Fig. 1.1
Genes involved in predisposition to brain tumors.
Cancer predisposing syndromes can be grouped in a variety of ways. Some syndromes will have many clinical manifestations, among which cancer is just a rare feature, while others have cancer as the only clinical manifestation. Other ways to divide these conditions include by pathogenesis or by age of onset. However, we will present the syndromes grouped by the specific tumors they cause, since this will allow clinicians involved in the care of patients with brain tumors to consider the appropriate differential diagnoses based on the specific tumor histology.
This chapter will focus on the most common tumor predisposition syndromes and will elaborate on the genetic background, pathogenesis, and clinical approach to these disorders. Details on additional syndromes are presented in Table 1.1.
Table 1.1
Genetic syndromes not described in this chapter and associated with brain tumors.
Syndrome | Inheritance | Genetic abnormality | Cardinal features | Brain tumors | References |
Aicardi syndrome | X-linked dominant lethal in males | Unknown | callosal agenesis, infantile spasms, chorioretinal lacunae | Choroid plexus papilloma | Frye RE, Polling JS, Ma LC. Choroid plexus papilloma expansion over 7 years in Aicardi syndrome. J Child Neurol. 2007 Apr;22(4):484-7. PubMed PMID: 17621535; PubMed Central PMCID: PMC2536525. |
Distal 22q11.2 deletion syndrome | Autosomal dominant microdeletion usually sporadic | 3.4 Mb deletion of chromosome 22q11.2, distal to the common DiGeorge syndrome (DGS) region encompassing the INI1/SMARCB1 | asymmetric face with preauricular sinuses and skin tags, cognitive disability | teratoid/rhabdoid tumor | Beddow RA, Smith M, Kidd A, Corbett R, Hunter AG. Diagnosis of distal 22q11.2 deletion syndrome in a patient with a teratoid/rhabdoid tumour. Eur J Med Genet. 2011 May-Jun;54(3):295-8. doi: 10.1016/j.ejmg.2010.12.007. Epub 2010 Dec 25. PubMed PMID: 21187175 Lafay-Cousin L, Payne E, Strother D, Chernos J, Chan M, Bernier FP. Goldenhar phenotype in a child with distal 22q11.2 deletion and intracranial atypical teratoid rhabdoid tumor. Am J Med Genet A. 2009 Dec;149A(12):2855-9. doi: 10.1002/ajmg.a.33119. PubMed PMID: 19938088 Jackson EM, Shaikh TH, Gururangan S, Jones MC, Malkin D, Nikkel SM, Zuppan CW, Wainwright LM, Zhang F, Biegel JA. High-density single nucleotide polymorphism array analysis in patients with germline deletions of 22q11.2 and malignant rhabdoid tumor. Hum Genet. 2007 Sep;122(2):117-27. Epub 2007 May 31. PubMed PMID: 17541642 |
Encephalocraniocutaneous lipomatosis | Isolated cases | unknown | Eye choristoma, nonscarring alopecia, nevus psiloliparus, subcutaneous fatty masses, nodular skin tags, and aplastic scalp defects, intracranial and intraspinal lipomas, congenital abnormalities of the meninges, seizures, developmental delays | low-grade glioma | Valera ET, Brassesco MS, Scrideli CA, de Castro Barros MV, Santos AC, Oliveira RS, Machado HR, Tone LG. Are patients with encephalocraniocutaneous lipomatosis at increased risk of developing low-grade gliomas? Childs Nerv Syst. 2012 Jan;28(1):19-22. doi: 10.1007/s00381-011-1601-z. Epub 2011 Oct 8. PubMed PMID: 21983849 |
Schimmelpenning-Feuerstein-Mims syndrome | Somatic mosaicism | HRAS, KRAS | sebaceous nevi, often on the face, associated with variable ipsilateral abnormalities of the central nervous system, ocular anomalies, and skeletal defects, seizures, developmental delays | Optic pathway glioma | Pavlidis E, Cantalupo G, Boria S, Cossu G, Pisani F. Hemimegalencephalic variant of epidermal nevus syndrome: case report and literature review. Eur J Paediatr Neurol. 2012 Jul;16(4):332-42. doi: 10.1016/j.ejpn.2011.12.004. Epub 2011 Dec 24. PubMed PMID: 22200538 |
Hypomelanosis of Ito | Somatic mosaicism | Chromsomal mosaicism | Skin macular hypopigmented whorls, streaks, and patches, developmental delays, seizures | choroid plexus papilloma | Morigaki R, Pooh KH, Shouno K, Taniguchi H, Endo S, Nakagawa Y. Choroid plexus papilloma in a girl with hypomelanosis of Ito. J Neurosurg Pediatr. 2012 Sep;10(3):182-5. doi: 10.3171/2012.5.PEDS11556. Epub 2012 Jul 13. Erratum in: J Neurosurg Pediatr. 2013 Jan;11(1):103. PubMed PMID: 22793165 |
Maffucci syndrome | Somatic mosaicism | IDH1 or IDH2 | multiple central cartilaginous tumors accompanied by soft tissue hemangiomas | Glioma, Pituitary adenoma, meningioma | Moriya K, Kaneko MK, Liu X, Hosaka M, Fujishima F, Sakuma J, Ogasawara S, Watanabe M, Sasahara Y, Kure S, Kato Y. IDH2 and TP53 mutations are correlated with gliomagenesis in a patient with Maffucci syndrome. Cancer Sci. 2014 Mar;105(3):359-62. doi: 10.1111/cas.12337. PubMed PMID: 24344754 |
