Rubella Virus
Tom C. Hobman
Rubella virus (RV) is the etiologic agent of rubella, a mild exanthematous disease associated with low-grade fever, lymphadenopathy, and a short-lived morbilliform rash. Rubella was first described as a distinct disease in the early 1800s.143 Prior to acquiring its more common name, the disease was named Rõtheln by the German physician de Bergen,65 leading to the common name of German measles, a milder form of the much more serious exanthem caused by the paramyxovirus, measles virus. Subsequently, in 1866 the disease name was anglicized to rubella by the British physician Veale.243
Predominantly a childhood disease, RV is endemic in many parts of the world. The introduction of comprehensive vaccination programs in most industrialized regions including the Americas, Western Europe, Japan, and Australia has drastically reduced the incidence of disease in these areas. Indeed, the indigenous circulation of rubella was officially declared eliminated in the United States in 2005. However, developed nations are still very much at risk for rubella outbreaks due to immigration from areas where RV vaccination is lacking. For example, in Africa and Southeast Asia, scheduled infant immunization against rubella is 0.1% and 4%, respectively.258 Because a very large proportion of the world’s population is still susceptible to RV (Fig. 24.1), we can expect that epidemics of rubella will still occur on a regular basis.
Until the keen observations of the ophthalmologist Norman Gregg, rubella was considered a relatively benign infection, associated with considerably less morbidity than measles. In 1941, Gregg encountered a large number of children with cataracts, many of whom had additional serious congenital defects. He noted that an apparent epidemic of congenital cataracts was directly preceded by a large rubella outbreak. Gregg proposed that the cataracts and the often associated congenital cardiac abnormalities were the consequence of maternal infection during pregnancy.83 Further studies by other investigators confirmed that the virus could have devastating effects on a developing fetus when acquired by the mother in early pregnancy.254 The realization that viruses can act as teratogenic agents spurred the efforts to develop an attenuated vaccine.
A key step in vaccine development was isolation and growth of RV in cultured cells.182,253 In 1969, the first rubella vaccine was licensed in the United States, 5 years after the last major epidemic in that country and shortly before the next significant outbreak was predicted to occur. Fortunately, no other major rubella epidemics occurred, largely in part because of the adoption of universal vaccination in infancy, which has been remarkably successful in controlling natural rubella and its devastating teratogenic effects.213
Infectious Agent
RV is an enveloped positive-strand RNA virus in the family Togaviridae. There are two genera that compose the Togaviridae: Alphavirus, which includes Sindbis and Semliki Forest viruses, and Rubivirus, whose sole member is RV. Togaviruses share a common genome organization and replication strategy (Fig. 24.2). Whereas alphaviruses employ animal reservoirs and arthropod vectors for transmission, humans appear to be the only natural host and reservoir for RV. Although only one serotype exists, there are at least 10 genotypes of RV (Fig. 24.3), which can be grouped into two clades.264 Circulation of clade 2 viruses, which contain three genotypes, is limited to Eurasia, while clade 1 viruses are more widely distributed. The genome of RV is a 5′ capped positive-sense RNA molecule of approximately 10 kilobases (kb) with a poly (A) tail. There is no serologic cross-reactivity between the alphaviruses and RV, and only limited genome sequence similarity, predominantly within the nonstructural genes in regions that encode functional domains such as the polymerase and protease activities.
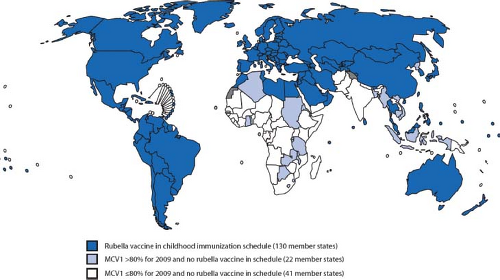 Figure 24.1. World map showing countries that have comprehensive rubella vaccination strategies. (From Progress toward control of rubella and prevention of congenital rubella syndrome—worldwide, 2009. MMWR Morb Mortal Wkly Rep 2010;59[40]:1307–1310.) |
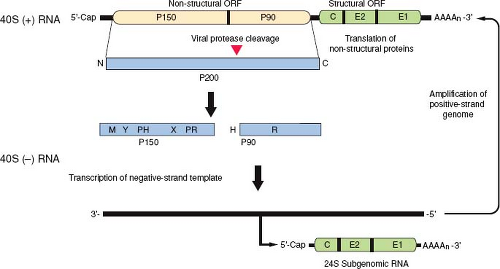 Figure 24.2. Organization and expression of the rubella virus (RV) genome. The genome is a single-stranded 40S RNA containing two open reading frames (ORFs). The 5′ proximal ORF encodes the nonstructural proteins p150 and p90. The 40S genome serves as a messenger RNA (mRNA) for translation of the nonstructural protein precursor p200, which is cleaved by a virus-encoded protease to produce p150 and p90. The relative positions of methyltransferase (M), Y, proline hinge (PH), X, protease (PR), helicase (H), and RNA-dependent polymerase (R) domains within p150 and p90 proteins are shown. These two proteins form the viral replicase that synthesizes a negative-sense 40S RNA, which then serves as a template for synthesis of more genomic RNA and the 24S subgenomic RNA. Both the 40S and 24S positive-sense RNAs are capped and polyadenylated. |
