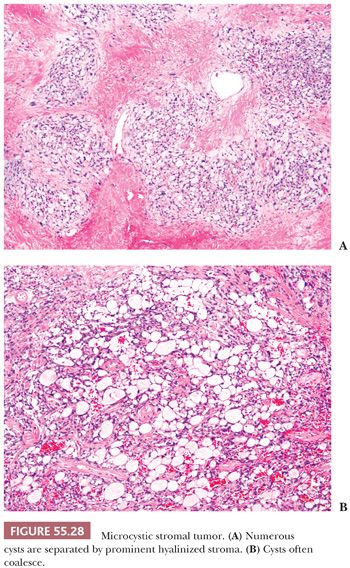
This is a rare neoplasm that occurs in adults and it is typically nonfunctioning. Tumors have a mean size of 10 cm and typically have a cystic or solid and cystic cut surface. All tumors reported to date have been confined to the ovary and associated with an excellent prognosis (81). Microscopic examination reveals lobulated cellular masses separated by hyaline bands and fibrous plaques (Fig. 55.28A). There is a variably prominent microcystic pattern (Fig. 55.28B) characterized by small cysts that anastomose with each other. The tumor cells contain finely granular, faintly eosinophilic cytoplasm and have generally round to oval nuclei with small nucleoli and only rare mitoses. Bizarre nuclear atypia of the degenerative type is common. Tumors are typically inhibin negative (rarely weakly and focally positive) and CD10, WT1, Cyclin D1 and β-catenin diffusely positive. About one-third of the tumors are cytokeratin positive but they are ER, PR, and EMA negative. β-Catenin immunohistochemical positivity is frequently associated with β-catenin mutations in exon (3,82).
Differential Diagnosis
Thecoma commonly enters in the differential diagnosis due to the presence of hyaline plaques common to both tumors. The distinctive nature of the tumor cells and marked microcystic change in microcystic stromal tumor set it apart from thecoma. Furthermore, thecoma, in contrast to microcystic stromal tumor, is typically positive for inhibin and calretinin and often expresses ER and PR and lacks β-catenin nuclear expression. Many other tumors can have microcysts, but thorough sampling will usually show distinctive features and aid in their correct classification.
SERTOLI AND SERTOLI-LEYDIG CELL TUMORS
Sertoli Cell Tumors
Clinical Features. These uncommon neoplasms occur at an average age of 30 years (83,84) and often have a nonspecific clinical presentation. However, patients may have estrogenic manifestations (mostly in lipid-rich variant), causing menstrual irregularities or isosexual pseudoprecocity in children. Less commonly, tumors are androgenic. An association with the Peutz-Jeghers syndrome has been occasionally reported (85). The prognosis is excellent in the vast majority of patients as tumors are usually confined to the ovary at time of diagnosis, but approximately 10% of the tumors have an adverse outcome (83,84).
Gross Features. The tumors are typically unilateral with a uniform, solid, and yellow cut surface, and they are typically less than 10 cm in largest dimension.
Microscopic Features. At low power, these tumors may show either a nodular or diffuse architecture. Tightly packed hollow or solid tubules usually predominate (Fig. 55.29). Complex branching tubules and diffuse or trabecular patterns may be occasionally seen, whereas nests, endometrioid-like tubules, alveolar arrangement, retiform foci, and pseudopapillary and spindled areas are rare (83). The stroma is usually not striking but may be abundant in about 30% of the neoplasms; rare cases may be markedly hyalinized and compress the neoplastic tubules, imparting an appearance similar to that seen in testicular sclerosing Sertoli cell tumors (83). Tumors with a more or less diffuse pattern can have collagenous septa and an associated lymphocytic infiltrate (83). The hollow tubules are typically lined by columnar to cuboidal cells with moderate amounts of pale or slightly eosinophilic cytoplasm. The solid tubules are filled with pale cells that contain moderate to abundant lipid in the lipid-rich Sertoli cell tumor (Fig. 55.29). Some neoplasms are composed of cells with abundant eosinophilic cytoplasm (oxyphilic variant), and these may be associated with the Peutz Jeghers syndrome (85). The nuclei are typically round to oval with small nucleoli, usually lacking atypical features, whereas the mitotic rate is generally low. However, occasional tumors have malignant histologic features (marked cytologic atypia, increased mitotic index and/or necrosis) and may metastasize. Minor foci of GCT or annular tubules may be present.

