Fig. 15.1
Macroscopic features of a schwannoma: encapsulated mass, associated with a nerve, with homogenous yellow/tan cut surface
The cut surface of schwannomas appears tan and homogenous (Fig. 15.1). There may be white areas (fibrosis) or yellow areas which represent fat infiltration or sheets of lipid laden macrophages. In some cases (especially in large tumors) there may be areas of hemorrhage or cystic degeneration.
In some cases, especially in the skin and viscera, schwannomas may grow in a plexiform pattern, expanding and replacing the nerve of origin, in a similar pattern to plexiform neurofibromas.
Histological Features
Most schwannomas have the conventional (classic) histological features. However, other patterns of growth and histological patterns may be seen, especially in association with tumor syndromes (see below) and may be misdiagnosed as other tumor types.
Conventional Schwannomas
The histological appearance of conventional schwannomas is of a benign spindle cell tumor with sharply demarcated margins (encapsulated). Nerve axons and perineurial cells are often present at the periphery of the tumor, and can be highlighted with immunostaining for Neurofilament and Claudin 1, respectively. In contrast with neurofibromas, very few axons (if any) are present within the tumor.
A biphasic pattern with compact (Antoni A) and loose (Antoni B) areas is a characteristic feature (Fig. 15.2). The proportions of these areas vary, and in some tumors only one type will be present. Diagnosis based on the loose (Antoni B) areas can be difficult as the histological features are nonspecific. The hallmark of schwannomas is the formation of nuclear palisades around nuclear free areas (Verocay bodies) which may at times form elongated ribbons (Fig. 15.3) or clusters where tumor cells form groups of “streaming” elongated, narrow nuclei. Longstanding tumors may display degenerative changes such as cysts and sheets of lipid laden macrophages. Some schwannomas may have large clusters of “back to back” large hyalinized vessels mimicking vascular malformations, often associated with thrombosis and hemosiderin-laden macrophages. Another type of degenerative change in schwannomas is the presence of scattered large, atypical, hyperchromatic nuclei (“ancient change”) which are not indicative of malignant transformation.
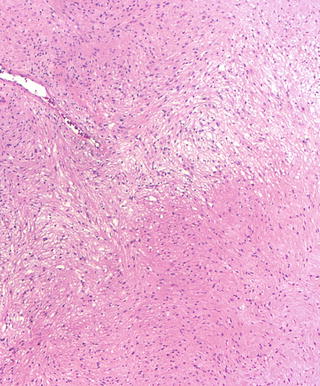
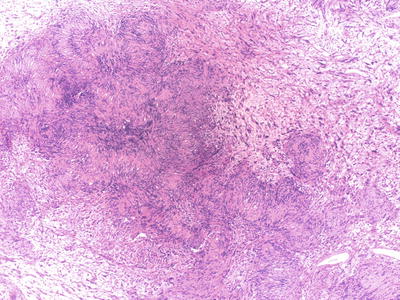
Fig. 15.2
Conventional schwannoma (H&E): Antoni A (compact) and Antoni B (loose, pale) areas are classic features in schwannomas
Fig. 15.3
Conventional schwannoma (H&E): Verocay bodies are characteristic of schwannomas and are nuclear palisades around nuclear free areas
Cellular Schwannomas
Cellular schwannomas are characterized by dense cellularity with intersecting fascicles or patternless growth, and lack of the histological hallmarks of conventional schwannomas. The characteristic biphasic (Antoni A/Antoni B) pattern and Verocay bodies are not present and the tumor is often composed of compact (Antoni A) areas only. In addition to hypercellularity, the tumors may display mitotic activity, nuclear atypia, and necrosis (Fig. 15.4). However, despite the presence of these worrisome histological features, cellular schwannomas are benign, and although recurrence rate is variable, they are slow growing and never metastasize [25–27].
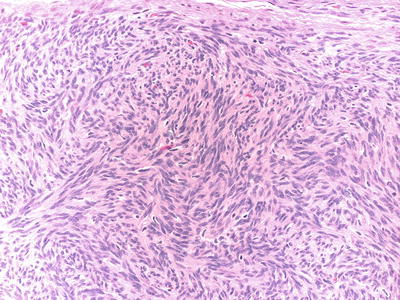
Fig. 15.4
Cellular schwannoma (H&E): Dense cellularity, fascicular growth pattern and scattered mitotic figs. in a cellular schwannoma
The lack of the classical histological features and the presence of dense cellularity and mitoses may prompt a diagnosis of malignancy. The differential diagnosis includes sarcomas such as leiomyosarcoma, or malignant peripheral nerve sheath tumor (MPNST). Helpful distinguishing features that support the diagnosis of cellular schwannoma include the presence of a peripheral capsule, diffuse S100 positivity and collagen IV expression.
