Fig. 16.1
Plexiform neurofibroma arising from the vagus nerve, involving the thymus gland
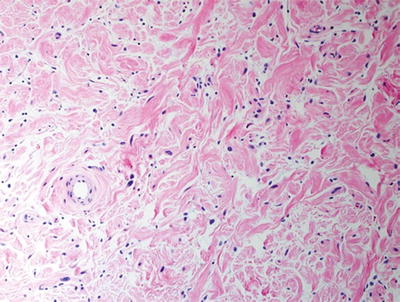
Fig. 16.2
Plexiform neurofibroma with low cellularity and “shredded carrot” collagen
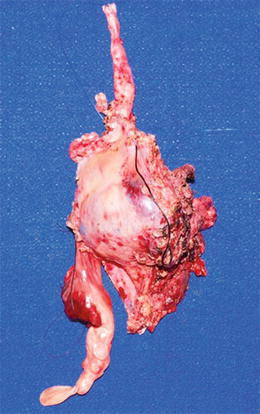
Fig. 16.3
Large mediastinal soft tissue mass (high grade MPNST) arising from vagus nerve (top) and encasing and eroding into the superior vena cava, with intravascular thrombosis (bottom) (Courtesy of Children’s Hospital of Wisconsin, Milwaukee, WI)
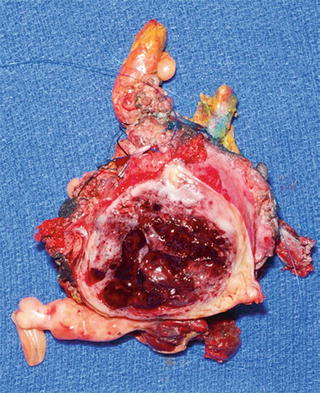
Fig. 16.4
Cut surface of MPNST depicted in Fig. 16.3 with peripheral rim of white-gray tumor and central hemorrhage and necrosis
Notable histological variation may be observed in MPNSTs. Common histological findings include fascicles of alternating cellularity (Fig. 16.5), whorls, palisading or rosette-like patterns, subendothelial condensation of tumor cells, and geographic necrosis [8, 11]. Occasionally, the tumors resemble primitive or undifferentiated sarcoma (Fig. 16.6). Less commonly, rhabdomyosarcomatous elements (malignant Triton tumor), angiosarcoma, melanin, neuroendocrine, or glandular structures are observed. The cell(s) of origin of these divergent features remain uncertain [7, 11].
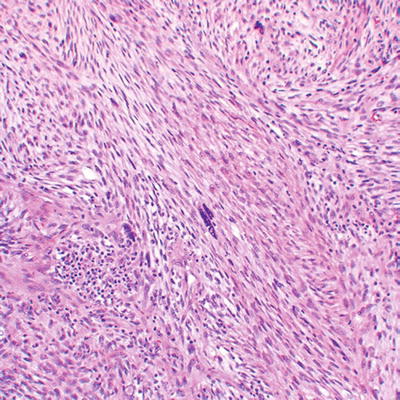
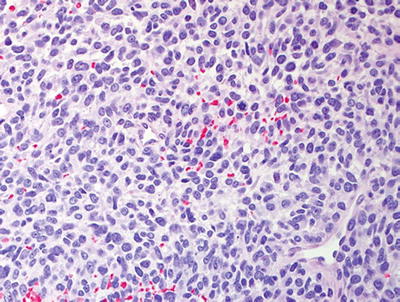
Fig. 16.5
High grade MPNST with spindled cells, fascicular pattern, and focal cytological atypia with nuclear enlargement and hyperchromasia
Fig. 16.6
High grade MPNST with appearance of undifferentiated, primitive sarcoma
MPNST grading is separated pathologically into low and high grade categories; the majority of MPNSTs are high grade [8]. Morphological criteria for a low grade MPNST include hypercellularity and nuclear enlargement (approximately 3× the size of a neurofibroma nucleus) and hyperchromasia, features also seen in high grade MPNST; however, low grade MPNSTs exhibit little necrosis and show fewer than five mitoses per 10 high power fields [18, 12]. The isolated presence of one of these features in a neurofibroma is not adequate for a malignant diagnosis. Diagnostic difficulties arise due to the lack of objective criteria for hypercellularity, hyperchromasia, and the extent of changes required for a malignant diagnosis. Features concerning for malignant transformation include increased cellularity and a fascicular pattern of growth not usually seen in conventional neurofibromas (Fig. 16.7). Histological grading systems include the US National Cancer Institute (NCI) system [13], based on the tumor histological type, location, and degree of necrosis, as well as pleomorphism, cellularity, and mitotic activity. The Federation National des Centres de Lutte Contre le Cancer (FNCLCC) is a French grading system which uses a score generated by degree of tumor differentiation, mitotic activity, and extent of necrosis [14]. Neither of these grading systems has proven entirely useful in distinguishing low versus high grade MPNST and predicting clinical behavior. Information regarding the molecular biology of MPNST is anticipated to prove important for not only diagnosis but possible targeted therapy options.