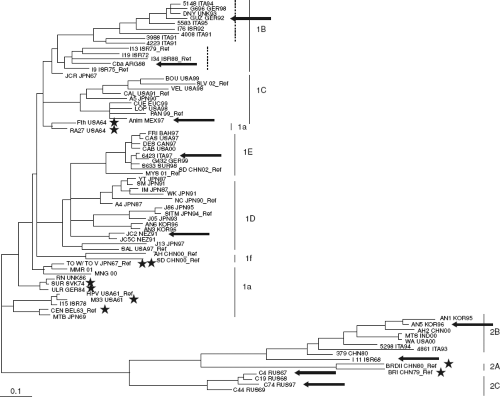 Figure 24.3. Phylogenetic tree of rubella virus (RV) genotypes based on analyses of E1 gene nucleotide sequences. Based on the analyses, there are two main clades 1 and 2, which are composed of seven and three genotypes, respectively. Reference strains for each genotype are indicated by “Ref.” Strains whose genomic sequence have been reported previously are indicated by stars, whereas those strains whose genomic sequences were determined by Zhou et al are indicated by arrows. Two sub-branches within genotype 1B are indicated by dashed lines. (From Zhou Y, Ushijima H, Frey TK. Genomic analysis of diverse rubella virus genotypes. J Gen Virol 2007;88[Pt 3]:932–941, with permission.) |
Morphology and Physicochemical Properties of the Virion
By transmission electron microscopy, RV virions appear as spherical particles with a mean diameter of 61 nm.160 Virus particles can often be observed budding into the Golgi complex of infected cells (Fig. 24.4). They have an electron-dense nucleocapsid core surrounded by a host-derived envelope.159 The nucleocapsid (30 to 40 nm in diameter) is composed of a single molecule of genomic RNA and multiple copies of capsid protein. Between the core and the envelope is an electron-lucent region giving the virions the appearance of having a ring or “toga” surrounding the nucleocapsid, from which the name is derived. Virus-encoded glycoprotein spikes project 6 to 8 nm outward from the lipid envelope236 and denote the virion surface. Early electron microscopic studies suggest that the RV nucleocapsids exhibit a T = 3 icosahedral symmetry,144 distinguishing them from those of alphaviruses that are arranged with a T = 4 symmetry; however, this has not been confirmed by subsequent high-resolution analyses.
The buoyant density of rubella virions in sucrose gradients is 1.18 to 1.19 g/mL.104 In comparison, the buoyant density of alphavirus virions is 1.20 g/mL, a difference that may be attributable to a wider electron-lucent zone between the core and envelope in rubella virions. The reported sedimentation coefficient of RV virions ranges from 240S and 350S.9 The reason for this heterogeneity is not known but may be due in part to contamination of the virus preparations by host cell–derived membranous material. The difficulty in obtaining highly purified homogenous preparations of RV virions is one of the numerous technical reasons that has impeded the progress of structural analysis.
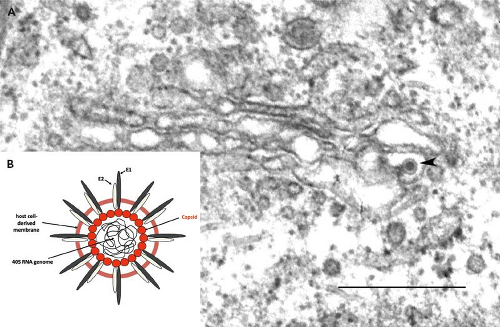 Figure 24.4. A: Electron micrograph of a rubella virion budding into the lumen of a Golgi cisterna in an infected Vero cell. Note the electron-lucent halo that surrounds the electron-dense nucleocapsid. Bar = 0.5 μm. (Courtesy of Dr. John Law and Ms. Honey Chan). B: Schematic of a rubella virus (RV) virion. |
The infectivity of RV virions is rapidly lost following exposure to protein denaturing agents (formaldehyde, ethylene oxide, and β-propiolactone) or treatments/reagents that cause lesions in nucleic acids (ultraviolet light or photodynamic dyes). Moreover, because it is an enveloped virus, exposure of RV preparations to nonionic or ionic detergents and lipid solvents abrogates the infectivity of virions.181 Virions are rapidly inactivated by incubation at 56°C for less than 20 minutes, and even at much lower temperatures such as 37°C, RV loses activity slowly (t½ = 48 hours).152 When stored at temperatures less than –60°C in the presence of protein stabilizers, RV preparations maintain their infectivity for many years. Fortunately, with respect to vaccine distribution, lyophilized RV preparations are stable for years at 4°C and for months at ambient temperatures.184
Biological Characteristics
RV has a restricted host range (only humans) in vivo but replicates in a wide variety of cultured mammalian cell lines, including primary African green monkey kidney cells, BHK21, RK13, and Vero cell lines. All strains of the virus (including wt+ strains) are slightly temperature sensitive, with higher yields of virus obtained when infected cells are cultured at 35°C rather than at 37°C. The vaccine strains RA27/3 and Cendehill are completely growth restricted at 39°C, while some wt+ isolates are still able to replicate at 41°C, albeit at lower levels.36
In comparison to the rapid lytic infection of mammalian cells by the alphaviruses, RV replicates more slowly, with an eclipse phase of 10 to 12 hours (c.f. 4 hours for Sindbis or Semliki Forest viruses), and peak titers of secreted virus are not observed until 36 to 48 hours.87,242 By comparison, peak viral titers for alphaviruses are observed within 8 hours. As well as exhibiting relatively slow replication kinetics, the amount of infectious virus progeny released per cell is estimated to be 1,000-fold less than for alphaviruses. Under optimal conditions, RV-infected BHK-21 and Vero cells can produce titers of 107 and 108 plaque-forming units (pfu) per mL, respectively.10,241 The relatively feeble replication of RV may be due to a number of factors including low uptake of virus and/or inefficiencies associated with the replicase/transcriptase complex or limitations of the assembly process. For example, RV buds into the Golgi complex, which is a much smaller membrane compartment than the endoplasmic reticulum or the plasma membrane. Another factor that may play a role is the high G + C content of the viral genome (69.5%), which is predicted to form stable secondary and tertiary structures that may impede replication/transcription or translation.68 Also related to the G + C content, the codon usage by RV is different from that of mammalian cells, requiring utilization of transfer RNAs (tRNAs) that may be of low abundance, a factor that has been suggested to limit the rate of protein synthesis.225