Immunohistochemical Features. Sertoli cell tumors like the vast majority of other sex cord stromal tumors express inhibin, calretinin, and SF-1 (15,19,20,86). They are often positive for CD99, WT1, AE1/3, Cam5.2, neuron-specific enolase (NSE), and CD56 (19,87) but negative for EMA and chromogranin. One tumor has been reported to be positive for FOXL2 showing a heterozygous mutational status (25).
Differential Diagnosis. Sertoli-Leydig cell tumors (SLCTs) are distinguished from Sertoli cell tumors by the finding of more than rare Leydig cells within the tumor. A number of non–sex cord–stromal ovarian tumors may have small tubular glands or solid tubular formations that may elicit the consideration of a Sertoli cell tumor. Endometrioid carcinoma and Krukenberg tumor are the two most common problematic tumors, but they have other patterns inconsistent with Sertoli cell tumor (including the finding of endometriosis, an adenofibromatous background, squamous differentiation or intraluminal mucin in the former and bilateral ovarian involvement, multinodular growth, as well as signet ring cells in the latter). A combination of inhibin, calretinin, and EMA should be helpful in this differential diagnosis. Occasionally, a tumor of probable wolffian origin may originate in the ovary and may simulate a Sertoli cell tumor. However, other patterns including sievelike and solid areas are noted, and the pseudotubular structures do not show a distinctive apical border as seen in Sertoli cell tumors. In contrast to Sertoli cell tumors, wolffian tumors if positive for inhibin typically show weak and focal staining (14,15). Furthermore, it has been recently reported that mesonephric proliferations including wolffian tumors are GATA-3 positive, although experience is limited. Sertoli cell tumor with a diffuse alveolar pattern, collagenous septa, and lymphocytes may rarely be confused with a dysgerminoma; however, dysgerminoma contains large, squared-off nuclei with one or multiple nucleoli and brisk mitotic activity. OCT 3/4 and c-kit (positive in dysgerminoma) and inhibin (positive in Sertoli cell tumors) will aid in this differential diagnosis if indicated. Carcinoid tumors may enter in the differential diagnosis of Sertoli cell tumor as both may show acini and a cordlike/trabecular growth. However, carcinoids typically have salt and pepper chromatin and sometimes cytoplasmic granules may be noted. The most helpful neuroendocrine marker in this differential diagnosis is chromogranin, as Sertoli cell tumors may be NSE and synaptophysin positive. Furthermore, carcinoids are inhibin negative. Sertoli cell tumors should be distinguished from the rare, large Sertoli cell tumors composed of uniform, solid tubules filled with immature Sertoli cells that may develop in the testes of patients with the complete form of the androgen insensitivity syndrome. Such patients have a normal female habitus; primary amenorrhea; no uterus; a contralateral testis, which may be composed predominantly of cellular stroma resembling ovarian stroma; absence of pubic and axillary hair; and a 46,XY karyotype (88).
Sertoli-Leydig Cell Tumors
Clinical Features. These neoplasms account for less than 0.2% of ovarian neoplasms (89–101) and occur at an average age of 25 years, except for the retiform variant that tends to present at a younger age (~15 years). Approximately half the patients show signs of hirsutism or virilization (less common in the retiform variant), but occasionally, there are estrogenic manifestations. Patients without endocrine manifestations have symptoms attributable to a pelvic or abdominal mass. Rarely, tumors have been associated with elevated serum α-fetoprotein (AFP) levels (100) but generally lower than seen with a YST. Most tumors are associated with a good outcome. Patients with well or intermediate differentiated tumors have an excellent to good prognosis, whereas poorly differentiated or those with heterologous mesenchymal elements (mostly rhabdomyosarcoma) are often associated with adverse outcome (100).
Gross Features. The vast majority of tumors are unilateral, but approximately 2% are bilateral. Size varies greatly, but mean diameter averages 10 cm. Typically, they form firm, lobulated, yellow or tan solid masses (Fig. 55.30). Cysts may be conspicuous, particularly if the tumor contains heterologous mucinous elements, and these cases may simulate conventional mucinous cystic tumors. Tumors with an extensive retiform component are often soft and spongy or have large, edematous intraluminal polypoid excrescences protruding into cysts. Areas of hemorrhage and necrosis are uncommon, except in the poorly differentiated variant.