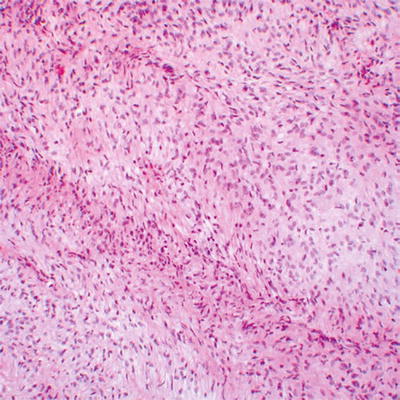
Fig. 16.7
Low grade MPNST with high cellularity and monotonous spindled cells arranged in long fascicles
Even more challenging is the separation of atypical neurofibroma (considered benign) from low grade MPNST (malignant), particularly in the setting of NF1. The term “atypical neurofibroma” has been applied to neurofibromas with degenerative nuclear changes [8]. This term, or alternatively, cellular neurofibroma, has also been used for nerve sheath tumors showing worrisome histological features, including high cellularity, few mitotic figures, monotonous cytomorphology, or fascicular growth, which do not fully meet criteria for malignancy. Atypical changes often develop in large, slowly growing neurofibromas [15]. Atypical neurofibromas have generally been regarded as benign. However, a study of NF1 patients suggests that atypical neurofibromas, defined as neurofibromas with increased cellularity and nuclear hyperchromasia and enlargement lacking mitotic figures (Figs. 16.8, 16.9, and 16.10), represent early malignant change in neurofibroma, with CDKN2A (p16) deletions (seen in MPNST) in the majority of studied cases [16, 17].
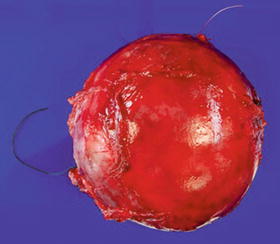
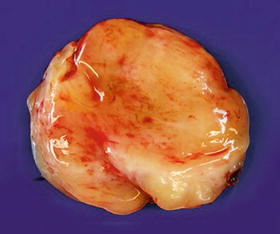
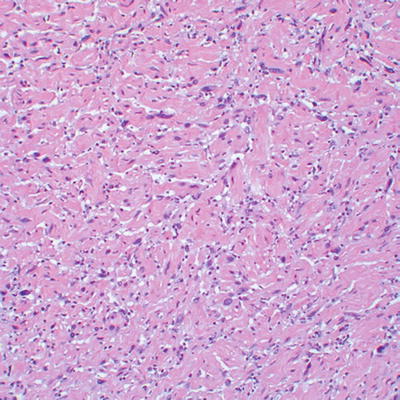
Fig. 16.8
Well-circumscribed, apparently encapsulated atypical neurofibroma
Fig. 16.9
Atypical neurofibroma shown in Fig. 16.8 with slightly gelatinous, yellow to white cut surfaces
Fig. 16.10
Atypical neurofibroma with wavy collagenous stroma with increased cellularity, nuclear enlargement, and hyperchromasia; no mitotic activity was appreciated
Histological examination of a soft tissue lesion in which the differential comprises MPNST should include routine-stained H&E sections and possibly reticulin, to clearly outline nerve fibers. In addition, immunohistochemical stains for S100β protein, the skeletal muscle markers desmin and myogenin, and a proliferation marker (MIB-1 or KI-67) may be useful [1]. Increased MIB-1 (Ki-67) and p53 nuclear labeling by immunohistochemistry are seen in high grade MPNST [6, 18]. Genetic loss of the CDKN2A locus, and therefore loss of p16 immunoreactivity, are not found in neurofibroma, but are common in MPNST [16, 17]. Both p16 and p27 expression are typically present in neurofibromas and low grade MPNSTs but absent in high grade MPNSTs [6]; loss of expression may highlight foci of malignant transformation in neurofibromas, as may molecular alterations, including EGFR amplification [19].
