SAR and QSAR in Drug Discovery and Chemical Design—Some Examples
Different in silico molecular modeling techniques innovate the process of developing new chemical entities with desired properties of interest. Since there are various possible reasons for failure while pursuing costly research paradigms like drug discovery, the implementation of rational strategies becomes inevitable to enhance efficiency. In this chapter, we present examples of drugs and other chemicals that have been designed using quantitative structure–activity relationship (QSAR) and other related in silico strategies.
Keywords
Lead optimization; clinical trial; approved drug; in silico success
11.1 Introduction
The design and development of new chemicals is a challenging task. The challenge becomes tougher while dealing with molecules of biological importance (e.g., drugs and pharmaceuticals). The act of developing new drug molecules, as well as modifying the existing ones, involves a multifaceted objective covering the aspects of desired pharmacological activity, undesirable side reactions, appreciable pharmacokinetic features, and suitable form of administration. Although it may be argued that many useful drugs, like acetylsalicylic acid, quinine, and penicillin, have come through serendipitous discovery as well as classical research, it should be considered that many other drug candidates have failed or been withdrawn due to undesirable outcomes. The major objective of the pharmaceutical and other chemical industries is to develop medicines (or other chemicals) that are suitably valued by regulatory authorities, patients, healthcare professionals and providers, and others to improve the quality of life of consumers. However, considering the existing economic conditions, the industry attempts to provide the best product possible at a suitable cost and time frame in order to cope with the market. Therefore, the criteria about adjustments comprise the quality, speed, and time parameters to make a product economically viable. However, it is definitely not possible to compromise with the quality of medicinal agents that we consume; hence, the need for less time-consuming, alternative, accurate, and economic methods becomes inevitable. Considering the huge cost incurred during the discovery of a drug molecule that comprises steps like initial synthesis and preliminary screening of biological activity, preclinical development (studies on animals for pharmacological effect and short- and long-term toxicities), phase I clinical trials (studies on healthy human volunteers), phase II clinical trials (studies on limited cohort of human volunteers with the specific disease), phase III clinical trials (studies on larger populations of affected human volunteers), phase IV clinical trials (post-marketing surveillance), and post-developmental quality control and quality assurance operations in order to get final approval for release, classical and empirical techniques seem impracticable. Hence, rational drug-design approaches are highly useful in providing a theoretical basis on the features of an investigational molecule addressing almost all possible aspects of its failure. Various in silico molecular modeling studies, including the quantitative structure–activity relationship (QSAR) methodology, enable us to gather sufficient information regarding the response of a chemical by comparing it with several other similar and different molecules, thereby allowing us to choose the one with optimized features. Furthermore, the use of different in silico methodologies also provides a good option for reducing animal experimentation, which involves ethical obligations. In this chapter, we present several success stories of representative approved and investigational drugs and other chemicals that have been designed and developed employing QSAR and related in silico molecular modeling operations.
11.2 Successful Applications of QSAR and Other In Silico Methods: Representative Examples
The research and development in the realm of QSAR and related molecular modeling techniques have traversed a long journey from which several successful outcomes have been obtained. We have attempted to present here a few success stories of computational methods in deriving some drugs and other chemicals [1–7] in Tables 11.1 and 11.2, respectively.