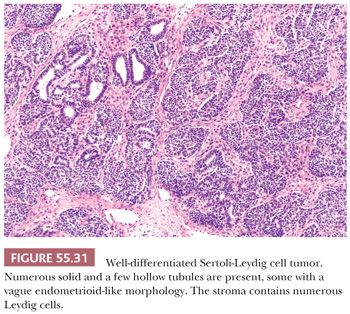
Microscopic Features. These tumors are divided into five subtypes. Well-differentiated SLCTs (99) are composed of hollow or solid tubules similar to those in well-differentiated Sertoli cell tumors (Fig. 55.31). The tubules are either hollow or solid, usually small and round to oval, but may be large and vary in shape. They may be lined by stratified cells and, rarely, resemble the glands of an endometrioid neoplasm (89) (Fig. 55.31). Sertoli cells show no cytologic atypia and minimal mitotic activity. However, in contrast to Sertoli cell tumors, more than a minor component of Leydig cells is present within the fibromatous stroma. These cells may contain crystals of Reinke and, less commonly, intracytoplasmic vacuoles.
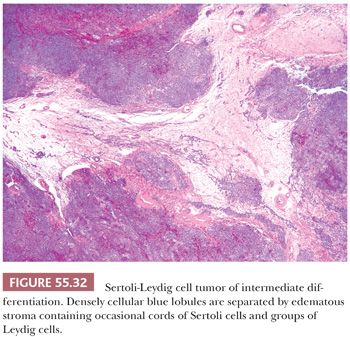
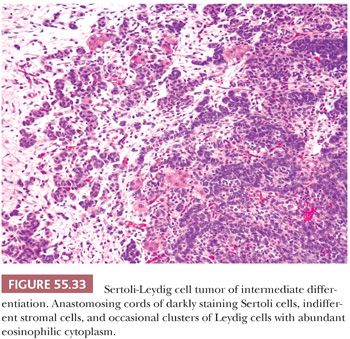
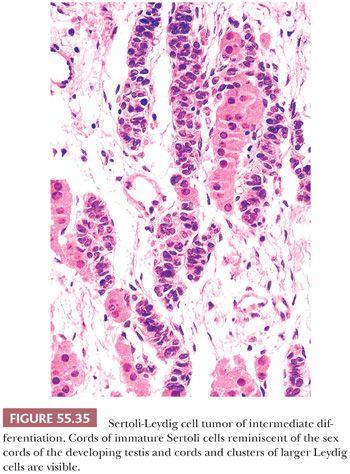
Tumors of intermediate differentiation characteristically have a lobulated (much less commonly diffuse growth) appearance on low magnification (Fig. 55.32), with densely cellular areas intersected by hypocellular typically edematous stroma. Within the cellular areas, there is typically a jumbled admixture of dark blue (but occasionally with pale/vacuolated or eosinophilic cytoplasmic) Sertoli cells growing in aggregates or nests and lesser numbers of Leydig cells with eosinophilic to pale, vacuolated cytoplasm (Figs. 55.33 to 55.35); Leydig cells are often most recognizable at the periphery of the cellular lobules. Sometimes, Sertoli cells are arranged in cords and/or trabeculae (Fig. 55.33). A tubular pattern may be seen recapitulating that seen in well-differentiated tumors (89). Cysts of varying sizes lined by flattened Sertoli cells, sometimes containing eosinophilic secretion, may also be present and may resemble thyroid follicles. In isolated instances, cells with bizarre nuclei may be seen (11). Tumors from patients in the third trimester of pregnancy typically show exaggerated edema with effacement of the lobular architecture and large sheets of Leydig cells (41).
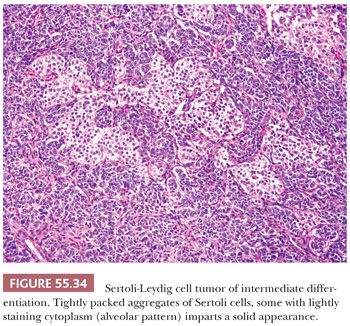
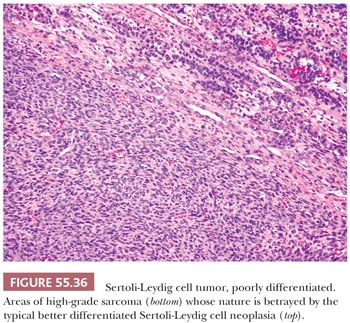
Poorly differentiated SLCTs are characterized by a diffuse growth of cells that are highly active mitotically and usually spindle-shaped, suggesting a sarcoma (Fig. 55.36), or less often rounded, simulating a poorly differentiated carcinoma. The finding of focal tubular or sex cord–like formation as well as Leydig cells or the presence of more distinctive patterns of Sertoli-Leydig cell neoplasia, which may be minor in extent, are necessary to establish the diagnosis.