Effects on Host Cell
Although RV can have devastating effects on the developing human fetus, infection of most cultured primate or rodent cell lines does not result in major cytopathology. However, in some cell lines (Vero, BHK-21, and RK13), when high multiplicities of infection (MOIs) are employed, the virus causes rounding and detachment of cells commencing at 24 hours postinfection
and increasing over the next 72 hours. Much of the RV-induced cytopathic effect is likely the result of apoptosis.61,149,195 Virions that are first inactivated by ultraviolet light do not induce apoptosis, indicating that viral replication is required to cause cell death.61,100 The cytopathic determinants of RV have been mapped to the nonstructural genes,193 and in agreement with this, expression of RV structural proteins alone in Vero cells does not induce apoptosis.100 Paradoxically, primary human embryonic fibroblasts seem to be particularly immune to RV-induced apoptosis.3
and increasing over the next 72 hours. Much of the RV-induced cytopathic effect is likely the result of apoptosis.61,149,195 Virions that are first inactivated by ultraviolet light do not induce apoptosis, indicating that viral replication is required to cause cell death.61,100 The cytopathic determinants of RV have been mapped to the nonstructural genes,193 and in agreement with this, expression of RV structural proteins alone in Vero cells does not induce apoptosis.100 Paradoxically, primary human embryonic fibroblasts seem to be particularly immune to RV-induced apoptosis.3
At low (less than 1) or moderate (1 to 3) MOIs, little virus cytopathic effect is observed in most cultured cells61,116 and chronic infections can readily be established (see Replication Strategy section). Induction of apoptosis in response to RV infection is undoubtedly an important antiviral defense mechanism that is employed by cells against many RNA viruses. Because viruses are obligate parasites, they require a living cell for replication. Depending on the apoptotic stimuli, cells can be killed in a matter of hours, a situation that would be seemingly problematic for RV, which has a long eclipse period and whose replication peak occurs between 2 and 4 days postinfection.87 However, the maximum RV-induced apoptosis does not occur until after 5 days postinfection,149 which is consistent with a scenario where the virus actively blocks apoptosis early in infection. Indeed, a recent study revealed that RV-infected cells are highly resistant to apoptotic stimuli.107 Mapping studies showed that the capsid protein is responsible for this process, which presumably provides a window of opportunity for the virus to replicate before apoptosis is initiated.
In most cell types, cellular macromolecular synthesis on a global level does not appear to be significantly affected, even following high-multiplicity infections (5 to 10 pfu per cell). For example, host cell RNA synthesis continues normally and protein synthesis is only slightly reduced at 72 hours postinfection in several highly permissive cell lines.87 However, in mitogen-stimulated peripheral blood mononuclear cells (PBMCs), host cell protein synthesis was reportedly inhibited by more than 90% at 48 hours postinfection.38 The capsid protein has been shown to block translation in vitro,108 but it has yet to be determined whether this viral protein is responsible for inhibition of protein synthesis in PBMCs or other cell types.
A number of characteristic morphologic changes occur in RV-infected cells. Similar to other togaviruses, RV infection is accompanied by drastic rearrangement of cellular membranes. For example, the endoplasmic reticulum (ER), Golgi complex, and mitochondria are often closely arranged around the virus replication complexes (Fig. 24.5), which are derived from endosomes and/or lysosomes.125,127 This arrangement of organelles could in theory facilitate the efficient transfer of virus genome from the site of RNA replication (endosomes) to the area of virus assembly (Golgi complex). Whereas organelle rearrangement is common in togavirus-infected cells, the formation of electron-dense plaques (22 to 25 nm in thickness) between organelles is unique to RV infection.123 The plaques (Fig. 24.6) and associated organelles have been termed confronting membranes or confronting cisternae and commonly involve outer membranes of mitochondria and rough ER, adjacent mitochondria, and adjacent ER membranes, respectively. Expression of capsid protein in the absence of other viral proteins induces mitochondrial clustering and formation of plaques,15 and immunoelectron microscopy revealed that capsid protein is a major component of the plaques.109 It has been known for years that a large pool of capsid protein is targeted to the surface
of mitochondria,16,126 and recently it was discovered that capsid affects mitochondrial import.109 The latter may explain the loss of cristae and dysmorphic (club-shaped) mitochondria in RV-infected cells.122 Together, these observations reflect the close link between RV replication and mitochondrial physiology.
of mitochondria,16,126 and recently it was discovered that capsid affects mitochondrial import.109 The latter may explain the loss of cristae and dysmorphic (club-shaped) mitochondria in RV-infected cells.122 Together, these observations reflect the close link between RV replication and mitochondrial physiology.
