Rhinoviruses
James E. Gern
Ann C. Palmenberg
History
The common cold has been recognized for millennia, and both the illness and prescribed remedies have been influenced by and engendered a broad body of folklore. In fact, the name of the illness stems from the belief that being chilled causes the illness, a concept that studies using experimental inoculation techniques have been unable to verify.53 In Chinese traditional medicine, colds were considered an illness of wind and cold, and the Roman physician Galen wrote “The white-colored substance (the phlegma) collects mostly … in those who have been chilled in some way”.14 Through the ages, ideas about pathogenesis have varied widely, and suggestions for common cold cures were creative and occasionally bizarre, but seldom helpful. Enthusiasm for a cure for the common cold remains quite high today; a web-based search yielded 4,120,000 hits in response to the terms “common cold cure.”
In modern times, Kruse in 1914139 demonstrated that cell-free filtrates of nasal secretions from affected individuals could transmit colds. In 1930, Dochez et al.50 confirmed these findings by transmitting colds to volunteers and apes using filtered nasal secretions that were free of bacteria, indicating a viral etiology. Progress in finding the cause for common colds was accelerated by the establishment of the Common Cold Unit (CCU) by the UK Medical Research Council in 1946.237 The building was originally a hospital established by Harvard University and the American Red Cross to support Great Britain in World War II, and after the war the building was donated to the British government. At the time one virus was presumed to cause colds, and another to cause influenza.237 The goals of the CCU, led by Christopher Andrewes and later David Tyrrell, were to identify the common cold virus, its means of transmission, and host characteristics that promoted more severe illness. From 1946 until the unit closed in 1989, more than 20,000 volunteers participated in these studies.
Rhinoviruses were discovered beginning in 1956 by two groups working independently.190,198 It was not long before researchers realized that several families of viruses caused common cold illnesses, and that human rhinovirus (HRV) serotypes were numerous. In 1967, a collaborative program classified the known rhinoviruses into 55 different serotypes,1 serotypes 56 through 89 were added in 1971,2 and the remainder of the classical serotypes were added in 1987.88
The development of molecular techniques for detecting HRV in a variety of clinical specimens led to a renewed period of discovery related to HRV classification and epidemiology. Recent findings include the discovery of the genetically distinct HRV-C species viruses that do not grow in standard tissue culture, and additional insights into the role of HRV not only in common colds, but also otitis, sinusitis, lower respiratory infections, asthma, and acute exacerbations of chronic respiratory diseases such as asthma.
Infectious Agent
Classification
The human rhinoviruses comprise the HRV-A, HRV-B, and HRV-C species of the Enterovirus genus in the Picornaviridae family. Classification is based on overt similarities in genome organization, capsid properties, and primary sequence conservation.185 Viruses are assigned to these species classifications if they share greater than 70% amino acid identity in the P1, 2C, and 3CD regions with other members. Within species, isolates are subdivided into numeric genotypes (Fig. 18.1). For the HRV-A and HRV-B, several historic clinical panels archived by the American Type Culture Collection, were combined and indexed into 101 original HRV types after assessment of antigenic crossreactivity or serotyping in rabbits. HRV-A87 was subsequently reassigned to the Enterovirus D (HEV-D68) after reevaluation of genetic, immunogenic, and receptor use (decay-accelerating factor as a receptor) properties.210 New HRV isolates, especially for the HRV-C, are now rarely tested for immunogenicity. Type assignments still respect the historic naming system, but rely more heavily on sequence comparisons, primarily of the VP1 protein or VP4/VP2. Strains within a common genotype generally share greater than 12% to 15% aligned amino acid identity in either or both of these regions. Full genome sequencing indicated that some historic types are more closely related than this (e.g., A09 and A95, or A29 and A44), and others such as A45 and A95 defining “clade D” may be sufficiently different as to warrant eventual designation as a fourth species (Fig. 18.1). Because many HRV-C’s are not yet fully sequenced, genotype assignments in this species still rely on comparative VP1 data. Nomenclature conventions for all HRVs cite both the species letter and genotype assignment (e.g., A16, B14, C01) to prevent ambiguity among assigned HRV-A (77 types), HRV-B (25 types), and HRV-C (49 types).