Table 11.1
| Sl. No. | Name of the drug agent | Chemical structure | Indication | Mechanism | US FDA approval/clinical trial status | Marketed by | Product | Trade name |
| Approved drugs | ||||||||
| 1 | Saquinavir | 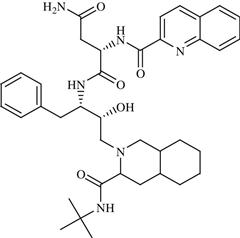 | AIDS | Inhibitor of HIV-1 protease enzyme | 1995 | Hoffmann-La Roche | Saquinavir mesylate | Invirase® |
| 2 | Amprenavir | 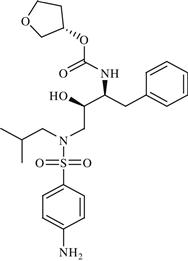 | AIDS | Inhibition of HIV protease enzyme | 1999 | GlaxoSmithKline | Amprenavir | Agenerase |
| 3 | Indinavir | 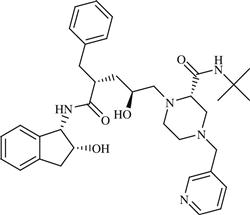 | AIDS | Inhibition of HIV protease enzyme | 1996 | Merck Sharp Dohme | Indinavir sulfate | Crixivan® |
| 4 | Lopinavir | 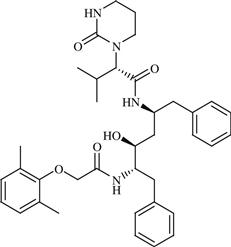 | AIDS | Inhibition of HIV protease enzyme | 2000 | Abott | Lopinavir/Ritonavir | Kaletra® |
| 5 | Nelfinavir | 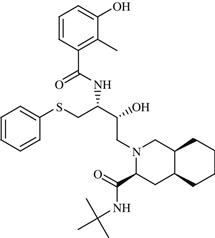 | AIDS | Inhibition of HIV protease enzyme | 1997 | Agouron | Nelfinavir mesylate | Viracept® |
| 6 | Raltegravir | 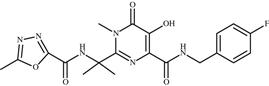 | AIDS | Inhibition of HIV-1 integrase enzyme | 2007 | Merck Sharp Dohme | Raltegravir potassium | Isentress |
| 7 | Ritonavir | 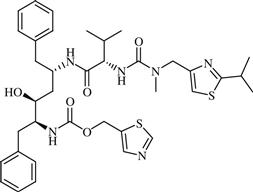 | AIDS | Inhibition of HIV protease enzyme | 1996 | Abott | Ritonavir | Norvir® |
| 8 | Dorzolamide | 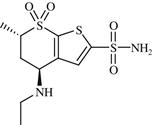 | Open-angle glaucoma and ocular hypertension | Inhibition of carbonic anhydrase II enzyme | 1994 | Merck | Dorzolamide hydrochloride | Trusopt® |
| 9 | Norfloxacin | 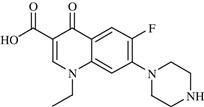 | Bacterial infection | Inhibition of bacterial DNA gyrase | 1986 | Merck | Norfloxacin | Noroxin® |
| 10 | Oseltamivir | 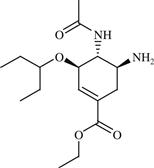 | Influenza | Inhibition of NA enzyme | 1999 | Hoffmann-La Roche | Oseltamivir phosphate | Tamiflu® |
| 11 | Zanamivir | 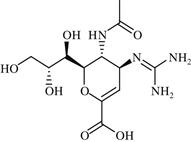 | Influenza | Inhibition of NA enzyme | 1999 | GlaxoSmithKline | Zanamivir | Relenza® |
| 12 | Boceprevir | 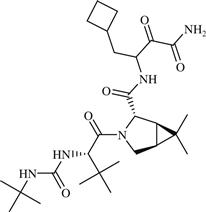 | Hepatitis C | Inhibition of NS3-NS4A serine protease of HCV | 2011 | Merck Sharp Dohme | Boceprevir | Victrelis® |
| 13 | Captopril | 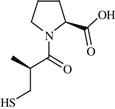 | Hypertension | Potent and reversible inhibition of ACE | 1981 | Bristol Myers-Squibb | Captopril | Capoten® |
| 14 | Aliskiren | 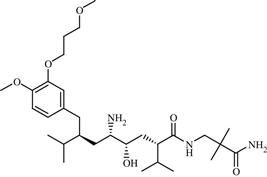 | Hypertension | Inhibition of rennin | 2007 | Novartis | Aliskiren hemifumarate | Tekturna® |
| 15 | Donepezil | 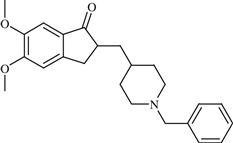 | Alzheimer’s disease | Inhibition of AChE enzyme | 1996 | Eisai Inc. | Donepezil hydrochloride | Aricept® |