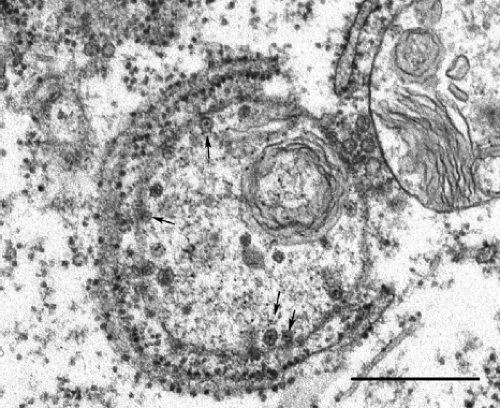 Figure 24.5. Electron micrograph of viral replication complex is associated with rough endoplasmic reticulum. Spherules are indicated by arrows. Bar = 0.5 μm. (Courtesy of Dr. John Law and Ms. Honey Chan.) |
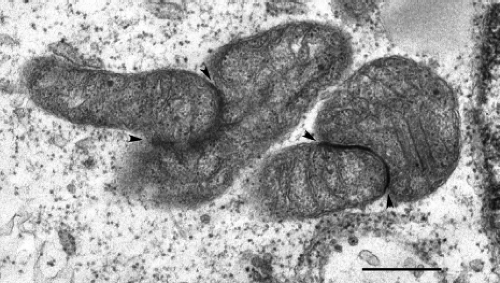 Figure 24.6. Aggregation of mitochondria and formation of electron-dense intermitochondrial plaques in rubella virus (RV)-infected cells. Bar = 0.5 μm. (Courtesy of Dr. John Law and Ms. Honey Chan.) |
Phenotypic Variation
Although there are now recognized to be at least 10 genotypes of RV (Fig. 24.3), there is only one serotype, and infection with one strain provides protection against all others recognized to date. However, the limited genetic differences seen (0.8% to 2.1% at the amino acid level221,263) are associated with differences in hemagglutination,133 plaque morphology,116 temperature sensitivity, virus yield, and cell tropism.36 Compared to natural RV infection, administration of vaccine strains is associated with milder acute symptoms and a lower incidence of complications such as joint and neurological symptoms,21,66,229 as well as teratogenic effects.13
The major neutralization epitopes appear to be highly conserved among RV strains,22,69 although differences have been noted in the kinetics of neutralization81 and in reactivity to certain monoclonal antibodies.36 However, both infection and immunization are believed to provide protection against all other strains for the duration of the immune response elicited, except in cases where incomplete immunity is induced.230
The vaccine strains RA27/3 and Cendehill display different tissue tropisms from wt+ strains, including growth restriction in both PBMCs and the B-cell lines, Raji and Cess cells.36 These two strains also show limited replication in cells derived from human joint tissue,152 with the Cendehill strain completely inhibited in synovial organ cultures and chondrocytes. In comparison, wt+ strains replicate to high titer (106 to 107 pfu/mL) in joint cells. The determinants that govern growth in joint tissue have been mapped to the 5′ end of the genome, which encodes the nonstructural proteins, and as such, it is likely that replication rather than binding and entry events determine tropism.135
Structure and Organization of the Genome
The genome of RV is a single molecule of positive-strand RNA of approximately 10 kb with a GC content of 69.5%, by far the highest of any RNA virus sequenced to date.6 The 5′ end of the RNA has a 7-methyl-guanosine cap,166 while at the 3′ end there is a poly (A) tract with a mean length of 53 nucleotides.246 The genome consists of two nonoverlapping polycistronic open reading frames (ORFs) separated by a nontranslated region of 123 nucleotides (Fig. 24.2).192 The 5′ proximal ORF (approximately 6,385 nucleotides) encodes the nonstructural proteins, while the 3′ proximal ORF (3,189 nucleotides) encodes the three structural proteins, capsid, E2, and E1. The structural proteins are translated from a 5′ capped and polyadenylated subgenomic RNA that is collinear with the 3′ one-third of the 40S genome.166 The complete nucleotide sequences have been determined for the genomes of several wild-type and vaccine strains of RV68 and are available in GenBank. In addition, infectious complementary DNA (cDNA) clones of several strains have been produced and used to map genetic elements involved in viral replication and attenuation.135,196,246,259
Cis-Acting Elements
The availability of infectious RV cDNA clones135,196,259 and replicons237 has enabled the application of reverse genetics to study the roles of cis-acting elements in genome replication. Both the 5′ and 3′ untranslated regions (UTRs) of the RV genome are believed to form secondary structures that influence transcription and translation events. For example, the predicted 5′ stem-loop (5′SL), which encompasses nucleotides 15 to 65, has a terminal loop as well as a bulge in the stem and a hinge region (Fig. 24.7). It is believed to form a pseudoknot,194 a structure that has been shown in a number of plant and animal viruses to enhance binding of proteins that function in RNA replication.57,110 Within the single-stranded leader sequence (nucleotides 1 to 14) and the stem-loop, there are three AUG codons starting at nucleotides 3, 41, and 57. Translation of the long ORF encoding the nonstructural genes is initiated at AUG41, while the first and third AUGs are in a different reading frame and are terminated at nucleotides 54 and 90, respectively. Indeed, site-specific mutagenesis studies suggest that AUG3 is not essential for viral replication,194 nor is it known whether short peptides that originate from either AUG3 or AUG57 play any role in virus biology. However, it is notable that the
three AUG codons are conserved in all strains sequenced to date.135,194 Analysis of the 5′ end sequences of a number of RV strains revealed that the stem-loop is largely conserved and that polymorphisms are restricted to the terminal loop or the hinge region.110,194 Mutations in the 5′SL are associated with differences in viral yield and plaque morphology,194 as well as tropism for joint tissue.135
three AUG codons are conserved in all strains sequenced to date.135,194 Analysis of the 5′ end sequences of a number of RV strains revealed that the stem-loop is largely conserved and that polymorphisms are restricted to the terminal loop or the hinge region.110,194 Mutations in the 5′SL are associated with differences in viral yield and plaque morphology,194 as well as tropism for joint tissue.135
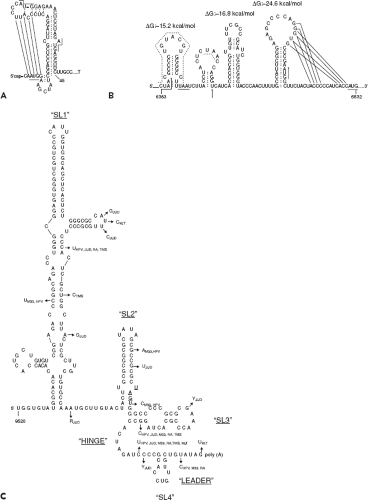 Figure 24.7. Predicted stem-loop structures in the (A) 5′ end sequence, (B) 5′ subgenomic end sequence, and (C) 3′ end sequence. Potential pairing of bases to form pseudoknots in structures A and B are shown with dotted lines. (A and B from Frey TK. Molecular biology of rubella virus. Adv Virus Res 1994;44:69–160; C from Chen MH, Frey TK. Mutagenic analysis of the 3′ cis-acting elements of the rubella virus genome. J Virol 1999;73[4]:3386–3403.) |
Between the nonstructural and structural genes is a 123-bp region that is predicted to form a series of stem-loops (Fig. 24.7). This region shares 58% identity with the equivalent region in the Sindbis virus genome and may be important for regulating synthesis of the subgenomic RNA. In addition, translation of the structural genes is affected by mutations in this region, which forms the 5′ end of the subgenomic RNA.180 In the 3′ UTR, there is a stretch of 59 nucleotides following the stop codon at nucleotide 9701. A complex secondary structure involving three major stem-loop structures43 has been predicted for the 3′ terminal 240 nucleotides (Fig. 24.7).