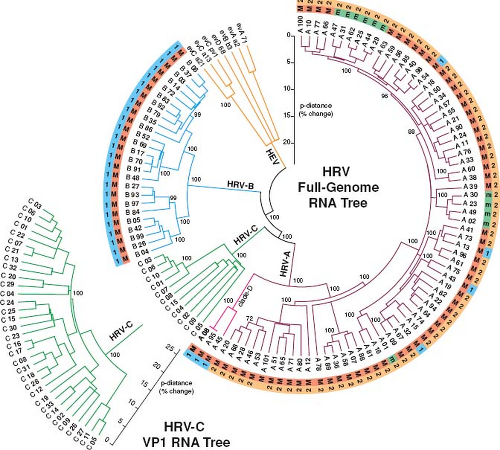 Figure 18.1. Circle phylogram relationships for known genotypes of human rhinovirus (HRV)-A, HRV-B, and HRV-C. The tree was calculated with neighbor-joining methods from aligned, full-genome RNA sequences, and rooted with data from human enterovirus (HEV)-A, HEV-B, and HEV-C species (similar to 186). The outer ring (“1” or “2”) indicates anticapsid drug group types, if known.7 The inner ring shows members of the Major (“M,” intercellular adhesion molecule 1 [ICAM-1]) and minor (“m,” low-density lipoprotein receptor [LDL-R]) receptor groups. The HRV-C receptor is unknown. Because few HRV-C are fully sequenced, relationships among these genotypes rely on partial VP1 RNA data (lower left). Bootstrap values (percent of 2,000 replicates) are indicated at key nodes. |
Physical Characteristics
As enteroviruses, the HRV have genome organizations (Fig. 18.2) and capsid structures (Fig. 18.3) similar to those of polioviruses, coxsackie viruses, and enteric cytopathic human orphan (ECHO) viruses. But unlike other enteroviruses that remain viable at pH 3.0, HRV particles are unstable at pH below 5 to 6. The icosahedral capsid (∼30 nm diameter) has 60 copies each, of proteins VP1, VP2, VP3, and VP4, named in order of descending electrophoretic mobility. The protein shell surrounds a densely packed, single-stranded, positive-sense RNA genome of 7079 (C01) to 7233 (B92) bases, a count that does not include the variable length 3′ poly(A) tail. Several HRV capsids have been resolved at the atomic level resolution, including A01 (1r1a), A02 (1fpn), A16 (1ayn), B03 (1rhi), and B14 (4hrv), with multiple structure variants showing receptor interactions (e.g., A16 with intercellular adhesion molecule 1 [ICAM-1], 1d3e) or antiviral drug interactions (e.g., A16 with pleconaril, 1c8m). Like poliovirus, the surfaces of HRV-A&B capsids are dominated by the three largest proteins (Fig. 18.3). VP4 is internal to the structure, centered near the fivefold axis. Around the exterior fivefold plateau, a symmetrical “canyon” provides receptor binding sites and immunogenic surfaces. There are no current structures for an HRV-C, but available sequences indicate significant deletions in the VP1 regions contributing to the fivefold plateau, so it is likely the HRV-C have unique topologies.161 Common to all HRV, the VP1 cores surround a hydrophobic “pocket” or cavity, which can uptake and bind antiviral drugs like pleconaril or other capsid-binding agents. Type-1 long (e.g., B14) or type-2 short (e.g., A16) pocket shapes are defined for most
HRV-A&B and determine whether a virus is susceptible to particular drugs aimed at inhibiting the uncoating process.7
HRV-A&B and determine whether a virus is susceptible to particular drugs aimed at inhibiting the uncoating process.7
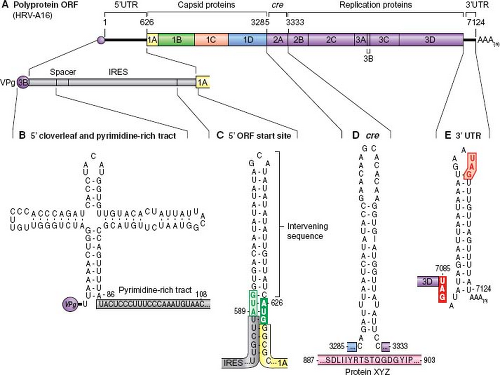 Figure 18.2. The genome of an HRV encodes a single polyprotein open reading frame (ORF) (A). Important RNA structural motifs include a 5′ cloverleaf (B), ORF start-site stem (C), a cre element (D), and 3′ stem motif (E). |
The HRV genome is messenger-sense, encoding the polyprotein open reading frame (ORF) and multiple important RNA structural motifs (Fig. 18.2). Adjacent to the 5′ cloverleaf, a regulatory feature for translation and replication, each HRV encodes a strain-specific pyrimidine-rich tract that may be involved in suppressing innate immunity triggers.186 The type-1 internal ribosome entry site (IRES) 3′ to this tract, includes a variable-length stem structure pairing the ORF start site (AUG) with an upstream AUG. Unlike poliovirus, intervening sequences between these AUGs are probably not scanned by initiating ribosomes.114 The picornavirus VPg uridylylation reaction, required for RNA synthesis, is templated by a special structure called the cre (cis-acting replication element) whose location varies in every species of picornavirus. For the HRV-A, the cre is in the 2A gene.225 For the HRV-B, the cre is in the 2C gene.225 The HRV-C cre has been proposed as one of two sites in the 1B gene.37,186,225 Neither has been confirmed experimentally. The short, 3′ untranslated sequences (untranslated regions, UTRs) are of highly variable sequence. Invariably, they configure as an inclusive stem motif displaying at least one bogus termination codon in the terminal loop. This codon may be in-frame or out-of-frame with the authentic ORF stop site, and has been proposed to play a role in the recruitment of translation termination factors.186
Pathogenesis and Pathology
Entry into the Host