Plexiform Schwannomas
Plexiform schwannomas grow within the nerve, expanding and replacing it along its course, so it appears grossly like a rope; a similar pattern as the better known plexiform neurofibromas, with which they may be confused (Fig. 15.5).
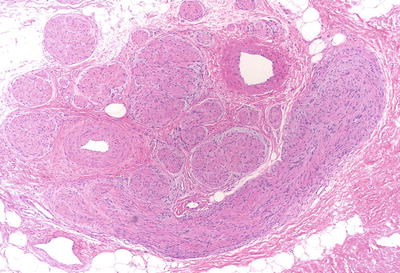
Fig. 15.5
Plexiform schwannomas (H&E): plexiform schwannomas grow along the nerve, expanding and replacing it
Plexiform schwannomas are most common in the skin but may also occur in soft tissue or major peripheral nerves [28–30]. In contrast to other schwannoma subtypes, plexiform schwannomas are often non-encapsulated and may infiltrate adjacent soft tissue, encasing nerves and skin adnexa. Furthermore, entrapped axons are often present within the tumor mass, features that mimic plexiform neurofibromas [31].
Plexiform schwannomas may be associated with NF2 or schwannomatosis; there is no association with NF1 [32, 33]. In contrast to plexiform neurofibromas, there is no risk of malignant transformation. Helpful for the histological diagnosis of plexiform schwannomas are the presence of conventional schwannoma features (Verocay bodies, Antoni A/Antoni B areas) and diffuse S100 positivity.
Melanotic Schwannomas
Melanotic schwannomas are rare and distinct tumors that are composed of neoplastic Schwann cells that contain melanin. The tumors appear as pigmented, circumscribed masses. Melanotic schwannomas are of two types: non-psammomatous and psammomatous, defined by the presence of psammoma bodies (concentrically laminated bodies that are PAS positive). Non-psammomatous melanotic schwannnomas are benign. In contrast, half of the psammomatous melanotic schwannomas are associated with Carney’s complex and may undergo malignant transformation [16, 17, 34]. Therefore, in the case of psammomatous melanotic schwannomas, the possibility of an underlying Carney’s syndrome should be investigated.
Melanotic schwannomas lack the classical features of conventional schwannomas (Antoni A/Antoni B areas, Verocay bodies, vascular clusters) and are often composed of large, epitheloid cells, with large round/oval nuclei and prominent nucleoli (Fig. 15.6). The differential diagnosis of melanocytic schwannomas is with melanocytic lesions; melanocytoma and melanoma (primary and metastatic). Electron microscopy may be helpful. Positive collagen IV immunostaining and a rich reticulin network can support the diagnosis of schwannoma.
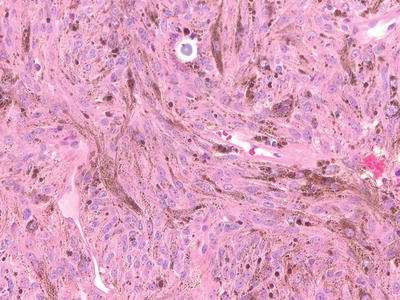
Fig. 15.6
Melanotic Scwhannoma: Melanotic schwannomas are composed of pigmented Schwann cells, and may contain psammoma bodies
Sporadic and Syndromic Schwannomas
The great majority (90 %) of schwannomas are single, sporadic tumors [35]. However, schwannomas may also occur as part of the clinical manifestations in patients with an underlying genetic predisposition (syndromic).
In a study in which the histological features of solitary sporadic schwannomas were compared to schwannomas associated with NF2; some histological features were found to be more common in sporadic/solitary schwannomas. In particular, solitary schwannomas were found to have prominent vascular clusters that mimic vascular malformations (“back to back” vessels with thick hyalinized walls or dilated sinusoidal vessels), thrombosis, and inflammation (Fig. 15.7) [36]. In addition, a more recent study found that in contrast to the mosaic pattern seen in NF-associated schwannomas (NF2 and schwannomatosis), most solitary/sporadic schwannomas retain INI1 expression and appear diffusely positive [37] (see below).