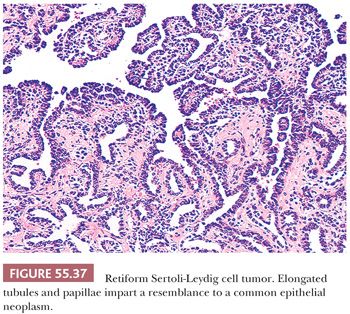
Patterns simulating those of the rete testis are found in approximately 10% of SLCTs, but only approximately 10% of these tumors are purely retiform and these patterns are absent in well-differentiated tumors. Retiform SLCTs are seen typically in patients who are younger (average age 15 years) than patients with other subtypes of SLCTs. The growth patterns include cysts containing polyps with edematous stroma, papillae containing fibrous to hyalinized cores, or cellular papillae that bud and simulate those of serous neoplasms. The papillae may be simple or have a complex branching pattern with a lining of stratified atypical cells (Fig. 55.37); glomeruloid structures are often present. Networks of slitlike tubules and cysts are also typical and broad, elongated ribbons of Sertoli cells often link up with the typical retiform areas. The stroma varies from densely cellular to hyalinized or edematous.
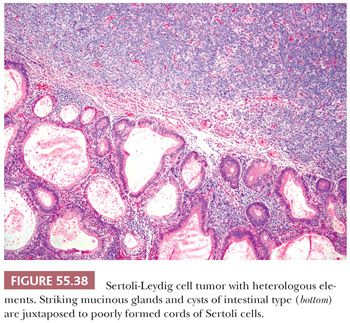
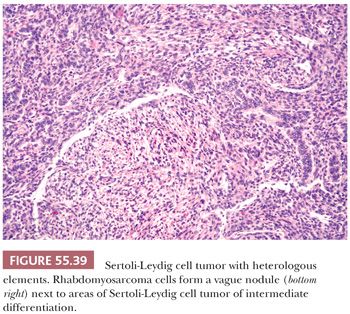
Heterologous elements are found in approximately 20% of SLCTs and have been encountered only in tumors of intermediate or poor differentiation. The most common component is mucinous epithelium of gastrointestinal type (97) (Fig. 55.38). Mucinous glands contain goblet cells and argentaffin cells in more than half and one-third of cases, respectively. The mucinous epithelium is usually benign but occasionally shows nuclear pseudostratification and cytologic atypia falling within either the borderline or low-grade carcinoma categories. In some of the tumors containing argentaffin cells, microscopic areas of carcinoid, including the insular and mucinous subtypes, are also present. Approximately one-fourth of SLCT with heterologous elements have foci of immature skeletal muscle (Fig. 55.39), cartilage, or both. Mesenchymal heterologous elements are usually found in tumors that are poorly differentiated (92). Occasionally, cells resembling hepatocytes and (exceptionally) neuroectodermal elements are seen (91).
Immunohistochemical Features. Inhibin and calretinin are typically positive in SLCT in both Sertoli and Leydig cells. However, of note, inhibin staining is always less extensive and less intense in Sertoli cells when compared to Leydig cells. Furthermore, staining may be minimal to absent in poorly differentiated areas. Other markers that can be used include WT1 and SF-1 (both cell types being positive) (14,15,17,19–21,102–106). Approximately 50% of SLCTs have been reported to express FOXL2, but positivity is restricted to the Sertoli cells, and positivity is only rarely associated with FOXL2 mutations (24,107). Sertoli cells can also express NSE and synaptophysin. Sertoli cells can be positive for keratins and rarely for EMA, thus a panel of antibodies that include inhibin and EMA is needed. Leydig cells are also positive for melan-A.
Molecular Features. Germline truncating mutations in DICER1, an endoribonuclease in the RNase III family that is essential for processing microRNAs, have been reported in approximately 60% of SLCTs (108).