The primary portal of entry for HRV infections is through inoculation of either the eyes or nose. Studies of seronegative infected volunteers have shown that very low doses of HRV, substantially less than the amount needed to infect cells in vitro (one tissue culture infectious dose50 [TCID50]), can cause infection when introduced via the conjunctiva or nasal mucosa.41 In contrast, approximately 10,000 times as much virus is needed to cause productive infection when the inoculation site is the tongue or external nares.41
Site of Primary Replication
The primary site of infection is the airway epithelium. In studies of airway tissues from either natural or experimentally induced colds, detection of viral protein or RNA is largely confined to the epithelial layer, along with an occasional cell in
the subepithelial layer (Fig. 18.4).11,23,175 Highly differentiated epithelial cells grown at air–liquid interface are more resistant to HRV infection compared to undifferentiated monolayers of cells.154 In addition, mechanical damage to well-differentiated cells significantly enhances viral replication in vitro.116 These findings suggest that the epithelial barrier function is an important contributor to the resistance against HRV infections. In addition, differentiation of epithelial cells in tissue culture in the presence of interleukin-13 (IL-13) in vitro causes increased numbers of goblet cells and greater HRV replication in vitro, suggesting that HRV replication is increased in goblet cells.143
the subepithelial layer (Fig. 18.4).11,23,175 Highly differentiated epithelial cells grown at air–liquid interface are more resistant to HRV infection compared to undifferentiated monolayers of cells.154 In addition, mechanical damage to well-differentiated cells significantly enhances viral replication in vitro.116 These findings suggest that the epithelial barrier function is an important contributor to the resistance against HRV infections. In addition, differentiation of epithelial cells in tissue culture in the presence of interleukin-13 (IL-13) in vitro causes increased numbers of goblet cells and greater HRV replication in vitro, suggesting that HRV replication is increased in goblet cells.143
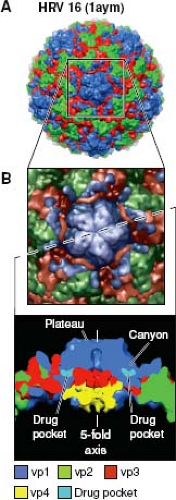 Figure 18.3. Capsid structure of a human rhinovirus (HRV) (A), illustrates the icosahedral surface topography of VP1, VP2 and VP3 proteins. The VP4 is internal to the capsid (B). Type 1 and type 2 antiviral capsid drugs bind in a pocket, internal to VP1. |
Spread
For years it was assumed that HRV infection was confined to the upper airway and did not affect the chest except under unusual circumstances. In fact, one of the breakthroughs that led to successful propagation of HRV in cell culture was the discovery that this virus replicated best at 33°C to 35°C. It was assumed that the lower airways were at core temperature (37°C), and this characteristic was thought to limit HRV replication to the cooler upper airways. Contrary to these initial assumptions, direct measurements in the lower airways have shown that large- and medium-sized airways are at the ideal temperature for HRV replication (Fig. 18.5).163 In addition, cultured lower airway epithelial cells support HRV replication in vitro at least as well, and perhaps even better, than cells derived from the upper airways.155,174
Following experimental inoculation of the upper airway, HRV has been detected in the lower airways of individuals with a variety of techniques. Secretions from the lower airways sampled by bronchoscopy and bronchial lavage were analyzed by reverse transcriptase–polymerase chain reaction (RT-PCR), and more than half of the lower airway specimens tested positive for HRV.73,214 These findings were extended by subsequent
studies demonstrating the presence of intracellular HRV RNA and protein using in situ hybridization and immunohistochemistry, respectively.175,187 Analysis of sputum has been used to provide an estimate of the quantity and kinetics of viral shedding from the large lower airways. After experimental inoculation of seronegative volunteers, viral shedding from the upper airway peaks 2 to 4 days later. Analysis of sputum specimens 3, 7, and 14 days after inoculation revealed that peak levels of virus in the sputum occurred 3 to 7 days after inoculation.175 In addition, about half of the volunteers had viral shedding in the sputum that was equal to or exceeded that found in the nasal secretions. Notably, only small amounts of virus were detected in bronchial lavage specimens, which originate from distal airways and alveoli.175 These findings suggest that HRV replication is greatest in the upper airway in most individuals, and that high-level replication can also occur in the large- and medium-sized airways.