These 3′ UTR sequences function in cis to regulate transcription, specifically, synthesis of plus-strand viral RNAs but not negative-sense RNA.44
These 3′ UTR sequences function in cis to regulate transcription, specifically, synthesis of plus-strand viral RNAs but not negative-sense RNA.44
Encapsidation Signal
Mapping studies revealed that capsid protein binds with relatively high affinity to a 29-nucleotide segment located in the 5′ nonstructural gene region between nucleotides 347 and 375.132
Nonstructural ORF and Protein Products
The nonstructural (NS) genes are located in the 5′ end of the viral genome within a long ORF commencing at AUG41. The NS ORF is translated as a single polyprotein greater than 200 kD that is cleaved into two products, p150 and p90,140 that function in replication and transcription of viral RNAs (Fig. 24.2). The gene order of the 5′ ORF is NH2-p150-p90-COOH. The two nonstructural proteins have several enzymatic activities including RNA polymerase, protease, and helicase and together form the viral replicase. Proteolytic processing of the p200 polyprotein marks the switch from synthesis of negative-strand genomic RNA to synthesis of plus-strand RNA.128
p150 contains several domains that are conserved among other RNA virus-encoded proteins. A protease domain located in the carboxyl portion of p150 (Fig. 24.2) is responsible for cleavage of the nonstructural precursor protein p200 and is critical for virus replication.42,260 Mutagenesis studies have shown that the RV protease is a metalloprotease that requires divalent cations for activity130,131 and functions in both cis and trans modes.129 A putative methyltransferase domain in the amino terminus is believed to have a role in capping the viral RNA.211 p150 also contains an X domain that has homology to the nonstructural proteins of alphaviruses, coronaviruses, and hepatitis E virus.80 The function of the X domain is not well characterized, but it is required for trans cleavage by the RV protease and in alphaviruses at least is important for replication.86 Interestingly, the X domain is the region that is most highly conserved between RV and alphaviruses.60 Two other domains of unknown function, Y and a proline hinge, are also present in p150. The order of these domains in p150 is NH2–methyltransferase–Y domain–proline hinge domain–X domain–protease–COOH. The second nonstructural protein, p90, comprises 905 amino acid residues and contains the replicase and helicase motifs. Based on comparison with global RNA-dependent RNA polymerase consensus sequences,111 the GDD tripeptide at amino acids 1965 to 1967 of p90 is the active site of the RV replicase. An RNA-stimulated nucleoside triphosphatase (NTPase) that promotes unwinding of the template during RNA replication is associated with the helicase motif.85
The localization of RV nonstructural proteins, p150 and p90, is less well studied than that of the structural proteins. This is because the nonstructural proteins are present at relatively low concentrations in infected cells, and furthermore, attempts to produce useful antibodies against these antigens has met with limited success. However, it has been reported that p150 associates with long tubular structures that correspond to sites of viral RNA synthesis.118 p90 has been detected in discrete cytoplasmic puncta that form linear chains.5 The identity of the p90-associated structures was not determined, but it is likely that they are replication complexes derived from endocytic vacuoles. Presumably, a large pool of p150 is also localized to the p90-positive foci. No information regarding sequences that target the nonstructural proteins to endocytic membranes has been reported.
Structural ORF and Protein Products
The structural genes are contained within the 3′ ORF of the 40S genomic RNA; however, they are actually translated from a subgenomic 24S RNA that is produced in infected cells. There are two in-frame AUG initiation codons in the subgenomic RNA separated by 21 nucleotides. The downstream AUG is in the more favorable context for translation initiation, but in vitro mutagenesis experiments indicate that both codons can be used.47 The 24S subgenomic RNA encodes three structural proteins, which are translated in the order NH2-C-E2-E1-COOH.165 Unlike alphavirus structural proteins, which require both host cell signal peptidase and a capsid-associated protease for complete processing,150 processing of the RV structural polyprotein (Fig. 24.8) into C, E2, and E1 only requires signal peptidase, which is encoded by the host cell.139,224
In the absence of genomic RNA, coordinated expression of the RV structural proteins in mammalian cells results in the assembly and secretion of rubella virus-like particles (VLPs).94,199 The VLPs, which resemble native virions in terms of morphology and antigenicity, have served as a useful tool for studying virus assembly, and as a consequence, the roles of RV structural proteins and their domains in the assembly process are relatively well understood. The fact that rubella VLPs are efficiently assembled and secreted in the absence of genomic RNA indicates that virus budding is not tightly linked to nucleocapsid assembly.