Pai syndrome | Autosomal dominant | unknown | Median cleft lip, corpus callosum lipoma, skin polyps | Midline central nervous system lipomas | Mishima K, Mori Y, Minami K, Sakuda M, Sugahara T. A case of Pai syndrome. Plast Reconstr Surg. 1999 Jan;103(1):166-70. Review. PubMed PMID: 9915178. |
Mupliple endicrine neoplasia type 1 | Autosomal dominant | MEN1 | tumors of parathyroids, pancreatic islets, duodenal endocrine cells, and the anterior pituitary | Meningioma and spinal cord ependymoma | Rogers L, Barani I, Chamberlain M, Kaley TJ, McDermott M, Raizer J, Schiff D, Weber DC, Wen PY, Vogelbaum MA. Meningiomas: knowledge base, treatment outcomes, and uncertainties. A RANO review. J Neurosurg. 2014 Oct 24:1-20. [Epub ahead of print] PubMed PMID: 25343186 |
Carney complex type 1 | Autosomal dominant | PRKAR1A | cardiac, endocrine, cutaneous, and neural myxomatous tumors, as well as a variety of pigmented lesions of the skin and mucosae | Pituitary adenoma, schwannoma | Gadelha MR, Trivellin G, Hernández Ramírez LC, Korbonits M. Genetics of pituitary adenomas. Front Horm Res. 2013;41:111-40. doi: 10.1159/000345673. Epub 2013 Mar 19. Review. PubMed PMID: 23652674 |
Oral-facial-digital syndrome type VI | Autosomal recessive | C5orf42, TMEM216 | Cerebellar malformations (molar tooth sign, tongue hamartoma, additional tongue frenula, upper lip notch, mesoaxial polydactyly of hands or feet | hypothalamic hamartoma | Lopez E, Thauvin-Robinet C, Reversade B, Khartoufi NE, Devisme L, Holder M, Ansart-Franquet H, Avila M, Lacombe D, Kleinfinger P, Kaori I, Takanashi J, Le Merrer M, Martinovic J, Noël C, Shboul M, Ho L, Güven Y, Razavi F, Burglen L, Gigot N, Darmency-Stamboul V, Thevenon J, Aral B, Kayserili H, Huet F, Lyonnet S, Le Caignec C, Franco B, Rivière JB, Faivre L, Attié-Bitach T. C5orf42 is the major gene responsible for OFD syndrome type VI. Hum Genet. 2014 Mar;133(3):367-77. doi: 10.1007/s00439-013-1385-1. Epub 2013 Nov 1. PubMed PMID: 24178751 |
Pallister-Hall syndrome | Autosomal dominant usually sporadic | GLI3 | central polydactyly, anorectal malformations | hypothalamic hamartoma | Kang S, Graham JM Jr, Olney AH, Biesecker LG. GLI3 frameshift mutations cause autosomal dominant Pallister-Hall syndrome. Nat Genet. 1997 Mar;15(3):266-8. PubMed PMID: 9054938 |
Proteus syndrome | Somatic mosaicism | AKT1 | disproportionate, asymmetric, and distorting overgrowth, bone abnormalities; characteristic cerebriform connective tissue nevus; epidermal nevi; vascular malformations of the capillary, venous, or lymphatic types; dysregulated adipose tissue; bullous lung alterations; intellectual disability; seizures; brain malformations | Meningioma | Cohen MM Jr. Proteus syndrome review: molecular, clinical, and pathologic features. Clin Genet. 2014 Feb;85(2):111-9. doi: 10.1111/cge.12266. Epub 2013 Oct 23. PubMed PMID: 23992099 |
Rubinstein-Taybi syndrome | Autosomal dominant usually sporadic | CREBBP | mental retardation, postnatal growth deficiency, microcephaly, broad thumbs and halluces, dysmorphic facial features | Meduloblastoma, meningioma | Roelfsema JH, Peters DJ. Rubinstein-Taybi syndrome: clinical and molecular overview. Expert Rev Mol Med. 2007 Aug 20;9(23):1-16. Review. PubMed PMID: 17942008 |
Short-rib thoracic dysplasia 12 | Autosomal recessive | unknown | constricted thoracic cage, short ribs, shortened tubular bones, ‘trident’ appearance of the acetabular roof | Hypothalamic hamartoma | den Hollander NS, van der Harten HJ, Laudy JA, van de Weg P, Wladimiroff JW. Early transvaginal ultrasonographic diagnosis of Beemer-Langer dysplasia: a report of two cases. Ultrasound Obstet Gynecol. 1998 Apr;11(4):298-302. PubMed PMID: 9618859 |
Wiskott-Aldrich syndrome | X-linked recessive | WAS | Immunodeficiency, thrombocytopenia, eczema, and recurrent infections | Primary reticulum cell sarcoma of the brain | Model LM. Primary reticulum cell sarcoma of the brain in Wiskott-Aldrich syndrome. Report of a case. Arch Neurol. 1977 Oct;34(10):633-5. PubMed PMID: 334130 |
Xeroderma pigmentosum | Autosomal recessive | XPA, ERCC1, ERCC3 (XP-B), XPC, ERCC2 (XP-D), DDB2 (XP-E), ERCC4 (XP-F), ERCC5 (XP-G), POLH (XPV) | severe sunburn with blistering, persistent erythema on minimal sun exposure, marked freckle-like pigmentation of the face, (photophobia, keratitis, atrophy of the skin of the eyelids, increased risk of cutaneous neoplasms | astrocytoma, medulloblastoma, schwannomas | Rapin I, Lindenbaum Y, Dickson DW, Kraemer KH, Robbins JH. Cockayne syndrome and xeroderma pigmentosum. Neurology. 2000 Nov 28;55(10):1442-9. Review. PubMed PMID: 11185579 |
Syndromes Associated with Glioma
Gliomas are by far the most common group of brain tumors associated with cancer predisposition syndromes. These syndromes should always be considered if an index patient presents with glioma. Each syndrome has unique features regarding clinical manifestations and personal or family history.