studies demonstrating the presence of intracellular HRV RNA and protein using in situ hybridization and immunohistochemistry, respectively.175,187 Analysis of sputum has been used to provide an estimate of the quantity and kinetics of viral shedding from the large lower airways. After experimental inoculation of seronegative volunteers, viral shedding from the upper airway peaks 2 to 4 days later. Analysis of sputum specimens 3, 7, and 14 days after inoculation revealed that peak levels of virus in the sputum occurred 3 to 7 days after inoculation.175 In addition, about half of the volunteers had viral shedding in the sputum that was equal to or exceeded that found in the nasal secretions. Notably, only small amounts of virus were detected in bronchial lavage specimens, which originate from distal airways and alveoli.175 These findings suggest that HRV replication is greatest in the upper airway in most individuals, and that high-level replication can also occur in the large- and medium-sized airways.
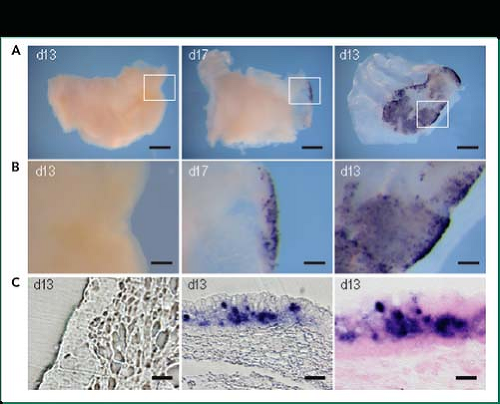 Figure 18.4. Human rhinovirus (HRV)-C15 replication within epithelial cells in sinus organ culture. A: Sinus tissue in organ culture was inoculated with HRV-C15 (center and right) or medium alone (left), and whole mounts of the tissue were analyzed for HRV-C15 RNA by in situ hybridization (purple stain). Scale bars, 1 mm. B: Higher magnification view of the areas boxed in panel a, showing uninfected cells (left) or cells containing viral RNA (center and right). Scale bars, 0.15 mm. C: Sections of mock (left) or HRV-C infected (center and right) sinus tissue. Right panel is counterstained with eosin (pink). Scale bars, 15 μm. (From Bochkov YA, Palmenberg AC, Lee WM, et al. Molecular modeling, organ culture and reverse genetics for the emerging respiratory pathogen human rhinovirus C. Nat Med 2011;17:627–632; with permission.) |
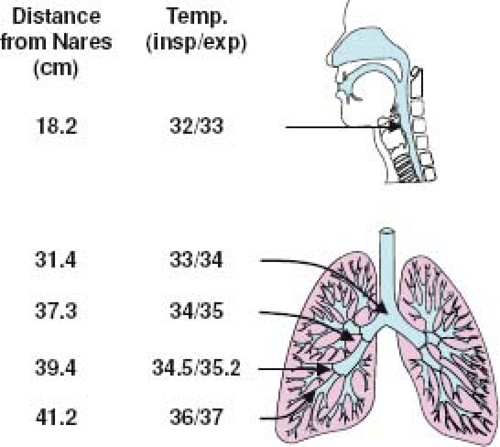 Figure 18.5. Temperatures in the lower airways are ideal for human rhinovirus (HRV) replication. Direct measurements of temperature in lower airways have been recorded at measured distances from the nares using a bronchoscope equipped with a small thermistor.163 Even when the inspired air is at room temperature (26.7°C), airway temperatures in the medium and large airways are in the range of 33°C to 35°C. In contrast, small airway temperatures approach core temperature (37°C). insp, inspiration; exp, expiration. |
Data from natural cold studies also provide evidence of lower airway infections with HRV. Viral recovery from sputum during colds can exceed that obtained from upper airway samples.106 In epidemiologic studies, HRV is frequently detected in children who are hospitalized with wheezing illnesses and in infants with pneumonia.166,167,193 In wheezing infants, HRV has been detected in lower airway biopsies, and HRV detection was associated with reduced lung function in these infants.158 In children with tracheostomies, samples of nasal mucus can be obtained directly from the lower airway without contamination from nasal secretions, and HRV detection rates from upper versus lower airway specimens are similar.217
In addition to infecting the nasopharynx, conjunctiva, and lower airways, HRV has also been recovered in specimens obtained from the middle ear and sinuses.29,117,194,196 The respiratory epithelium in each of these two locations is contiguous with that of the nasopharynx, and the virus presumably spreads via local extension. Rhinovirus viremia has been detected in infected children by PCR but not by recovery of infectious virus,260 and a study of experimentally infected adults showed no evidence of circulating HRV RNA.45 Therefore, systemic infections do not occur in immune competent individuals. HRV are inactivated at pH less than 6, thus preventing swallowed virus from replicating in the gastrointestinal tract.