Capsid
The capsid protein is a phosphoprotein with an apparent molecular mass of 33 to 35 kD.139 The protein contains 300 amino acid residues and is rich in arginine and proline residues, particularly at its amino terminus, which gives it a net positive charge46 and facilitates its interaction with genomic RNA during nucleocapsid formation. Capsid protein often migrates as a doublet on SDS-PAGE gels. The reason for this remains unknown, but it is not due to the use of an alternate translation start site.47,139 Virion-associated capsid protein can be isolated as disulfide-linked dimers, but intermolecular disulfide bonding is not necessary for formation of virus particles.12,124
Cleavage of the RV capsid protein from the polyprotein precursor is distinct from that of alphavirus capsid proteins, which contains an autoprotease that separates the capsid protein from the polyprotein.150 As a consequence, alphavirus capsids are free in the cytoplasm of infected cells. RV capsid protein, in contrast, lacks autoprotease activity, and separation from E2 is carried out by host cell signal peptidase.46 The E2 signal peptide is therefore retained as the carboxyl terminus of capsid protein (Fig. 24.8), where it serves to confer membrane association of the protein.91,139,224 The membrane association of RV capsid protein is unique among togaviruses and may account for some of the unusual morphogenetic features of the virus. For example, retention of the E2 signal peptide on capsid protein is necessary for assembly of VLPs and presumably infectious virions.120
Role of the Capsid Protein in Virus Assembly
The primary function of capsid protein during virus assembly is to homo-oligomerize and bind viral genomic RNA to form
the nucleocapsid. The RNA-binding domain of capsid protein resides within amino acid residues 28 to 56 and binds to a packaging signal (nucleotides 347 to 375) in the genomic RNA.132 Posttranslational modification of capsid protein may play an important role in regulating the formation of nucleocapsids because phosphorylation of serine 46 within the RNA-binding domain negatively regulates RNA binding.119,121 This is believed to prevent nonspecific binding of cellular RNAs to capsid protein and to delay binding of genomic RNA until the virion components are targeted to the budding site. In vitro, capsid protein is a substrate for protein phosphatase IA, an enzyme that has been implicated in Golgi-associated functions.121 Accordingly, targeting of capsid protein to the Golgi complex, followed by dephosphorylation at this site, could explain how nucleocapsid assembly is synchronized with virus assembly (Fig. 24.9). Supporting this scenario is the observation that virion-associated capsid protein has a higher affinity for viral RNA and contains significantly less phosphate than cell-associated capsid protein.119,121 Similar to alphaviruses,223 it is thought that capsid protein drives budding of RV through interactions with the envelope glycoproteins; however, direct binding between RV capsid protein and E2 and/or E1 has yet to be demonstrated.
the nucleocapsid. The RNA-binding domain of capsid protein resides within amino acid residues 28 to 56 and binds to a packaging signal (nucleotides 347 to 375) in the genomic RNA.132 Posttranslational modification of capsid protein may play an important role in regulating the formation of nucleocapsids because phosphorylation of serine 46 within the RNA-binding domain negatively regulates RNA binding.119,121 This is believed to prevent nonspecific binding of cellular RNAs to capsid protein and to delay binding of genomic RNA until the virion components are targeted to the budding site. In vitro, capsid protein is a substrate for protein phosphatase IA, an enzyme that has been implicated in Golgi-associated functions.121 Accordingly, targeting of capsid protein to the Golgi complex, followed by dephosphorylation at this site, could explain how nucleocapsid assembly is synchronized with virus assembly (Fig. 24.9). Supporting this scenario is the observation that virion-associated capsid protein has a higher affinity for viral RNA and contains significantly less phosphate than cell-associated capsid protein.119,121 Similar to alphaviruses,223 it is thought that capsid protein drives budding of RV through interactions with the envelope glycoproteins; however, direct binding between RV capsid protein and E2 and/or E1 has yet to be demonstrated.
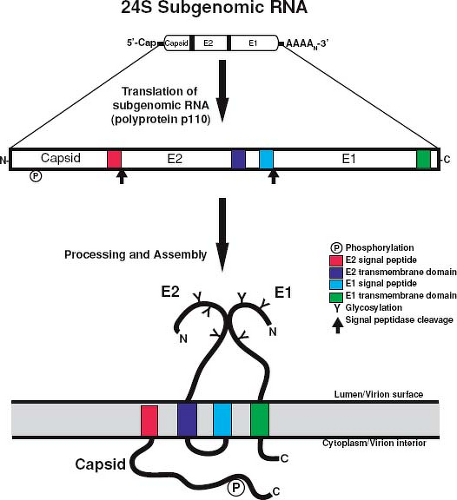 Figure 24.8. Processing of rubella virus (RV) structural proteins. The 24S RNA serves as the messenger RNA (mRNA) for translation of a structural polyprotein precursor, which is then cleaved by host signal peptidase to produce capsid, E2 and E1. The relative membrane orientations of the structural proteins immediately following translocation into the endoplasmic reticulum (ER) are shown. The amino (N) and carboxyl (C) termini of the proteins are indicated. |
Nonstructural Roles of the Capsid Protein
In addition to nucleocapsid assembly, the RV capsid protein plays a role in regulating viral transcription and replication. The first indication of this nonstructural function came from the observation that expression of capsid protein rescues the replication of an RV replicon containing an in-frame deletion of p150.238 Further analyses indicated that the capsid protein is involved in modulating viral genomic and subgenomic RNA synthesis.239 In the absence of capsid protein expression, the ratio of genomic RNA to subgenomic RNA is substantially lower. It is believed that a pool of capsid protein that associates with the replication complexes is responsible for regulating synthesis of viral RNAs. Interestingly, the effects of capsid protein on transcription depend on the levels of replicon RNA.45 At low levels of RNA, capsid protein expression enhances replication of viral RNA, but with higher levels of RNA, capsid protein is inhibitory for this process.