Neurofibromatosis Type I (NF1, von Recklinghausen’s Disease)
NF1 is by far the most common CNS tumor predisposition syndrome. It is an autosomal dominant condition with a worldwide incidence of 1 per 2,500–3,000 individuals [6]. Importantly, this is a multisystem condition and diagnosis is generally made based on clinical criteria [6–8]. The following criteria are sensitive and specific in adults but affected children may not fulfil the criteria and genetic testing may aid the diagnosis [8]:
1.
Six or more café-au-lait macules over 5 mm in greatest diameter in prepubertal individuals and over 15 mm in greatest diameter in postpubertal individuals
2.
Two or more neurofibromas of any type or one plexiform neurofibroma
3.
Freckling in the axillary or inguinal regions
4.
Optic glioma
5.
Two or more Lisch nodules (iris hamartomas)
6.
A distinctive osseous lesion such as sphenoid dysplasia or tibial pseudarthrosis
7.
A first-degree relative (parent, sib, or offspring) with NF1 as defined by the above criteria
Individuals with NF1 can have significant morbidity unrelated to cancer predisposition [9]. The nervous system is commonly affected in NF1, and most cancers are of nervous system origin including gliomas, benign neurofibromas, and malignant nerve sheath tumors (MPNSTs). However, other cancers including chronic myelomonocytic leukemia, breast cancer, certain endocrine tumors, rhabdomyosarcoma, and neuroblastoma are reported with this condition [10].
Molecular Pathogenesis
NF1 results in loss of function of the tumor suppressor protein Neurofibromin. This large protein is a key negative regulator of the RAS pathway by catalyzing the hydrolysis of active guanosine triphosphate-bound RAS to inactive guanosine diphosphate-bound RAS [11]. Dysfunctional neurofibromin results in constitutive activation of downstream oncogenic pathways including MAPK and mTOR. Mutations or deletions in the NF1, gene can be identified in more than 95 % of individuals with NF1 [13]. However, since the gene is very large and difficult to analyze, diagnosis and management can be made based on clinical criteria. RAS/MAPK pathway activation is seen in almost all pediatric low-grade astrocytomas [13] and in 88 % of adult malignant gliomas [14]. Actual somatic mutations in NF1 occur in 20 % of adult gliomas.
Mouse models of gliomas frequently alter the RAS/MAPK pathway. However, additional alterations in major tumor suppressor pathways such as TP53, RB, and PTEN are required to generate tumors [15].
Indeed, NF1 deficient mice do not have tumors but hyperplasia mimicking the optic pathway gliomas (OPG) commonly seen in these individuals [16]. Taken together, the benign nature of tumors seen in the CNS in patients with NF1 support the concept of oncogene induced senescence as a mechanism to explain the spontaneous growth arrest of these tumors when the RAS pathway is constitutively active [17].
Gliomas
The most common CNS tumor in NF1 are optic pathway gliomas (OPG) affecting up to 15 % of individuals with the syndrome. Conversely, up to a third of children with OPG have germline mutations in NF1. Bilateral optic nerve gliomas exist almost exclusively in children with NF1 (Fig. 1.2).
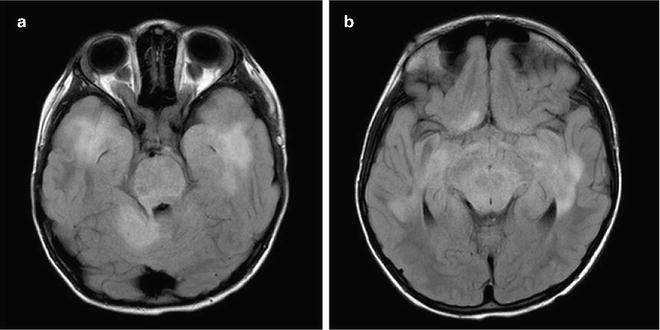
Fig. 1.2
Pathognomonic MRI findings in NF1. Bilateral optic nerve glioma (a) and FASCI (b) are almost exclusively seen only in children with NF1.
NF1 related OPG typically have an indolent course with spontaneous growth arrest. Indeed, the vast majority of these OPG will not progress after initial diagnosis. Up to 15 % of these tumors, however, do progress, resulting in visual loss or other symptoms and requiring intervention. High-grade gliomas are relatively uncommon, but have been reported and should be considered in patients whose tumors arise in an uncharacteristic location or demonstrate particularly aggressive behavior [18, 19].