Differential Diagnosis. SLCTs are difficult to distinguish from GCTs on gross inspection, except that they almost never form unilocular thin-walled cysts, rarely form multilocular thin-walled cysts, and much more often contain a mucinous cystic component. At the microscopic level, typical GCTs and SLCTs differ considerably, but characteristic features of one tumor type are often present as minor foci in the other. The prominent tubules and discrete clusters of Leydig cells in well-differentiated SLCTs readily distinguish them from GCTs, although rare GCTs contain small numbers of well-formed tubules and, in exceptional cases, Leydig cells with crystals of Reinke. The cellular lobules of SLCTs of intermediate differentiation impart a distinctive low-power appearance that is rarely simulated by GCTs, which can have a lobulated growth but typically is punctuated by follicles. The cells of GCTs are more mature in appearance than those of most SLCTs, and although nuclear grooves may be seen in Sertoli cells, they are seldom as conspicuous as in GCTs. The stromal component of SLCTs frequently has a sarcomatous appearance, which is rare in GCTs which often have a thecomatous background. Heterologous elements (almost always) or retiform growth (always) are diagnostic of SLCT. Finally, Leydig cells of SLCTs tend to cluster in small groups, whereas in GCTs, luteinized theca cells are usually not as prominent. The retiform variant of SLCT may be misdiagnosed as YST because of the young age of the patient, but the two tumors have little in common in terms of microscopic characteristics. Pure or almost pure retiform tumors, however, may closely resemble surface epithelial tumors and have occasionally been reported as such. The papillae of retiform SLCT can closely simulate those of a serous borderline tumor, and if nuclear stratification is pronounced and glands and solid epithelial foci are present in the stroma, the appearance can simulate a serous carcinoma. Juxtaposition of epithelium and cellular stroma in retiform SLCTs has also suggested a carcinosarcoma. Clinical and pathologic features, including young age, androgenic manifestations in 20% of patients, and the usual finding of more typical patterns of SLCT provide clues to the diagnosis of retiform SLCT. Inhibin positivity as well as lack of EMA staining among other markers can be very helpful in difficult cases but rarely necessary. Confusion with the very rare primary ovarian Wilms tumor is also possible as there may be morphologic and immunohistochemical overlap, including glomeruloid/retiform architecture, rhabdomyoblastic differentiation, and WT1 positivity; inhibin should be negative in Wilms tumor.
SLCTs are sometimes misinterpreted as primitive germ cell tumors, particularly during pregnancy, when striking stromal edema may efface preexistent architecture imparting a loose, reticular appearance similar to that seen in YST. However, the nuclei of SLCTs are not as primitive as those of YST, and the various patterns of these two tumors are generally distinctly different. It should be noted that SLCTs may contain hyaline bodies. Immunohistochemistry may be helpful as SLCTs are negative for SALL-4 (49).
Heterologous SLCTs, in contrast to teratomas, almost never contain neuroectodermal tissue, and squamous epithelium and skin appendages have not been reported in SLCTs. Heterologous SLCTs containing glands and cysts lined by gastrointestinal-type epithelium may be confused on gross examination with pure mucinous cystic tumors of the ovary. Although a history of virilization is much more suggestive of SLCT, mucinous tumors containing luteinized stromal cells may also be masculinizing. The diagnosis of heterologous SLCT rests on finding a Sertoli-Leydig cell component, which is usually of intermediate differentiation, between the glands and cysts or at the periphery of the tumor.
Because the carcinoid components of SLCTs are usually of microscopic size, they should not be confused with usual carcinoid tumors. Heterologous tumors with mesenchymal elements may be mistaken for ovarian sarcomas when there are few recognizable typical SLCT elements. Before a pure ovarian sarcoma is diagnosed in a young woman with signs of virilization, heterologous SLCT should be excluded by adequate sampling. Criteria for the differentiation of SLCTs from endometrioid carcinomas, tubular Krukenberg tumors, and carcinoids are discussed under those headings. Although it is most common for an endometrioid carcinoma to simulate an SLCT, occasional SLCTs, particularly when well differentiated, may have a prominent component of tubules that have an endometrioid-like morphology (89).
GYNANDROBLASTOMA
Tumors containing Sertoli-stromal cell and granulosa-stromal cell elements in varying proportions have been reported as gynandroblastoma. Because minor components of one tumor frequently occur in an otherwise typical tumor of the other type, the diagnosis of gynandroblastoma should be reserved for neoplasms containing at least 10% of the second tumor type, and both components should be clear-cut to avoid a lack of reproducibility of diagnosis. If this strict definition is used, gynandroblastoma is rare (109). When unusual components, such as JGCT and SLCT of intermediate differentiation coexist, the pathologist’s report is more meaningful if the specific designation of each component is included as diagnostic terms.