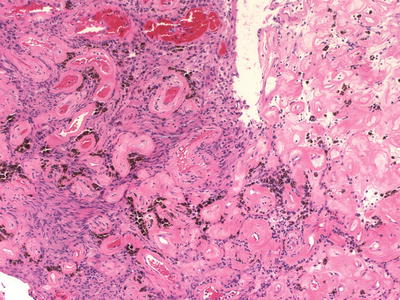
Fig. 15.7
Sporadic/solitary Schwannoma: Clustered thick hyalinized blood vessels mimicking vascular malformations are a common finding in solitary/sporadic schwannomas
Syndromic Schwannomas
The syndromes associated with multiple schwannomas include NF2, schwannomatosis and Carney’s Syndrome.
Neurofibromatosis Type 2 (NF2)
NF2 is an autosomal dominant disorder characterized by neoplastic and dysplastic lesions of Schwann cells, meningothelial cells, and glial cells. Patients are predisposed to develop multiple schwannomas, and the hallmark of the disease is bilateral VS. In addition, NF2 patients are predisposed to develop other tumors; multiple meningiomas and spinal ependymomas. Non-neoplastic lesions associated with the syndrome include meningioangiomatosis, glial hamartomas, retinal hamartomas, posterior subcapsular cataracts, epiretinal membranes, and polyneuropathies [1, 38].
The disease is rare, with an estimated incidence of 1 per 40,000 newborns [35]–1:25,000 [39] and is caused by a germline mutation in the NF2 gene on chromosome 22q that encodes the protein Merlin. De novo mutations (patients without family history) occur in 30 % of the patients. Particularly difficult to diagnose are patients with mosaic NF2 in which clinical manifestations may overlap with other forms of neurofibromatosis (NF1 or schwannomatosis) or may not fulfill the clinical criteria for the diagnosis of NF [40]. In these scenarios, the pathological diagnosis of nerve sheath tumors may be particularly helpful in supporting a suspected clinical diagnosis. Schwannomas associated with NF2 often present at an earlier age than sporadic, non-syndromic schwannomas [41, 42].
NF2-associated schwannomas frequently have a multilobular appearance (“bunch of grapes”) which may be apparent macroscopically and/or microscopically [36]. In some cases, meningioma and schwannoma form a collision tumor, in which the two components are seen on the same slide (Fig. 15.8). Schwannoma/meningioma collision tumors are pathognomonic for NF2. In contrast to sporadic solitary schwannomas, the pattern of INI1 immunostaining of schwannomas associated with NF2 or schwannomatosis is a mosaic pattern, in which there is partial loss, with mixed positive and negative cells (Fig. 15.9); [37]. Therefore, the pattern of growth (multinodular) and the mosaic INI1 expression pattern may support the diagnosis of an NF-associated schwannoma in some cases. Plexiform cutaneous schwannomas may be seen in childhood in NF2 patients and should not be confused with neurofibromas (which would lead to the clinical diagnosis of NF1).
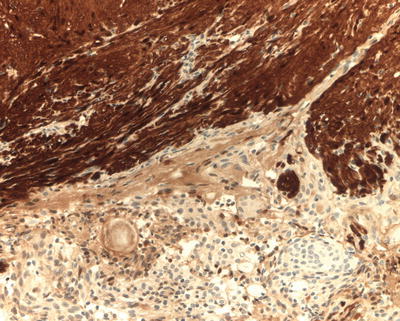
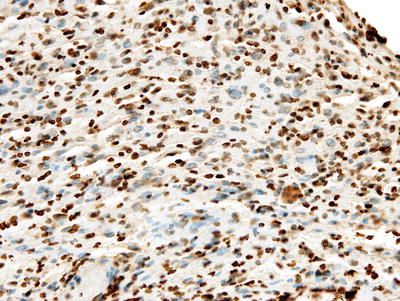
Fig. 15.8
Schwannoma/meningioma collision tumor (S100 immunostain): a collision tumor composed of schwannoma (on the right, immunopositive for S100) and meningioma (on the left, S100 negative) is pathognomonic of NF2
Fig. 15.9
Mosaic INI1 immunostaining (INI1 immunostain): partial loss (mosaic staining) of INI1 expression is common in NF-associated schwannomas
Schwannomatosis
Histologically, schwannomatosis-associated tumors often have prominent myxoid stroma, which may lead to a misdiagnosis of a neurofibroma [8, 11–13, 43–45]; (Fig. 15.10). Peripheral nerve sheath tumors with mixed features of schwannoma/neurofibroma are referred to as “hybrid tumors” [46] and may represent a myxoid schwannoma or a “Schwann cell rich” neurofibroma. Hybrid tumors are more common in the context of the neurofibromatoses [47] and misdiagnoses can be avoided by immunohistochemical stains that highlight the different components of the tumor (axons, Schwann cells, perineurial cells). Many of the schwannomatosis-associated tumors (especially familial schwannomatosis) have a mosaic (partial lack) pattern of expression of INI1 protein [37].