Patients with NF1 often exhibit multiple lesions, mainly in the basal ganglia and brainstem which are difficult to assess. These include T2 bright lesions on MRI, without significant mass effect, termed FLAIR (fluid attenuated inversion recovery) associated sub-cortical intensities or FASCI (Fig. 1.2). These lesions tend to disappear spontaneously after initial growth and rarely cause symptoms. Differentiating between FASCI and low-grade gliomas in NF1 patients may be challenging.
Clinical Implications
Since NF1 is a multisystem condition, careful monitoring is recommended in multidisciplinary clinics [6]. Due to the marked variability in clinical manifestations, including tumor occurrence, strategies for surveillance and follow-up must be tailored to each individual patient. Optic gliomas affecting both optic nerves and/or coexistence of FASCI should raise a suspicion of NF1 even in the absence of typical neurocutaneous findings such as café-au-lait macules, and genetic counseling is recommended.
Unfortunately, surveillance neuroimaging in asymptomatic children with NF1 has not been shown to reduce the incidence of visual loss in this population, and frequent neuro-ophthalmologic examination remains standard of care [20]. For FASCI and other atypical brain lesions, close monitoring is recommended and treatment should be reserved for patients with progressive clinical symptoms and in some cases, radiological progression.
Individuals with NF1 are particularly sensitive to the damaging effects of ionizing irradiation, leading both to an increased incidence of irradiation-induced cancers [21], as well as to cerebrovascular damage (Moyamoya syndrome) [22, 23]. Cranial irradiation in NF1 patients with OPG in particular and brain tumors in general, should be avoided as long as reasonable alternative treatment options exist.
Molecular Targeted Therapies
Inhibitors targeting the RAS and mTOR pathways are of great interest for the potential treatment of NF1 and NF1-related tumors, and are being investigated as novel therapeutic approaches in preclinical and clinical studies. Furthermore, targeting the microenvironment believed to be necessary for NF1 tumor growth may allow for additional NF1 specific therapies for these patients [24, 25]. For example, inhibition of c-KIT has shown encouraging efficacy for peripheral NF1 related neurofibromas in subsets of patients [26, 27], although it remains to be seen whether c-kit represents a molecular target of value in NF1 related CNS tumors.
Li–Fraumeni Syndrome
The Li–Fraumeni syndrome (LFS) is the prototype cancer predisposition syndrome, causing cancer in multiple sites at different ages. LFS is an autosomal dominant condition affecting 1 in 5,000–10,000. Individuals with the disorder have a lifetime risk of 85–100 % of developing cancer. Originally described by Frederick Pei Li and Joseph F. Fraumeni, Jr. in 1969 [28] as a familial breast, soft tissue sarcoma and brain tumor predisposition syndrome, it is now known that these individuals have a risk of developing cancer in many additional organs, including rare tumors such as adrenocortical carcinomas, as well as hematologic malignancies [29].
According to Li et al. 1988 [30], who described the syndrome, the diagnosis is clinically established in families with a proband with a sarcoma diagnosed before age 45 years and a first-degree relative with any cancer before age 45 years and a first- or second-degree relative with any cancer before age 45 years or a sarcoma at any age. The association of LFS with germline mutations in the TP53 prompted the formation of criteria to enhance the yield of TP53 clinical genetic testing. The following criteria were published by Chompret et al. [31] and revised and evaluated by Gonzalez et al. [32], Tinat et al. [33], and Ruijs et al. [34] According to these studies the risk of a TP53 mutation exceeds 20 % in any individual with:
1.
A tumor belonging to the LFS tumor spectrum (e.g., soft tissue sarcoma, osteosarcoma, brain tumor, pre-menopausal breast cancer, adrenocortical carcinoma, leukemia, lung bronchoalveolar cancer) before age 46 years and at least one first- or second-degree relative with a LFS tumor (except breast cancer if the proband has breast cancer) before age 56 years or with multiple tumors; or
2.
Multiple tumors (except multiple breast tumors), two of which belong to the LFS tumor spectrum and the first of which occurred before age 46 years; or
3.
Adrenocortical carcinoma or choroid plexus tumor, regardless of family history.
Molecular Pathogenesis
In 1990 the association between LFS and germline mutations in the tumor suppressor gene TP53 was made [35]. TP53 is located at chromosome 17p13.1 and has been called the “gatekeeper of the genome,” since it represents one of the key proteins that maintain genome integrity after DNA damage, hypoxia, and other stressors. TP53 activation results in cell cycle arrest, senescence, and apoptosis. TP53 is also involved in key metabolic pathways in the cell including cell metabolism and mitochondrial function [36]. TP53 represents one of the most commonly mutated tumor suppressors known. Molecular genetic evidence of TP53 pathway disruption can be found in more than 50 % of tumors from adult cancer patients, as well as in 80 % of adult [13] and 50 % of pediatric high-grade gliomas [37].
Development of faithful preclinical models of LFS has been hindered by the fact that TP53 alteration in animal models generally fails to recapitulate the tumor phenotypes of the human disorder, and brain tumors are not a part of the phenotype even in some of the newer models [38]. Nevertheless, many glioma and medulloblastoma mouse models utilize TP53 alterations in conjunction with other cancer genes to mimic the human disease [39].