SEX CORD TUMOR WITH ANNULAR TUBULES
Clinical Features
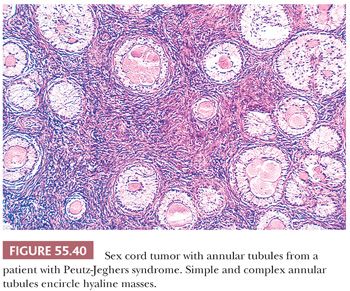
One-third of sex cord tumors with annular tubules (SCTATs) are associated with Peutz-Jeghers syndrome (PJS; gastrointestinal polyposis, oral and cutaneous melanin pigmentation, and, rarely, adenoma malignum [minimal deviation adenocarcinoma] of the cervix) and are almost always benign (110). In patients with PJS, the tumor has always been an incidental finding at operation or autopsy, whereas in patients without the syndrome, it is detected clinically and pursues a malignant course in approximately one-fourth. Estrogenic manifestations, including menstrual disturbances and isosexual precocity, are present in approximately 40% of patients, with or without PJS. An unusual number of SCTATs in patients without the syndrome produce progesterone, which may be sometimes associated with decidual changes in the endometrium (111). Müllerian-inhibiting substance may also be produced by the tumor (112). Patients with the PJS often display STK11 germline mutations (113).
Gross Features
The appearance differs depending on whether the patient has PJS. In patients with the syndrome, the tumors are typically bilateral, multifocal, calcified, and less than 3 cm in diameter. Tumors from patients without PJS are almost always unilateral and usually larger, solid, and yellow and are rarely calcified. Cysts are occasionally present and rarely predominate.
Microscopic Features
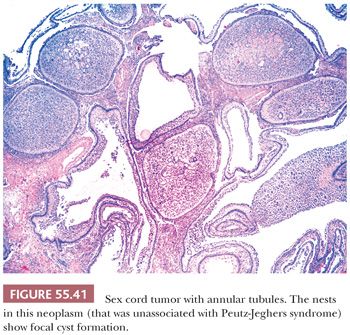
These tumors are characterized by sharply circumscribed, ring-shaped tubules encircling nodules of hyalinized basement membrane–like material (Fig. 55.40). Simple tubules encircle single hyaline nodules; the more common complex tubules surround several nodules. Focal cystic degeneration of the more complex tubular structures can be seen in tumors not associated with the PJS (Fig. 55.41). The lumens of the tubules are filled with pale cytoplasm, and the nuclei are located in an antipodal arrangement along the margins of the hyaline nodules and at the periphery of the simple and complex tubules. In some areas, the cells proliferate toward the centers of the tubules and may form solid aggregates. Isolated tubules are elongated, as in typical Sertoli cell tumors. In the tumors associated with PJS, a few nests may be composed of cells containing large, lipid-laden vacuoles; there is often focal to extensive calcification of the tubules. Cytologic atypia and mitotic activity is only increased in SCTAT unassociated with the PJS.
In large tumors unassociated with PJS, foci of typical GCT (114), solid tubular Sertoli cell tumor (81), or both may be present. Because bundles of Charcot-Böttcher filaments have been occasionally found on ultrastructural examination, some investigators regard SCTAT as a type of Sertoli cell tumor. On the other hand, others have considered SCTAT a variant of GCT because that component is seen in some cases. Because of its distinctive clinicopathologic features, it is preferable to consider SCTAT a specific subtype of sex cord–stromal tumor, with a potential for bidirectional differentiation.
Immunohistochemical Features
These tumors are typically positive for inhibin, calretinin, and WT1 and negative for epithelial markers (104).
Differential Diagnosis
These tumors should be distinguished from pure granulosa or Sertoli cell tumors as they may show focal areas, particularly in the non–syndrome-associated cases, compatible with those diagnoses. To make a diagnosis of SCTAT, the vast majority of the tumor should display the typical microscopic appearance of this tumor, which has a distinctive pattern in the form of its annular tubules that is different from other sex cord tumors. The differential diagnosis with gonadoblastoma is only at low-power magnification and results from both having a discrete nested pattern and basement membrane material surrounded by neoplastic cells. However, gonadoblastoma is typically seen in a setting of gonadal dysgenesis, and the nests of tumor cells are composed not only of sex cord cells but also germ cells.