| 16 | Imatinib | 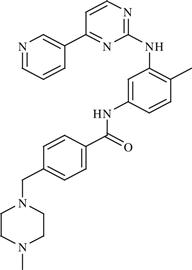 | Cancer: Chronic myelogenous leukemia (CML), gastrointestinal stromal tumors (GISTs), etc. | Inhibition of BCR-Abl tyrosine kinase enzyme | 2003 | Novartis | Imatinib mesylate | Gleevec® |
| 17 | Tirofiban | 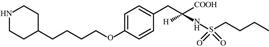 | Thrombosis | Inhibition of fibrinogen | 2000 | Medicure | Tirofiban hydrochloride | Aggrastat® |
| Investigational drugs under clinical trial | ||||||||
| 18 | PRX-00023 | 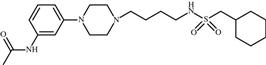 | Depression, anxiety | Agonism of 5-HT1A | Phase III completed | Epix Pharmaceuticals, Inc. | PRX-00023 | − |
| 19 | LY-517717 | 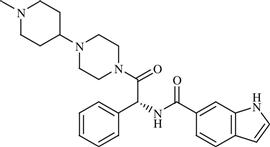 | Venous thromboembolism following hip or knee replacement | Inhibition of factor Xa serine protease enzyme | Phase II completed | Eli Lilly/Protherics | LY-517717 | − |
| 20 | TMI-005 | 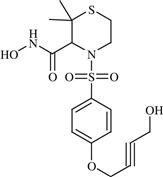 | Rheumatoid arthritis | Inhibition of TNF-α convertase enzyme (TACE) | Phase II completed | Wyeth (Pfizer) | TMI-005 | − |
| 21 | Nolatrexed | 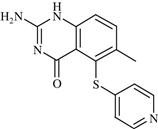 | Cancer: Unresectable Hepatocellular Carcinoma (HCC) | Inhibition of thymidylate synthase enzyme | Phase III completed | Agouron | Nolatrexed dihydrochloride | Thymitaq |
| 22 | Raltitrexed | 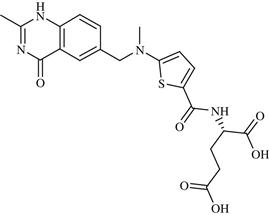 | Cancer: malignant neoplasm of colon and rectum | Inhibition of thymidylate synthase enzyme | Phase II completed | Astrazeneca | Raltitrexed | Tomudex |
| 23 | AUY922 (NVP-AUY922) | 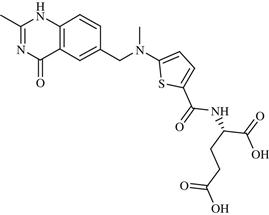 | Cancer | Inhibition of HSP90 | Phase II completed | Novartis | AUY922 | − |
| 24 | Rupintrivir (AG7088) | 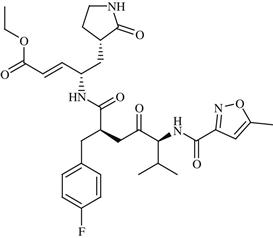 | Rhinovirus infection | Inhibition of HRV 3C protease enzyme | Phase II completed | Agouron | Rupintrivir | − |
Table 11.2
Representative list of registered agrochemicals (pesticides) designed using QSAR techniques
| Sl. No. | Name | Chemical structure | Mechanism | EPA registration | Pesticide type |
| 1 | Bifenthrin | 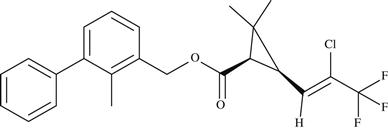 | Acts by interfering with the nervous system of insects | 1989 | Conventional chemical |
| 2 | Ipconazole | 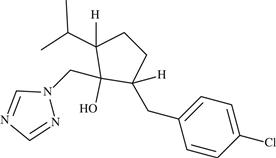 | Acts by inhibiting C-14 demethylation during ergosterol biosynthesis | 1993 | Conventional chemical |
| 3 | Metconazole | 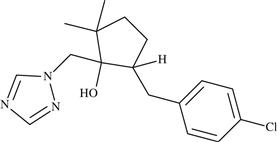 | Acts by inhibiting C-14 demethylation during ergosterol biosynthesis | 2007 | − |
11.2.1 Examples of some approved drugs
Example 1
a. Name of the drug: Captopril
b. Disease indication: Used for the treatment of hypertension.