Three types of brain tumors are associated with LFS: high-grade gliomas, choroid plexus carcinoma, and medulloblastoma. Choroid plexus tumors affect LFS carriers in the first decade of life and medulloblastomas usually in the second, while malignant gliomas can occur throughout childhood, but more commonly in young adults.
Gliomas have been recognized as part of LFS from the earliest reports [28]. Although TP53 expression is associated with worse outcome in childhood glioblastoma [40], currently, no data exist regarding the significance of germline TP53 mutations in pediatric high-grade gliomas. The surveillance protocol [41] developed by our group uncovered several low-grade gliomas suggesting that some of LFS associated glioblastomas arise as secondary glioblastomas and may benefit from early intervention.
Choroid plexus carcinomas are one of the most common presentations of LFS in young children and were recently added to the criteria for the diagnosis of the syndrome [33]. Furthermore, a significant number of patients with choroid plexus carcinoma will harbor germline TP53 mutations. Somatic mutations in TP53 are observed in up to 50 % of choroid plexus carcinomas, and this confers a poorer chance of survival for these patients [2]. This phenomenon may be caused by increased resistance of TP53 mutant tumors to radiation and chemotherapy [42].
Medulloblastomas harbor somatic TP53 mutations in 5–10 %, and appear strictly confined to the WNT (wingless-related integration site) and SHH (sonic hedgehog) subgroups of tumors [43]. Remarkably, TP53 mutations do not alter the excellent survival of patients with WNT medulloblastomas, while TP53 mutant SHH medulloblastomas are commonly seen in the second decade of life and have unfavorable outcome. Interestingly, these are commonly individuals with LFS. SHH medulloblastomas from LFS patients have a unique molecular genetic profile, suggesting chromothripsis (“chromosome shattering”) as the initiating event [44].
Clinical Implications
Current recommendation is to screen for germline TP53 mutations, i.e., LFS, in all individuals presenting either with a high-grade glioma and a family history of LFS tumors, or patients diagnosed with a choroid plexus carcinoma or medulloblastoma harboring somatic TP53 mutations [33]. Cancer surveillance protocols developed specifically for individuals with LFS have revealed a high rate of early tumor detection [45]. Recently, a striking survival benefit for children has been observed using these protocols, mainly due to improved early detection of brain tumors (Fig. 1.3). Although no molecular targeted therapy for TP53 mutated tumors is currently available, detection of a germline TP53 mutation has significant prognostic and therapeutic implications for the patient. Both children and adults with LFS have been considered to be at an increased risk for developing radiation therapy-induced secondary malignant tumors [46, 47], as well as secondary myelodysplastic syndrome following specific chemotherapies [48].
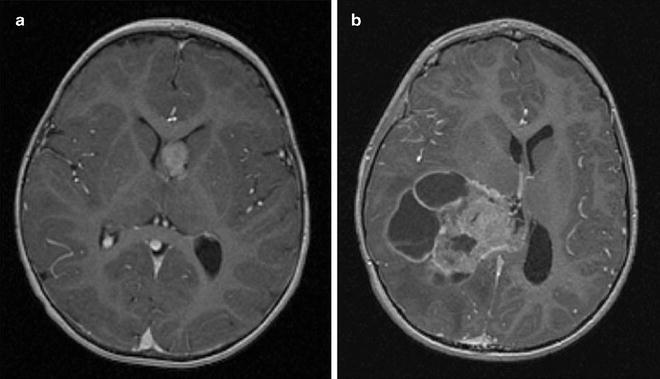
Fig. 1.3
Early detection of CPC after implementation of the surveillance protocol for LFS. Early detection of asymptomatic choroid plexus carcinoma in an LFS patient undergoing a surveillance protocol (a). A tumor from a patient with sporadic symptomatic choroid plexus carcinoma (b). The LFS patient underwent complete tumor resection followed by chemotherapy and is alive 7 years later. The patient with sporadic tumor did not survive despite radiation therapy and multiple courses of chemotherapy.
Constitutional Mismatch Repair Deficiency Syndrome
Constitutional Mismatch Repair Deficiency Syndrome (CMMR-D) is a rare familial cancer predisposition syndrome that has a unique clinical phenotype. This syndrome frequently presents with cafe-au-lait macules like NF1 [49, 50], resulting in occasional misdiagnosis and inappropriate management. CMMR-D is due to germline biallelic (homozygous or compound heterozygous) mutations in one of the mismatch repair (MMR) genes. Germline monoallelic mutations in MMR genes cause hereditary non-polyposis colon cancer, Lynch syndrome and brain tumor-polyposis syndrome type 1 (BTPS1 or Turcot type 1) [51, 52]. The brain tumor in BTPS1 is glioblastoma multiforme.
Individuals with CMMR-D are predisposed to different and more aggressive cancers than Lynch syndrome. Children with CMMR-D are usually affected within the first two decades of life and present with hematological malignancies (most commonly T-cell lymphomas), malignant brain tumors, and gastrointestinal cancers.
Molecular Pathogenesis
Germline mutations in MLH1, MSH2, MSH6, and PMS2 have been reported in association with CMMR-D. These mismatch repair genes are critical in repairing single base pair mismatches and misalignments [49]. In the absence of such genes, high mutation rates are observed, including in cancers which are described as “mutator phenotype” [14]. CMMR-D is inherited in an autosomal recessive fashion and is found mostly in consanguineous families. Some patients may have NF1 in addition to CMMR-D, which is thought to be caused by “secondary” early or germline mutations in the NF1 gene as a part of the mutator phenotype [53].