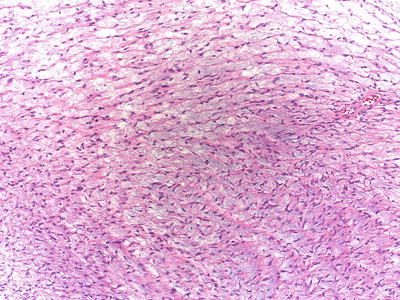
Fig. 15.10
Myxoid schwannoma (hybrid tumor): Schwannomas with abundant myxoid stroma may mimic neurofibroma (hybrid tumor) and are common in schwannomatosis
Carney’s Complex
Schwannomas from patients with Carney’s complex are pigmented and often calcified [18]. Histologically, they contain melanin and psammoma bodies and lack the classic features of conventional schwannomas. There is a risk of malignant transformation in 10 % of the cases. Although the histological criteria are not well defined, large nuclei with prominent nucleoli, brisk mitotic activity, and necrosis are worrisome signs of aggressive biological behavior.
Cytogenetics and Molecular Genetics
A number of studies to date have examined the cytogenetics and molecular genetics of schwannomas, including sporadic schwannomas and schwannomas associated with NF2 and schwannomatosis. A number of early studies demonstrated that loss of heterozygosity (LOH) is common in NF2-associated and sporadic schwannomas [48, 49], and subsequent work showed that both sporadic and NF2-related VS harbor mutational inactivation or loss of both alleles of the NF2 gene [5, 50], consistent with Knudson’s two hit model of tumorigenesis [51]. In the case of schwannomatosis, a four hit mechanism has been proposed, involving NF2 and either SMARCB1 [12] or LZTR1 [15].
Studies in schwannomas using comparative genomic hybridization (CGH) [52–55] have identified loss on chromosome 22 (NF2) as the most common hit by far, detectable in up to 56 % of sporadic and 62 % of NF2-associated schwannomas, and LOH was caused by mitotic recombination in a subset [53]. Other genetic aberrations observed in subsets of tumors included gains involving 9q, 10q, 17q, 19p, and 19q, as well as losses involving 9p. Of note, and perhaps not surprising, some of the data indicate that genetic aberrations outside of chromosome 22 predominantly occur in tumors that were previously treated with radiotherapy [53]. Other investigators have looked at CpG island hypermethylation of the NF2 gene as an alternate mechanism of gene silencing, but the results have been largely negative [56, 57].
Gene Expression Profiling
Several studies have been published on gene expression profiling in schwannomas, showing evidence of deregulation in the proto-oncogene MET, as well as ITGA4, PLEXNB3/SEMA5, and CAV1 [58, 59]. In addition, upregulation of osteopontin (SPP1), a gene involved in the protein degradation of the NF2 gene product Merlin, was observed [58]. Gene regulation at the posttranscriptional level has been examined in a recent study, focusing on miRNA profiling of schwannomas [60]. In that study, 12 miRNAs were found to be significantly deregulated in tumors, including miR-7. Targets of miR-7 include several oncogenes relevant to schwannoma biology, including epidermal growth factor receptor (EGFR), p21-activated kinase (Pak1), and associated cdc42 kinase1 (Ack1).
Prognostic Stratification
Extent of resection is the strongest predictor of recurrence free survival. According to published data, recurrence risk for vestibular schwannomas ranges from 0 to 4 % after gross total resection, 9–29 % after near-total resection and 25–65 % after subtotal resection [61–63].
In NF2, a genotype-phenotype correlation exists and is of prognostic value. Compared to other hereditary disorders, NF2 has an unusually high rate of mosaicism of greater than 30 % amongst de novo patients [64], and clear associations between type of mutation and disease severity has been recognized. For example, while 5′ truncating mutations are associated with a high tumor burden, severe disease course and early mortality, missense mutations have been linked to a relatively mild phenotype [65, 66]. For individual tumors, it is presently not known whether the type of NF2 mutation present in the tumor, or any other genetic or molecular characteristics are prognostic or predictive of tumor aggressiveness or risk of recurrence after surgical resection.