Given its multiple roles, it is not surprising that the RV capsid protein is associated with multiple organelles. Consistent with its function in virus assembly, pools of capsid protein are localized to the Golgi.11,94 Transport of capsid to the Golgi region from the site of its synthesis, the ER, depends on E1 and E2.74,120 Capsid also localizes to mitochondria and to virus
replication complexes.16,123 In contrast, the glycoproteins E1 and E2 are targeted only to the virus budding site (Golgi), suggesting that they do not have major nonstructural roles. Together with p150, replication complex–localized capsid protein118 is thought to modulate replication of viral RNA, whereas the mitochondrial pool of capsid plays one or more critical roles in virus–host interactions (Fig. 24.10). For example, it potently blocks apoptosis by sequestering the proapoptotic host protein Bax into nonfunctional complexes.107 RV capsid also interferes with mitochondrial import,109 a critical process that is linked to apoptosis in mammalian cells.183 Finally, it has been known for more than a decade that capsid interacts with the mitochondrial matrix protein p32.16 The precise function of this interaction is not clear, but several lines of evidence suggest that it is important for virus replication. First, ablation of the p32-binding site in capsid protein reduces the replication efficiency of RV by 1,000-fold.15 Second, depletion of cellular p32 pools by RNA interference results in a 10-fold reduction in virus replication.49 Capsid protein–p32 interactions may also be important for synthesis of subgenomic RNA and/or translation of structural proteins. Finally, loss of stable binding between capsid protein and p32 is correlated with reduced mitochondrial clustering in
the perinuclear region. Together, these results suggest that interactions between capsid protein and p32 are important for RV-induced rearrangement of mitochondria, a phenomenon that is associated with optimal replication.
replication complexes.16,123 In contrast, the glycoproteins E1 and E2 are targeted only to the virus budding site (Golgi), suggesting that they do not have major nonstructural roles. Together with p150, replication complex–localized capsid protein118 is thought to modulate replication of viral RNA, whereas the mitochondrial pool of capsid plays one or more critical roles in virus–host interactions (Fig. 24.10). For example, it potently blocks apoptosis by sequestering the proapoptotic host protein Bax into nonfunctional complexes.107 RV capsid also interferes with mitochondrial import,109 a critical process that is linked to apoptosis in mammalian cells.183 Finally, it has been known for more than a decade that capsid interacts with the mitochondrial matrix protein p32.16 The precise function of this interaction is not clear, but several lines of evidence suggest that it is important for virus replication. First, ablation of the p32-binding site in capsid protein reduces the replication efficiency of RV by 1,000-fold.15 Second, depletion of cellular p32 pools by RNA interference results in a 10-fold reduction in virus replication.49 Capsid protein–p32 interactions may also be important for synthesis of subgenomic RNA and/or translation of structural proteins. Finally, loss of stable binding between capsid protein and p32 is correlated with reduced mitochondrial clustering in
the perinuclear region. Together, these results suggest that interactions between capsid protein and p32 are important for RV-induced rearrangement of mitochondria, a phenomenon that is associated with optimal replication.
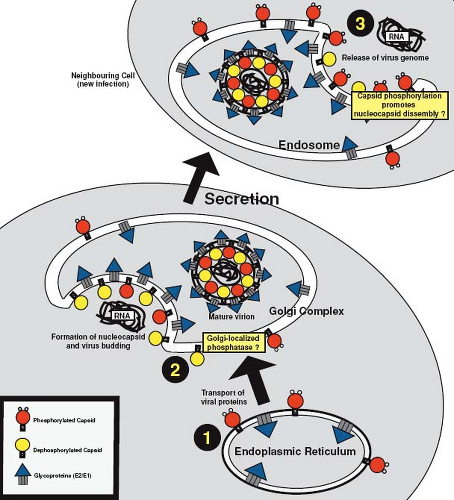 Figure 24.9. Model to illustrate the putative roles of dynamic phosphorylation of capsid protein in virus replication. 1: Phosphorylation of newly synthesized capsid protein prevents nonspecific binding of RNA and premature formation of nucleocapsid at the early stages of virus assembly. 2: Capsid protein is subsequently targeted to the Golgi complex and dephosphorylation of the protein at this stage allows interaction with the genomic RNA, formation of the nucleocapsid, and subsequent virus budding. 3: Timely rephosphorylation of capsid protein before or during virus entry promotes the disassembly of nucleocapsid. |
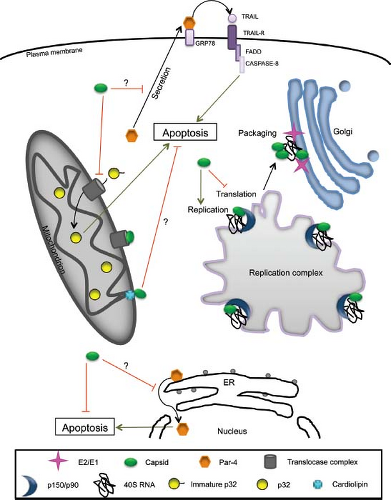 Figure 24.10. Integrated model of rubella virus (RV) capsid nonstructural functions. At the replication complex, capsid associates with nonstructural proteins (p150 and p90) and regulates transcription and replication of viral RNA. Late in the infection cycle, local sequestration of translation initiation factors such as poly(A) binding protein (PABP) may modulate the switch from translation to packaging of the genomic RNA, a mechanism that would leave the viral RNA available for packaging into nucleocapsids at the Golgi complex. Capsid prevents apoptosis by binding to Bax and inducing the formation of hetero-oligomers that are incompetent for pore formation at the mitochondria and potentially at the endoplasmic reticulum (ER) membranes. Capsid may also inhibit apoptosis by preventing translocation of pro-apoptotic proteins such as p32 into the mitochondria or by engaging in complexes with the mitochondrial lipid, cardiolipin. (Modified from Ilkow CS, Willows SD, Hobman TC. Rubella virus capsid protein: a small protein with big functions. Future Microbiol 2010;5[4]:571–584.) |
Envelope Proteins
E1 and E2 are type I membrane proteins that dimerize to form the spike complexes on the surface of the virion (Fig. 24.8). The major functions of these spikes are to bind receptors on the host cell and to mediate fusion with cell membranes.114 The envelope proteins, E1 in particular, are the major antigenic determinants against which neutralizing antibodies are directed.249
E1 is 481 amino acid residues in length and exhibits an apparent molecular mass of 58 kD when analyzed by SDS-PAGE. The mature protein contains three asparagine-linked oligosaccharide moieties.95 An amino terminal signal peptide facilitates translocation of E1 into the ER and a 22–amino acid hydrophobic sequence at its carboxyl terminus mediates membrane association.96 E1 also contains a carboxyl terminal tail of 13 amino acid residues that is exposed to the cytoplasm. E2 (282 amino acids residues) contains two stretches of hydrophobic amino acid residues near its carboxy terminus, an 18–amino acid residue transmembrane domain, and the E1 signal peptide.11,46 These two hydrophobic domains are separated by a 7–amino acid residue loop that is rich in basic amino acid residues. Similar to E1, translocation of E2 into ER membranes is mediated by an amino terminal signal peptide.91 E2 is heavily glycosylated and contains both asparagine-linked and O-linked carbohydrates.96,136 Finally, both glycoproteins are modified by the addition of palmitate prior to virus assembly.93,249 The role of this modification in spike protein function has not been investigated, but it is possible that acylation may serve to further stabilize and/or orientate E2 and E1 in the membrane. The membrane orientations of the RV structural proteins are shown in Figure 28.8.