Brain Tumors
Malignant gliomas are the most common type of tumor observed in individuals with CMMR-D, usually presenting in the second decade of life. Some patients are initially diagnosed with low-grade gliomas, but these tend to transform to high-grade tumors. Medulloblastomas and PNETs (primitive neuroectodermal tumors) are also seen, but may have glial markers suggesting an earlier cell of origin. Some of these patients with high-grade gliomas have been reported as long-term survivors, possibly suggesting a somewhat more favorable prognosis compared to other adults and children with high-grade gliomas [56, 57].
Clinical Implications
Diagnosis of Lynch-related cancers can be made by evidence of microsatellite instability [58]. However, this method has not been shown to be sensitive in CMMR-D, especially in brain tumors and lymphomas. Immunostain of tumor tissue for the MMR proteins is almost universally negative in CMMR-D cancers, and has the unique diagnostic feature of negative stain in the corresponding normal tissue.
Any patient with gliomas, T-cell lymphoma and either café-au-lait macules, consanguinity or a family history of colon cancer should be screened for any of the four mismatch repair genes. Similarly, high index of suspicion should be raised for “NF1” patients with malignant gliomas and consanguinity.
Individuals with Lynch syndrome benefit from a strict surveillance protocol (http://www.NCCN.org/) and from preventive colectomy. Therefore, early and accurate diagnosis may benefit parents and other family members. Since the risk of gliomas and lymphoma is extremely high for biallelic MMR patients, following a surveillance protocol may be beneficial for children with CMMR-D [59]. There are several reports of MMR tumors responding to specific agents including retinoic acid [60], which may be exploited for the treatment of these tumors.
Melanoma Astrocytoma Syndrome
In the mid-1990s, a syndrome of melanoma and other skin lesions associated with CNS malignancies was first described [61]. Since then, several other reports have delineated the association between familial melanoma and glioma. A common locus on the short arm of chromosome 9 was uncovered, and germline mutations were described in CDKN2A which codes for two proteins: p16(INK4) and p14(ARF) [62, 63]. Additional mutations in another gene in that location, PTPRD, were reported [64]. ARF and Ink4A are major tumor suppressors in the TP53 and RB1 pathways respectively, and are altered in the majority of sporadic gliomas [14]. Further data, however, will be needed before specific screening and surveillance recommendations can be developed.
Syndromes Associated with Medulloblastoma
Medulloblastoma is the most common malignant brain tumor in children, but is also seen in adults. Several syndromes were first described to be associated with childhood medulloblastoma, but further data supports involvement of some of these syndromes in adult tumors as well [43]. We summarize here the most common syndromes, while others are described in Table 1.1.
Gorlin Syndrome (Basal Cell Nevus Syndrome)
Basal cell nevus syndrome (BCNS) is an autosomal dominant condition associated with multiple developmental anomalies and predisposition to benign and malignant tumors. The hallmark of BCNS is development of basal cell carcinomas and medulloblastomas. The association of multiple nevoid basal cell “epithelioma,” jaw cysts and bifid ribs was first reported in 1960 [65].
Evans et al. [66] and Kimonis et al. [67] have published criteria for clinical diagnosis. Two major criteria and one minor or one major and three minor criteria are diagnostic of Gorlin syndrome. Some criteria require X-rays. Exposure to X-rays increases the risk for basal cell carcinoma and should be avoided.
Major Criteria
1.
Falx calcification ascertained by AP skull X-rays.
2.
Jaw keratocysts seen as translucencies on orthopantogram X-rays.
3.
Two or more palmar/plantar pits.
4.
Basal cell carcinoma before age 30 or multiple after age 30.
5.
A first-degree relative with Gorlin syndrome.
Minor Criteria
1.
Childhood medulloblastoma (primitive neuroectodermal tumor [PNET]).
2.
Lympho-mesenteric or pleural cysts.
3.
Macrocephaly (OFC >97th centile).
4.
Cleft lip/palate.
5.
Vertebral/rib anomalies (bifid vertebra), bifid/splayed/extra ribs ascertained by X-rays.
6.
Preaxial or postaxial polydactyly.
7.
Ovarian/cardiac fibromas.
8.
Ocular anomalies (cataract, developmental defects, and pigmentary changes of the retinal epithelium).
Molecular Pathogenesis
The gene responsible for BCNS is PTCH1 which is located on chromosome 9q22.3 [68]. PTCH1 is a protein that is a major suppressor of the sonic hedgehog (SHH) pathway by direct inhibition of SMO. Disruption of PTCH1 leads to constitutive activation of the pathway and induction of GLI target genes and cell proliferation and survival. SHH is involved in neural development and midline segregation, which can explain some of the syndromatic manifestations of BCNS.Germline mutations in Sufu, which is a direct inhibitor of GLI have been reported in familial and sporadic medulloblastoma [69] in up to 50 % of desmoplastic tumors [70].
Most mouse models of medulloblastoma utilize alterations in the SHH pathway [71]. While alteration of PTCH1 only results in a tumor incidence of 10–20 %, its combination with other alterations results in very efficient formation of aggressive tumors.