The differential diagnosis of MPNST includes sarcomas, including adult-type fibrosarcoma, synovial sarcoma, rhabdomyosarcoma, leiomyosarcoma, dedifferentiated liposarcoma, and clear cell sarcoma. One useful distinction from benign Schwann cell tumors is the partial or complete loss of S100β expression in MPNST (Fig. 16.11). Conversely, isolated expression of S100β should not necessarily be diagnostic of MPNST, as S100β expression has been reported in leiomyosarcomas, rhabdomyosarcomas, and synovial sarcomas [6, 8, 18].
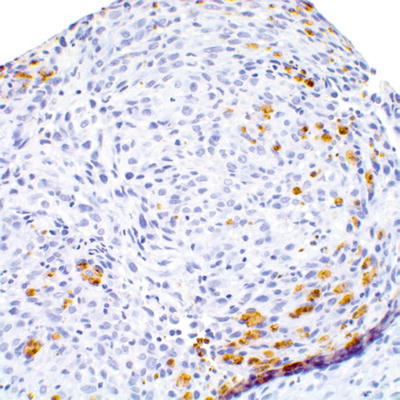
Fig. 16.11
Focal retention of S100β expression in high grade MPNST
Synovial sarcoma, a high grade sarcoma of undetermined cell lineage, may occur in soft tissues in either biphasic (which includes a spindle cell component with interspersed glandular structures) or monophasic (spindle cell component only) forms. The monophasic variant may closely resemble MPNST, and may involve nerves or exhibit a plexiform growth pattern. Both synovial sarcoma and MPNST may show glandular differentiation. The only definitive histological feature used in the distinction of MPNST from synovial sarcoma is the presence of pleomorphic cells, not seen in synovial sarcoma (Fig. 16.12). Demonstration of SS18-SSX1 or SS18-SSX2 gene fusions, usually resulting from a characteristic X;18 translocation, may be required for definitive diagnosis of intraneural synovial sarcoma, as these gene fusions are limited to synovial sarcoma [8]. No specific chromosomal rearrangements in MPNST have been revealed by conventional cytogenetics, although a complex karyotype is characteristic (see below for details) [20].
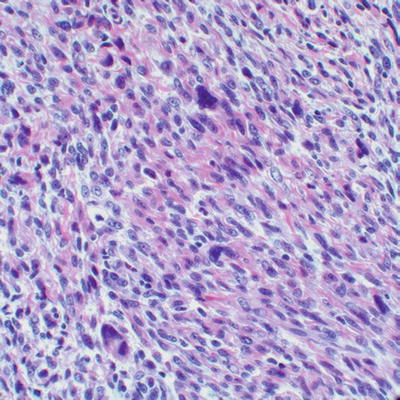
Fig. 16.12
High grade MPNST with eosinophilic stroma, elongated cells with vesicular nuclei, and cytological atypia
Epithelioid MPNST, a rare subtype of MPNST, is characterized by a predominance of large epithelioid cells. Epithelioid MPNSTs are more frequent in superficial sites and exhibit strong and usually diffuse expression of S100β protein [8]. The majority of MPNSTs arising within preexisting schwannomas, which occurs very rarely, are of epithelioid type [10]. The differential diagnosis of epithelioid MPNST includes epithelioid sarcoma, clear cell sarcoma, melanoma, and carcinoma. The absence of expression of melanocytic markers (MelanA, HMB45, MART-1) is useful in the differentiation of epithelioid MPNST from melanoma and clear cell sarcoma [8]. Absent cytokeratin expression distinguishes epithelioid MPNST from epithelioid sarcoma and carcinoma. Both epithelioid MPNST and epithelioid sarcoma may show loss of SMARCB1/INI1/BAF47 protein expression [21], a potential diagnostic pitfall in the consideration of rhabdoid tumor [6, 8, 13–15, 17, 18, 22].
Most MPNST are frankly high grade, aggressive tumors by histology and clinical behavior, and carry a dismal prognosis. Adverse prognosticators include truncal location, size >5 cm, incomplete resection, local recurrence, young age [7, 9], and high grade. According to some authors, histological grade is the most important prognostic factor for soft tissue sarcomas [13, 23] including MPNSTs. Past literature portended a worse prognosis for NF1-associated tumors as compared to sporadic MPNSTs [7, 24]. However, a large recent study indicates that while NF1 patients with MPNSTs demonstrate overall increased mortality compared to those patients with non-NF1-associated MPNSTs, decreased survival did not appear to be related to inherent tumor behavior [24].