UNCLASSIFIED SEX CORD–STROMAL TUMORS
Approximately 5% to 10% of sex cord–stromal tumors have patterns and cell types intermediate between those of SLCTs and GCTs or unusual patterns that do not permit reproducible placement in either category (115). Rare tumors are so poorly differentiated that they cannot be readily categorized as being in the sex cord family on routine evaluation, but immunostains may aid in their placement in the sex cord group. For example, an undifferentiated tumor in a young female that cannot be otherwise classified should be evaluated for inhibin, calretinin, CD56, CD99, FOXL2, and SF-1 expression, although staining may be of variable intensity and minimal to absent in unclassified sex cord–stromal tumors. Talerman et al. (116) designated as “diffuse nonlobular androblastoma” those tumors characterized by a diffuse fibrothecomatous or granulosa cell–like proliferation, with minor foci of typical sertoliform tubules. Rare tumors of this type that we have seen have looked more like a GCT than SLCT but are sufficiently unusual that we designate them as unclassified sex cord–stromal tumors and write a notation.
Sex cord–stromal tumors from pregnant patients are not infrequently placed in the unclassified sex cord category. In one study, 17% were placed in this category, and others diagnosed as GCT or SLCT had large areas with an indifferent appearance (41). Major features that led to difficulty in classification included the presence of prominent edema in the tumor, prominent luteinization in GCTs, and extensive Leydig cell maturation in SLCTs during the third trimester of pregnancy.
Another rare sex cord–stromal tumor has been reported in two girls with PJS and isosexual pseudoprecocity (117). The unusual microscopic features included tubular differentiation, a retiform pattern, and a diffuse proliferation of cells, some of which contained abundant eosinophilic cytoplasm.
STEROID CELL TUMORS
These neoplasms account for approximately 0.1% of ovarian tumors and are composed of large round or polyhedral cells that resemble lutein, Leydig, and adrenocortical cells. The terms lipoid cell tumor and lipid cell tumor had been used for many years (118) to describe them but, although most tumors in this category contain abundant intracellular fat, approximately 25% do not, leading to the paradox of lipid-free lipid cell tumors. To avoid this incongruity, the designation steroid cell tumor was proposed and adopted. These tumors are divided into two major categories: Leydig cell tumor (119) and steroid cell tumor, not otherwise specified (NOS) (120). The term stromal luteoma (121) has been used for small tumors that arise within the ovarian stroma and are associated with stromal hyperthecosis (121). However, it is now favored that many larger steroid cell tumors are also of stromal origin and that stromal luteomas are best considered small steroid cell tumors and should not be separately classified.
LEYDIG CELL TUMORS
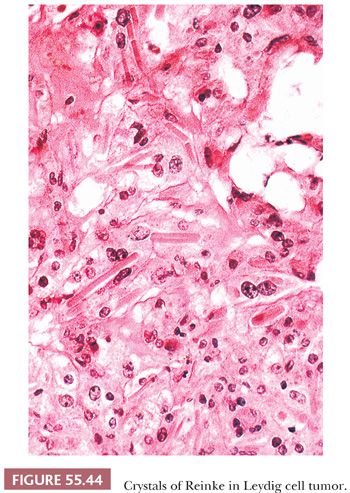
The Leydig cell nature of a steroid cell tumor can be confirmed by the identification of cytoplasmic crystals of Reinke on light or electron microscopy. However, because only about one-third of testicular Leydig cell tumors are found to contain Reinke crystals on light microscopic examination, some ovarian steroid cell tumors, NOS are most probably Leydig cell tumors lacking crystals. Leydig cell tumors (with crystals) account for approximately 15% of steroid cell tumors.
These tumors can originate in the hilus or within the ovarian stroma (Leydig cell tumor, nonhilar type) (122). Hilus cell tumors, which are by far the most common, arise from normal hilus cells that are usually located in the hilus adjacent to nonmedullated nerve fibers. Only rare Leydig cell tumors evolve from ovarian stromal cells. An ovarian stromal cell derivation for these tumors is tenable because they lie within the ovarian stroma and because intracytoplasmic crystals of Reinke can be found in isolated instances in the steroid cells of what appears to be otherwise typical stromal hyperthecosis.