Sequence analysis of PTCH1 yields a mutation in 50–80 % of patients with Gorlin syndrome [72, 73]. Partial and whole-gene deletions are found in 6–21 % of patients [74]. In patients with mental retardation chromosome analysis and chromosomal microarray may reveal the 9q22.3 microdeletion. The 9q22.3 microdeletion syndrome is associated with cognitive impairment, metopic synostosis, obstructive hydrocephalus, macrosomia and seizures, in addition to the features of Gorlin syndrome [75].
Medulloblastoma
The first report of a brain tumor, medulloblastoma, in association with this hereditable syndrome [76] was published in 1963. The development of medulloblastoma in the setting of Gorlin syndrome occurs earlier compared to sporadic tumors. Most patients are younger than 3 years of age, and almost all tumors have a specific pathological subtype termed desmoplastic variant [77]. More specifically, desmoplasia with extensive nodularity (MBEN) is almost pathognomonic for the syndrome in young children [78, 79]. Of note, desmoplastic medulloblastomas in young children usually have a favorable outcome even without radiation therapy [80].
Meningioma
Several reports have documented the development of intracranial meningiomas in patients with Gorlin syndrome with or without prior craniospinal irradiation [81], although strength of association remains unknown.
Clinical Implications
Individuals with the clinical manifestations of BCNS, a family history of basal cell carcinomas, or other manifestations of the syndrome should be screened for germline mutations in PTCH1 and SUFU.
Patients with medulloblastoma and any of the above should also be screened, since radiation therapy is associated with an increased risk of developing basal cell carcinomas within the irradiated fields [82] in almost all patients. Furthermore, since the rate of germline mutations in the SHH pathway is extremely high in young children with desmoplastic medulloblastoma (Fig. 1.4) [70], children less than 3 years of age with desmoplastic tumors should be screened even without clinical manifestations of the syndrome. The current consensus recommends yearly brain MRI scans for all patients with BCNS until the age of 8 years [83].
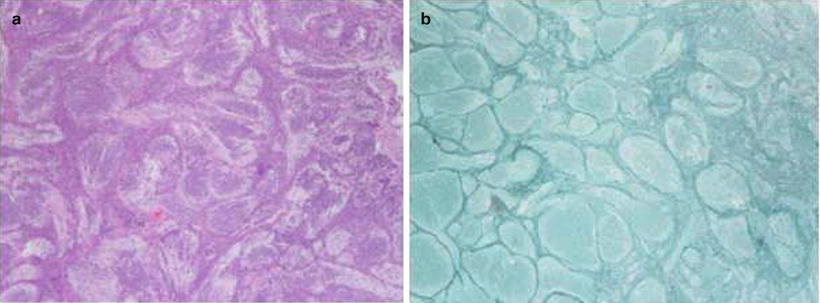
Fig. 1.4
Desmoplastic medulloblastoma. (a) Pale areas surrounded by densely packed hyperchromatic cells. (b) Same tumor reveals fine reticular areas with islands lacking reticulin
The recent development of novel SHH pathway inhibitors [84] may lead to future targeted therapies for individuals with both Gorlin and SUFU syndromes, possibly including primary tumor prevention strategies.
Brain Tumor-Polyposis Syndrome 2 (BTPS 2 or Turcot Type 2; Familial Adenomatosis Coli)
Familial adenomatous polyposis (FAP) is an autosomal dominant cancer predisposition syndrome. Although the hallmark of this syndrome is the development of multiple gastrointestinal tract polyps and subsequent cancers, children with FAP are also at risk of developing medulloblastoma, hepatoblastoma, and aggressive fibromatoses.
The association between colorectal carcinoma and medulloblastoma was first reported in 1949 [85] and medulloblastoma is the only tumor observed in children with this syndrome. These patients and/or their family members display numerous (>100) small colonic polyps with later onset of malignant transformation to adenocarcinoma later in life. The pattern of inheritance here, distinct from BPTS type 1 (see glioma syndromes), is autosomal dominant and has been shown to be due to a heterozygous germline mutation in the adenomatous polyposis coli (APC) gene. Although the risk of developing medulloblastoma in patients with FAP is estimated 92-fold that of the general population [86], it is still a rare phenomenon among carriers.
Molecular Pathogenesis
FAP is caused by germline mutations in the gene adenomatosis polyposis coli (APC) [87]. APC is located on chromosome 5q21-22 and is a major regulator of the WNT pathway, which plays a paramount role in controlling embryonic development, stem cell viability and proliferation. Hyperactivation of the WNT pathway is reported in 5–10 % of medulloblastomas, usually as a result of mutations in CTNNB1 [88, 89]. APC mutations are rare in sporadic medulloblastoma. Interestingly, no current mouse models exist for APC-driven medulloblastoma.
Clinical Implications
Since medulloblastoma is rare in FAP, carriers are not routinely screened for these tumors. However, a patient with medulloblastoma and FAP should undergo GI cancer surveillance, since the development of medulloblastoma in PBTS 2 may precede the development of colonic adenocarcinoma. Indeed, patients are reported with simultaneous diagnoses of medulloblastoma and colonic adenocarcinoma. It is important to note that APC mutated medulloblastomas are distinct from most WNT activated medulloblastomas. WNT tumors will have CTNNB1 mutations that can be diagnosed by nuclear staining of the gene product. Although WNT pathway activation generally confers favorable survival in sporadic medulloblastomas, the prognosis of APC mutated tumors is still uncertain, and FAP patients therefore should not be treated with less aggressive protocols.