Molecular Signaling Pathways
The molecular signaling pathways that drive tumor initiation and progression associated with loss of Merlin have been subject to intense research efforts over the past decades. It has become evident that rather than acting through a single pathway or at a single cellular compartment, Merlin regulates a wide variety of cellular processes, including contact inhibition, tumor suppression and growth through signaling at the cellular cortex and nucleus. Loss of Merlin has been linked to loss of contact inhibition and activation of a number of pro-growth signaling cascades, as recently reviewed by Li et al. [7]. These include the Rac-PAK [67–70], mTORC [71–73], EGFR/PDGFR/c-kit RAS-RAF-ERK [74–82], PI3K-Akt [83], FAK-Src [84], and Hippo pathways [85–87]. In addition, Merlin has been shown to interact with α-catenin and Par3 at adherens junctions [88], and with the scaffold and signaling protein Angiomotin at tight junctions [89]. Recent studies showed that in addition to cortical functions, Merlin also translocates to the nucleus to alter gene expression through inhibition of the E3 ubiquitin ligase CRL4DCAF1 [90, 91]. Several of these pathways have been validated in preclinical studies involving in vivo and/or in vitro schwannoma models.
The tumor microenvironment, including angiogenesis, has been recognized as an important aspect of schwannoma biology. VEGF and its receptors are expressed in schwannomas, and expression levels are associated with increased rates of tumor growth [92, 93]. Anti-VEGF(R)-directed therapy with bevacizumab and vandetanib normalized the vasculature of NF2−/− schwannoma xenografts in nude mice and decreased tumor growth [94]. Recent data suggest that Merlin regulates angiogenesis in schwannomas through Rac1-dependent semaphorin 3F expression [95].
Molecular Targeted Therapies
VEGF
The first “molecular targeted” therapy to show clinical success in treating VS in NF2 patients has been bevacizumab, a monoclonal anti-VEGF antibody. Based on retrospective data, bevacizumab may result in radiologic responses and/or hearing improvement in approximately 50 % of patients, although treatment effect is only maintained with continuous administration [96–99]. Recently completed and ongoing prospective clinical trials with bevacizumab (ClinicalTrials.gov identifiers NCT01207687 and NCT01767792) will provide additional data on the efficacy and safety of this therapy, including in children.
ErbB Receptor Family
Preclinical data implying overexpression and activation of ErbB family receptors in promoting schwannoma growth led to a clinical trial using lapatinib, a small-molecule inhibitor targeting EGFR and ErbB2. In this phase 2 study, 24 % of evaluable patients experienced a radiologic response. Median time to overall progression (i.e., volumetric progression or hearing loss) was 14 months, but only one transient hearing response was observed [100].
mTOR
Based on the observation that loss of Merlin leads to activation of mTORC1 signaling [71, 72], several phase 2 clinical trials with everolimus (RAD001) have been conducted. Results of one of these trials have been published recently, suggesting that everolimus is not clinically effective in treating NF2-related VS [101].
PDGFR and c-kit
Schwann cells express PDGFRα and PDGFRβ [76]. Signaling through these receptors activates the RAS-RAF-MEK-ERK and PI3K-AKT signaling pathways, and is important for Schwann cell proliferation in vivo and in vitro [77–79]. Overexpression of PDGFRβ has been observed in VS [74, 80, 81], and PDGFR inhibitors including AG1296, imatinib, and nilotinib are effective in preventing PDGFR-driven proliferation when tested in VS in vitro models [74, 82]. VS cells express activated c-KIT and are growth-inhibited by imatinib [81] and nilotinib [82]. Based on this preclinical data, a phase 2 clinical trial with nilotinib is ongoing (ClinicalTrials.gov identifier NCT01201538).
Other Targets and Future Outlook
Some of the key tumorigenic signaling pathways associated with loss of Merlin, such as the Hippo signaling pathway and activation of the E3 ubiquitin ligase CRL4DCAF1, are not directly targetable with currently approved drugs, but of interest for future therapeutic development. Although it appears that loss of NF2 may be sufficient for tumor formation and progression, it is conceivable that other oncogenic drivers may cooperate with loss of Merlin. To identify such molecular genetic drivers in schwannomas, future studies using next-generation sequencing approaches, such as whole-exome/whole-genome sequencing and RNA-seq, could provide valuable information.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