Clinical Features
Hilus cell tumors are diagnosed at an average age of 58 years (typically postmenopausal), causing hirsutism or virilization in 80% of patients. The androgenic manifestations are often milder and of longer duration than those associated with SLCTs, but they may be abrupt. Patients may have elevated testosterone serum levels and 17-ketosteroids urinary levels. Occasionally, there are estrogenic manifestations. Tumors are associated with an excellent prognosis.
Gross Features
Hilus cell tumors range up to 15 cm in diameter, but the great majority are less than 5 cm. Almost all are unilateral. They are well circumscribed; lobulated; fleshy; and brown, black, orange-red, or yellow; hemorrhagic mottling is common.
Microscopic Features
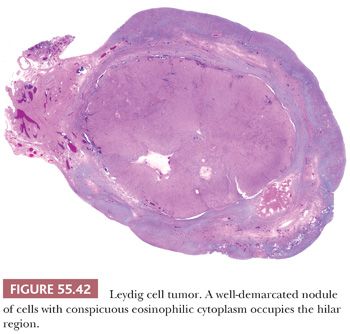
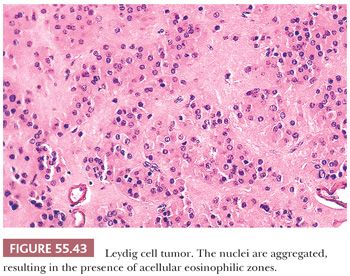
Tumors are typically centered in the hilus, pushing against the surrounding ovarian cortex (Fig. 55.42). Sheets, cords, or nests of uniform rounded or polyhedral cells with large central nuclei containing one or more prominent nucleoli are characteristic (119); occasional nuclei may contain cytoplasmic pseudoinclusions, and spotty nuclear atypia of the degenerative type is common. Areas with clustering of nuclei alternating with nuclei-free perivascular zones are not common but distinctive (Fig. 55.43). The cytoplasm is eosinophilic and finely granular, although small cytoplasmic lipid vacuoles may be present. Lipochrome pigment is seen in varying amounts. Crystals of Reinke (Fig. 55.44) are often present, but their detection may require a prolonged search. Fibrinoid degeneration of large blood vessel walls is a typical and seemingly distinctive feature. Although these tumors typically lack prominent stroma, some may have a nodular appearance due to thick collagenous septa. The morphologic features of Leydig cell tumors of the nonhilar type are similar to those of hilus cell tumors except for their location. An almost certain diagnosis of hilus cell tumor can be made if a steroid cell tumor lacking crystals of Reinke has more than one of the following features: a hilar location, a background of hilus cell hyperplasia, a close association with nonmedullated nerve fibers, a uniform nucleus-free zone around blood vessels, and fibrinoid degeneration of the vessels within the tumor.
Immunohistochemical Features
The tumor cells are typically positive for vimentin, inhibin, calretinin, melan-A, microphthalmia-associated transcription factor (MiTF), SF-1, and CD99 and often express androgen receptor but only rarely AE1/3 (15,19,20,123–126). WT1 is often negative and FOXL2 has been reported to be negative (24).
STEROID CELL TUMOR, NOT OTHERWISE SPECIFIED
Clinical Features
Steroid cell tumors that cannot be diagnosed as Leydig cell tumor are most common (120). They occur at any age, with an average age of 43 years. Patients frequently present with virilization due to androgen excess (41%) but occasionally with estrogenic or no endocrine manifestations. In children, they may induce isosexual pseudoprecocity or result in Cushing syndrome (127). The tumors are almost always unilateral and International Federation of Gynecology and Obstetrics (FIGO) stage I (118,120,128), but approximately 20% of patients have extraovarian extension of their tumors at the time of diagnosis (particularly when associated with Cushing syndrome).
Gross Features
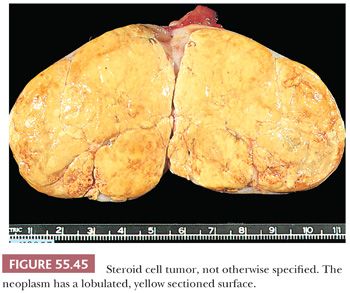
Tumors are often well-circumscribed and usually range from 3 to 10 cm, being on average, larger than Leydig cell tumors (128). Most are yellow (Fig. 55.45) or orange due to abundant intracytoplasmic lipid, but they may be red to brown if they are lipid-poor or dark brown to black if they contain abundant lipochrome pigment. Necrosis, hemorrhage, and cystic degeneration are occasionally observed.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