Fanconi Anemia Cancer Predisposition Disorders
Fanconi anemia is a cancer predisposition syndrome with bone marrow failure and characteristic malformations. The malformations are seen in about 60 % of affected individuals and include low birth weight, short stature, pigmentary abnormalities of the skin, abnormal thumbs, and hypoplastic radii [90]. There is genetic variability in Fanconi anemia (FA): patients may have mutations in one out of 15 known genes that cause the syndrome. One of the genes, FANCB, is associated with X-linked recessive inheritance and the rest with autosomal recessive [90].
The following features of FA suggest the clinical diagnosis and genetic testing [91]:
1.
Characteristic congenital malformations as well as growth and developmental delays
2.
Bone marrow failure in childhood which is usually progressive
3.
Aplastic anemia in adults
4.
Unexpected bone marrow failure after chemotherapy or radiation
5.
Myelodysplastic syndrome or acute myelogenous leukemia
6.
Solid tumors in young age including squamous cell carcinomas of the head and neck, esophagus, and vulva
If Fanconi anemia (FA) is clinically suspected the next step is chromosome breakage studies using diepoxybutane or mitomycin C as clastogenic agent [92]. If increased chromosome breakage is ascertained then DNA sequencing and deletion/duplication tests are available for all the known genes: FNCA, FANCB, FANCC, BRCA2, FANCD2, FANCE, FANCF, FANCG, FANCI, BRIP1, FANCL, FANCM, PALB2, RAD51C, and SLX4 [93]. If the patient is Ashkenazi Jewish with no history of carrier testing in the patient or the parents then testing for the FANCC mutation (c.456 + 4A > C) should be the first step [94].
Bone marrow failure states and/or myeloid dysplasias or leukemias (median age for onset is 14 years) develop in most individuals with FA [91]. In addition, a variety of solid tumors have long been recognized to develop with increasing frequency, particularly liver adenomas (in association with prior androgenic steroid use for the bone marrow failure) [90], and gastrointestinal and gynecological carcinomas, with a median age at diagnosis of about 29 years [95]. The median age for onset of the leukemias is 14 years. It has been estimated that, by theoretically removing the competing risks of marrow failure and leukemias, individuals with FA have an estimated cumulative probability of developing a solid tumor of 76 % by the age of 45 years [96].
Although brain tumors in FA patients were reported in the past [97, 98], the involvement of medulloblastoma [99] and glioma [100] in specific germline mutations has not been suggested until recently.
Molecular Pathogenesis
FA is a genetically heterogeneous disorder associated with either biallelic mutations in any of the known 14 autosomal genes or a mutation in an X-linked gene [101]. Individuals in the FA complementation group FANCD1 are estimated to represent no more than 3 % of all individuals with FA, and it is this group in whom biallelic mutations with BRCA2 are found. BRCA2 mutations are well known to be associated with familial predisposition to breast and ovarian cancer. Brain tumors have also been reported in such families [102]. These individuals may present a more severe phenotype with early onset of cancer. In particular, the cumulative probability of developing a brain tumor (almost always medulloblastoma) could be high as 85 % in the first decade [99]. This knowledge has prompted a search for other genes in the pathway and familial childhood cancers. Recently, germline mutations in PALB2, another gene in the Fanconi pathway, were reported to be associated with medulloblastomas and other pediatric cancers [103].
Clinical Implications
The rare individuals who develop medulloblastoma in the setting of FA do so at a very early age, often before a diagnosis of FA has been made. Individuals with FA undergoing treatment for cancer are known to be highly sensitive to both irradiation and chemotherapy, with increased susceptibility for treatment-associated toxicities, especially from alkylator-based chemotherapy [106]. Thus, any early onset pediatric brain tumor with cutaneous, skeletal, or neurological abnormalities consistent with a diagnosis of FA or in case of severe unexpected toxicity from chemotherapy, genetic counseling is recommended.
The concept of synthetic lethality is being exploited in the use of PARP inhibitors in BRCA1/2 deficient breast, ovarian, and pancreatic cancers [107, 108], which could be of value in FA as well. To date, however, no data exist on the use of PARP inhibitors in the treatment of FA-related medulloblastoma.
Meningioma
Meningiomas are slow growing CNS lesions accounting for roughly a third of CNS tumors in adults [109]. While most of these tumors are sporadic or occur as a result of prior radiation therapy, familial cases of meningiomas are well reported. The most common genetic syndrome associated with meningiomas is neurofibromatosis type 2 (NF2). This syndrome has additional clinical features, and will be discussed in the section of tumor specific syndromes. However, several kindreds with familial meningioma lack linkage to the NF2 locus [110] are diagnosed with other known syndromes such as Li–Fraumeni, Cowden, Gorlin, and multiple endocrine neoplasia (MEN) [111]. Recently, genetic analysis of familial meningiomas uncovered involvement of the SWI/SNF family members SMARCB1 and SMARCE1 in several families diagnosed with schwannomatosis [112] with meningiomatosis [113]. Furthermore, as described above, patients with Gorlin syndrome including SUFU mutations [114], carry an increased risk of meningiomas.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