Cytogenetics and Molecular Genetics
Primary among MPNST initiating mutations are mutations in the NF1 gene. NF1 patients carry a constitutional mutation of the NF1 tumor suppressor gene located on the long arm of chromosome 17 (17q11.2) [25], and mutation, or loss of the second allele, was found in 40 % of NF1 MPNST [26]. MPNST are particularly prevalent in NF1 patients whose constitutional mutations involve whole gene deletion, which can include contiguous genes that may contribute to tumor formation [27]. NF1 mutations are also present in 41 % of sporadic MPNST [26], explaining why expression signatures and genomic changes overlap in NF1 and sporadic tumors [28–30]. Mutations in RAS or RAS pathway genes may also cause MPSNT tumor initiation in MPNST lacking NF1 mutations; activating mutations in N-RAS(1/11), K-RAS (1/11) [31], and B-RAF (1/13 MPNST) [26] were identified in sporadic MPNST.
As discussed above, atypical neurofibromas represent an early stage in MPNST transformation from neurofibroma. Atypical neurofibromas (15/16) showed homozygous loss of the CDKN2A locus on chromosome 9p21.3 [17], and deletions of the CDKN2A locus are present in about 50 % of MPNSTs [16, 32]. The CDKN2A locus encodes two proteins: p16INK4A, which inhibits the cyclin-dependent kinases 4 (CDK4) and 6 (CDK6), and p14ARF, which inhibits the MDM2 ubiquitin ligase resulting in stabilization of tp53 [16, 32]. Mouse models support the importance of this locus in MPNST, as Nf1+/−; Ink4a/Arf−/− mice develop GEM-PNSTs resembling human MPNST [33].
Another tumor suppressor commonly inactivated in MPNST is TP53. An “inactivated p53-associated proliferation” gene expression signature was identified in 18/20 MPNST, and p53 inactivation caused downregulation of miR-34a, preventing MPNST cell apoptosis in tissue culture [34]. Estimates of MPNST with TP53 alterations (mutations or stabilized TP53) vary between 24 and 75 % [35–37], which is likely due to the variable sensitivity and specificity of different assays for assessing p53 expression and mutations, as well as intra-tumor heterogeneity [38–41]. TP53 stability can also be regulated through p14ARF so that if p14ARF is retained, TP53 is stabilized without TP53 mutation. While biallelic inactivation of the TP53 locus is rare in MPNSTs [42] in mouse models, only complete loss of tp53 and Nf1 correlates with Genetically engineered mouse-Malignant Peripheral Nerve Sheath Tumor (GEM-MPNST) formation [43, 44].
The PTEN gene is an “off signal” for phosphoinositide 3-kinase (PI3K) signaling, and PTEN inactivation generally leads to activation of PI3K. Frequent monosomy of the PTEN locus was identified in MPNST without PI3KCA or PTEN mutations [31, 45]; PTEN methylation is detected in 45 % of MPNSTs, though not neurofibromas, and associated with early metastasis [46]. Co-deletion of Nf1 and Pten or expression of RasG12D or EGFR in combination with Pten deletion also resulted in GEM-PNST [47, 48]. In addition, expression of the retinoblastoma (RB) tumor suppressor, a molecule that impedes cell cycle progression, is lost in 25 % of MPNSTs [49, 50].
As in most sarcomas, chromosomal gains, losses, and rearrangements in MPNSTs are numerous and variable [51], and MPNSTs commonly have hypodiploid or near-triploid karyotypes. Combined genomic somatic copy number alteration (CNA) and loss of heterozygosity (LOH) analysis on sets of neurofibromas and MPNSTs verified that recurrent or overlapping copy number variations (CNVs) or CNAs are absent from neurofibromas, while MPNSTs showed 232 CNAs (encompassing >2,900 genes) and more than 500 genes showed consistent LOH [52]. The microRNA miR-10b can target NF1 messenger RNA [53]; in principal miRs that target NF1 might also contribute to NF1 tumorigenesis.