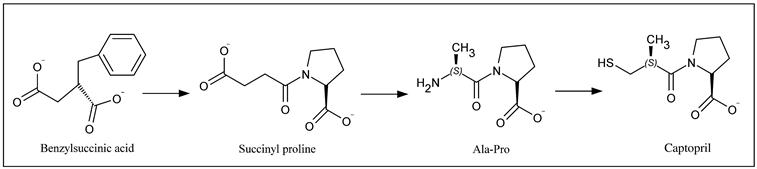
d. Brief developmental history: Captopril was designed using the approach of structure-based drug design in the late 1970s [8]. The basic concept came from the inhibition of the enzyme carboxypeptidase A. Among the first developments were L-benzylsuccinic acid [9], a potent inhibitor of carboxypeptidase A and the pentapeptide BPP5a [10], an inhibitor of ACE (which is isolated from the Brazilian viper, Bothrops jararaca). The N-terminal fragment of BPP5a, including dipeptide, tripeptide, and tetrapeptide fragments, possesses the ACE inhibitory activity. Benzylsuccinic acid was considered as a model compound, assuming that succinyl amino acids act as the by-product inhibitors of ACE. Structure–activity relationship (SAR) studies on succinyl-proline moiety led to the design and development of Captopril characterized by an IC50 value of 23 nM [8]. Two essential structural modifications were incorporated in succinyl-proline. In order to establish a stronger binding interation with the zinc ion of ACE, the carboxylic acid residue of succinyl-proline was replaced with the mercapto group, and a stereospecific R-methyl group was added to the succinyl moiety emulating the methyl group that is similar to that of Ala-Pro (L-Ala residue). Figure 11.1 shows the structural developmental phases of Captopril.
e. Approval status: Approved by the US Food and Drug Administration (FDA) in 1981.
Example 2
a. Name of the drug: Dorzolamide
b. Disease indication: Used for the treatment of open angle glaucoma and ocular hypertension.
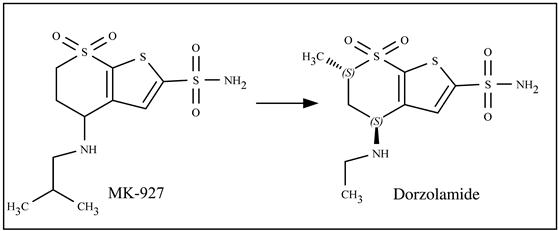
d. Brief developmental history: The discovery was directed by exploring the binding of compounds at the active site of the CA II enzyme using suitable tools. MK-927 was considered as the prototype chemical for reducing IOP. From chiral analysis, the S-enantiomer of MK-927 was observed to be more active than the R isomer, and X-ray crystallographic studies using the human CA II (HCA II) enzyme showed binding interaction between the zinc ion of HCA II and the deprotonated (presumably) sulfonamide nitrogen of MK-927, while the thiophene ring was reported to be placed between hydrophobic and hydrophilic walls of the active site [11,12]. Dorzolamide was developed from MK-927 through the conformational optimization of its enantiomers. At first, the pseudoequatorial conformation between the R and S enantiomers (MK-927) was observed to be preferable by employing ab initio studies at the 6–31 G* level. One difference between the two enantiomers was in the thiophenesulfonamide N-S-C-S dihedral angle, the ideal being 72°. The S-form (150°) showed a preference over the R form (170°) since the latter showed an additional twist of the thiophene ring. The second difference was in the geometry of the 4-isobutylamino substituent; an ab initio study at the 3–21 G* level showed that the trans geometry in the S-form is preferable than the gauche form in the R [12]. Two structural modifications were performed to enhance the inhibitory potency. With the aim of reducing the pseudoaxial energy penalty, a methyl group at the thienothiopyran ring was introduced; and to reduce the lipophilic effect due to the methyl group, ethylamino moiety was used in place of the 4-isobutylamino group. The final structure was termed dorzolamide, and from X-ray crystallographic analysis using HCA II, the S,S configuration was observed to be the best characterized by a favorable thiophenesulfonamide N-S-C-S dihedral angle of 140° [12]. Figure 11.2 presents the chemical structures of MK-927 and dorzolamide.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