Replication Strategy
The replication strategy of RV is similar to that of alphaviruses in many respects, but there are several quantitative and qualitative differences. As discussed previously, RV replication is less robust and much slower than that of alphaviruses.87 In addition, it is not possible to obtain a uniformly infected population of cells within 24 hours, even at high multiplicities of infection.214 The reason for this phenomenon is not clear, but
given the fact that eventually all cells within a permissive culture become infected, it is possible that a cell cycle–dependent factor is required for RV entry or early viral replication events. Another major difference from the alphaviruses is that a large pool of the nascent RV capsid protein remains membrane associated.120,224 This may be one of the reasons that RV nucleocapsid assembly differs from that of alphaviruses, which are not membrane associated. Free nucleocapsids are rarely observed in RV-infected cells but rather assembly is coincident with virus budding at the Golgi complex, a process that is hypothesized to be regulated by reversible phosphorylation of the capsid protein.121 A schematic diagram of how RV enters, replicates, and exits from host cells is shown in Figure 24.11.
given the fact that eventually all cells within a permissive culture become infected, it is possible that a cell cycle–dependent factor is required for RV entry or early viral replication events. Another major difference from the alphaviruses is that a large pool of the nascent RV capsid protein remains membrane associated.120,224 This may be one of the reasons that RV nucleocapsid assembly differs from that of alphaviruses, which are not membrane associated. Free nucleocapsids are rarely observed in RV-infected cells but rather assembly is coincident with virus budding at the Golgi complex, a process that is hypothesized to be regulated by reversible phosphorylation of the capsid protein.121 A schematic diagram of how RV enters, replicates, and exits from host cells is shown in Figure 24.11.
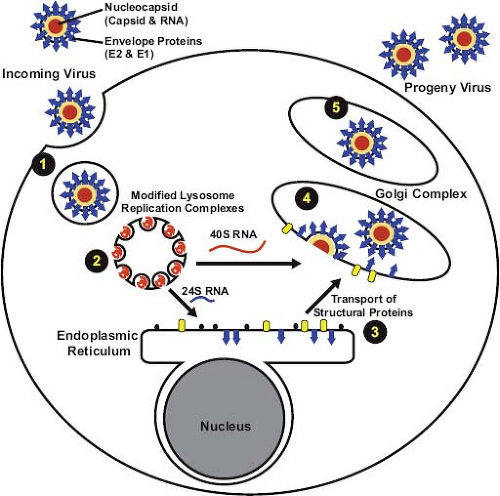 Figure 24.11. Entry and exit strategy of rubella virus (RV). 1: Uptake of RV is dependent upon receptor-mediated endocytosis. The low pH of the endosome/lysosome induces virus uncoating. 2: RNA replication occurs at the cytopathic vacuoles that originate from endosomes/lysosomes. Genome amplification and synthesis of the 24S subgenomic RNA also occurs at these sites. 3: Synthesis of structural proteins takes place at the endoplasmic reticulum and processing by the host cell signal peptidase separates the polyprotein into individual structural proteins. Subsequently, the structural proteins assemble and are transported to the Golgi complex. 4: Nucleocapsid assembly and virus budding occur at the Golgi complex. 5: Rubella virions undergo a series of maturation events before exocytosis. |
Virus Entry
Host cell–encoded proteins and lipids are important for virion binding,50,142 after which the virus is internalized by receptor-mediated endocytosis.186 At low pH, the envelope glycoproteins become fusogenic, and capsid undergoes a conformational change and becomes hydrophobic,114,145 indicating that the acidic environment within endosomes induces fusion of the viral envelope with cellular membranes followed by release of genomic RNA from the nucleocapsid. The type I membrane protein myelin oligodendrocyte glycoprotein binds to the E1 glycoprotein and was recently identified as a host receptor for RV.50 This protein is most highly expressed in the central nervous system, but in epithelial and lymphoid cells, which serve as the portals of entry and initial sites of replication, myelin oligodendrocyte glycoprotein levels are much lower. As such, it cannot be ruled out that the virus uses different receptors in different cell types.
Replication Complexes
Similar to all other plus-strand RNA viruses that infect mammalian cells, replication of RV RNA occurs in association with cellular membranes. Cytoplasmic vacuoles (Fig. 24.5) with regularly shaped invaginations or spherules (60 nm in diameter) are observed in RV-infected cells.127 The spherules are connected to the vacuolar membranes by thin membranous necks and can be stained with antibodies against p150 and double-stranded RNA, suggesting that they are the sites of viral RNA synthesis.118,125 These vacuoles also co-localize with lysosomal markers, indicating that they are derived from endosomes and/or possibly lysosomes.138
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