Amplification of the epidermal growth factor receptor gene EGFR is frequent in MPNST [45, 54]. Perrone et al. found that EGFR was amplified in all sporadic MPNST and half of NF1 MPNST [31]. EGFR overexpression was correlated with a worse prognosis in one study [55], but not in another [56]. No activating mutations in EGFR have been detected in MPNST. Ligands that activate the EGFR including transforming growth factor (TGF)α and heparin binding epidermal growth factor (HBEGF) are expressed in 90 % of MPNST, suggesting the presence of an autocrine loop in MPNST cells [45, 57]. In 15 % of a small series of MPNST, the amplicon including PDGFRA, KIT, and VEGFR-2/KDR was present. Among the three genes, PDGFRA is most frequently amplified [49, 58, 59] and rarely mutant [59]. Hepatocyte growth factor is expressed and its c-Met receptor is expressed in 82 % of MPNST, and the MET gene is amplified in MPSNT [49, 60]. Short hairpin RNAs targeting MET and XL184, a multi-kinase inhibitor targeting MET and VEGFR2, decreased MPNST tumor growth and metastasis in tumor xenografts [61]. While in vitro studies in cell lines support roles for these receptors in MPNST, several histology-specific clinical trials with agents targeting PDGFR, C-KIT, and EGFR were completed, all without achieving responses or meaningful disease stabilization as single agents [62–65]. Possibly blocking one or more of these receptors will be useful in combination with other therapeutic agents.
Gene Expression Profiling
Sporadic and NF1 MPNST are indistinguishable by transcriptome analysis [30]. Transcriptome analysis comparing Schwann cells to MPNST found that expression of markers of neural crest cells is a prominent theme in human MPNST cell lines and tumors [29, 66]. Neural crest markers include SOX9 and TWIST1, which are dramatically upregulated in MPNST [29, 66–68]. MPNST cells are dependent on expression of these genes, as downregulation of SOX9 caused cell death and downregulation of TWIST1 decreased cell migration [29, 66]. Increased expression of the neural crest markers FOXD3, PAX7, SOX5, and AP-2α in MPNST compared to neurofibroma was described in a series of 34 MPNSTs [68]. The placodal markers EYA/SIX are also upregulated in MPNST cells and tumors [69], and shRNA to diminish EYA4 expression prevented tumor formation and caused necrosis. EYAs are phophatases that could in principle be targeted therapeutically.
Whole genome microRNA analysis of MPNST tumors identified downregulation of 14 miRs, and upregulation of two (miR-210 and miR-339-5p) [70]. There was no overlap with serum microRNAs in MPNST patients [71]. Serum miR expression distinguished patients with MPNST from those without MPNST. The authors identified miR-24 as upregulated in NF1 and MPNST, and MiR-214 and miR-801 as upregulated in serum of individuals with sporadic or NF1-related MPNST. The sensitivity (0.820) and specificity (0.844) of a three miR panel to identify NF1 MPNST supports a potential role in helping to diagnose MPNST and/or as a possible indicator of response to therapy [71].
On two-dimensional gel analysis of proteins, MPNST most closely resembled synovial sarcoma and clear cell carcinoma [72]. For this reason, a goal remains to identify the markers that distinguish MPNST from these tumors, and from surrounding neurofibroma. Several markers, each analyzed in relatively few tumors, may distinguish neurofibromas from MPNST. These include Tenascin-C and NNAT [73]; Cathepsin K [74]; and markers of an angiogenic switch: SMA, vWF, VEGF, and VEGF receptors Flt1 and Flk1 [75]. Many growing or atypical neurofibromas and MPNST stained positive for CD10 [76]. Some neurofibromas and MPNST express hTERT [77].
Prognostic Stratification
Complete surgical resection is the only known curative MPNST therapy, and predicts favorable prognosis in all MPNST patients [23, 78, 79]. In addition, survival is significantly better in female versus male MPNST patients [80, 81]. Gain/amplification of the CDK4 gene on chromosome 12q14.1 and upregulation of the FOXM1 gene on chromosome 12p13.3 were significant independent predictors of poor survival in 87 MPNST patients [82]. Chromosomal losses of 10q and Xq and gain of 16p were also associated with reduced MPNST patient survival [28]. In a large series, 93 % of MPNST showed positive staining for phospho-MEK, while about half expressed phospho-S6K, phospho-mTOR and/or phospho-AKT, and immunoreactivity toward all three mTOR pathway markers predicted significantly worse outcomes than in patients with tumors negative for the three markers [83]. Intriguingly, a single nucleotide polymorphisms in the microRNA biogenesis pathway gene DROSHA (rs1991401) significantly increased MPNST risk in NF1 patients, while SNPs in AGO2 and GEMIN4 in this pathway decreased risk [71]. To date, none of these indicators have been used to stratify patients for clinical trials.
Treatment of MPNSTS
Only complete MPNST surgical resection has been shown to be curative, and remains the cornerstone of therapy, but is rarely feasible due to tumor location or nerve association [23, 78, 79, 84, 85]. Radiotherapy is commonly used for local control in inoperable or incompletely resected MPNSTs, but when used as primary treatment, high doses of radiation are needed (median 50 Gy) [3]. The role of chemotherapy for adult and pediatric soft tissue sarcomas, including MPNSTs, is controversial. Only doxorubicin, dacarbazine, and ifosfamide are agents consistently associated with response rates of 20 % or more in patients with soft tissue sarcomas [23, 86, 87], and the combination of ifosfamide and doxorubicin has produced response rates as high as 46 % in these tumors [87, 88]. The response rate of MPNSTs to chemotherapy is unknown. Some investigators have suggested that they have intermediate chemosensitivity, less responsive than synovial sarcoma, but more responsive than refractory diseases such as alveolar soft part sarcoma [86]. However, recently others have questioned whether MPNSTs are at all chemosensitive [84]. Carli et al. summarized the 25-year experience of pediatric MPNSTs in German and Italian Groups [3]. The patients described encompass a span of three decades and were treated on standard sarcoma protocols. First, response to ifosfamide was significantly better than to cyclophosphamide (65 % vs. 17 %). Second, while chemotherapy increased overall and event-free survival over no chemotherapy, the 5-year overall survival for patients with unresectable and metastatic MPNST remained approximately 30 %. It may be that the addition of targeted agents to chemotherapy will improve response rate, and potentially improve outcome without undue morbidity.
Molecular Signaling Pathways
NF1 is an off signal for Ras GTPases [89]. Therefore, NF1 loss activates signaling pathways downstream of Ras-GTP, and the Raf-MEK-ERK and mTOR-S6K-Akt pathways have been explored as potential therapeutic targets (Fig. 16.13). Targeting MEK with PD0325901 in a xenograft and in a genetically engineered mouse model transiently delayed MPNST growth, correlating with suppression of tumor vasculature and tumor cell proliferation [90, 91]. Using rapamycin or its analog RAD001 to target the mTOR/S6K pathway also transiently blocked MPNST growth in xenografts and a mouse model [92–94]. This effect of rapamycin was converted to cytotoxicity in combination with agents that promote proteotoxic/endoplasmic reticulum (ER) stress in a genetically engineered mouse model [95]. Based on these data, combinatorial clinical trials are being considered.
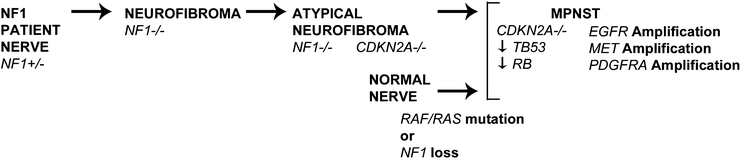
Fig. 16.13
Schematic illustration of some of the multiple genetic changes believed to contribute to NF1-related and sporadic MPNST. In NF1 patients, benign neurofibromas form when NF1 haploinsufficient cells in the Schwann cell lineage lose remaining functional NF1. Subsequent progression toward MPNST is via an atypical neurofibroma intermediate, and is associated with loss of the tumor suppressor gene CDKN2A. MPNST also show mutation of additional tumor suppressor genes and amplification of several growth factor receptors. The bottom row shows that mutations in RAS genes, RAF genes, and NF1 were recently identified in sporadic MPNST
We have chosen to omit discussion of a host of studies focusing on effects in MPNST cell lines, pending confirmation of significant effects in in vivo model systems. In xenografts, hyaluronan oligomers suppressed drug transporter activity and inhibited growth of MPNST tumor growth, with synergy between oligomers and doxorubicin [96]. The effect of 4-hydroxytamoxifen on K-Ras degradation and MPNST cell autophagy correlated with decreased MPNST growth [57, 97]. Inhibition of Aurora kinases using MLN2036 caused prolonged MPNST growth arrest in the G2/M phase of the cell cycle [98]. The combination of histone deacetylase inhibitor PC-24791 (which promotes autophagy) and autophagy blockade with chloroquine abrogated MPNST xenograft growth and promoted cell apoptosis, although the durability of the response is not known [99]. Blocking STAT3 with FLLL32 or shSTAT3 prevented growth of MPNST xenografts but did not arrest growth of established tumors [100]. Whether these xenograft studies will translate to effects in immune-competent models or clinical trials remains to be tested.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