Cell and Tissue Tropism
Biopsies of the upper airway from infected volunteers show a patchy pattern of infection with small foci of infected cells.10,195,256 Point cultures of the airway have demonstrated high levels of HRV shedding in the nasopharynx and especially in the adenoidal region. Examination of biopsies obtained during experimentally induced colds suggest that a specific type of nonciliated adenoidal epithelial cell, resembling the intestinal M cell, expresses high levels of ICAM-1 and supports high-level viral replication.255 It is possible that these cells play a sentinel role in the detection of viral respiratory infections.
The receptors for HRV are also expressed by airway cells other than epithelial cells. Besides epithelial cells, HRV can bind to macrophages, monocytes, eosinophils, and fibroblasts.72,75,90 Macrophages and monocytes are good sources of type I and type III interferons,131,136 which may explain why there is little or no HRV replication in these cells. Airway fibroblasts75 and possibly smooth muscle cells140 support HRV replication in tissue culture, but it has not been established whether these cells, which are located several cell layers under the epithelial surface, are infected in vivo.
Receptors
All HRV-A&B use ICAM-1 (88 “major” types) or alternatively, low-density lipoprotein receptor (LDL-R, 11 “minor” types) for recognition and attachment to cells.78,243 In addition, the major group virus HRV-A89 binds to heparan sulfate in the absence of ICAM-1.242 The external cellular receptor(s) used by the HRV-C are most certainly different but yet to be identified.23,161 Studies utilizing atopic force spectroscopy indicate that after initial HRV binding, multiple receptors are rapidly recruited (within 200 ms).205
Species Tropism
Rhinoviruses primarily infect humans. Chimpanzees can be infected, but although viral shedding can be detected, there are few or no signs or symptoms of illness.108 HRV replication in rabbit smooth muscle cells has been reported by a single group.86 HRV do not bind to murine ICAM-1, but can bind to murine LDL-R.261 Accordingly, minor group viruses can replicate in murine epithelial cells,24,236 and strains have been selected for this property by serial passage in murine cell lines.17,91 In addition, epithelial cells from transgenic mice engineered to express human ICAM-1 support replication of major group viruses.17
The demonstration that HRV can grow in murine epithelial cells has led to the development of murine models of HRV infection.17,24,178,180,236 Several features of these models resemble infection of the human, including replication in the respiratory epithelium, induction of type I interferons and neutrophil chemokines, and neutrophilic airway inflammation.17,180 In addition, HRV infection of mice increases inflammation in response to allergen exposure, suggesting that these models could be informative for asthma.17 Limitations of the models include the requirement of a large inoculating dose,
and short duration (<24 hours) of significant viral replication and shedding. A second rodent model of picornavirus respiratory infection was developed using a genetically altered strain of Mengovirus, a natural mouse pathogen that causes a polio-like syndrome.206 Intranasal inoculation of rats with attenuated Mengovirus causes infection of the airway epithelium with similarities to HRV infections in humans. The ability to investigate HRV host–cell interactions in vivo using the genetic tools available in rodent models should yield important new insights into the pathogenesis of infection in humans.
and short duration (<24 hours) of significant viral replication and shedding. A second rodent model of picornavirus respiratory infection was developed using a genetically altered strain of Mengovirus, a natural mouse pathogen that causes a polio-like syndrome.206 Intranasal inoculation of rats with attenuated Mengovirus causes infection of the airway epithelium with similarities to HRV infections in humans. The ability to investigate HRV host–cell interactions in vivo using the genetic tools available in rodent models should yield important new insights into the pathogenesis of infection in humans.
Immune Response
Immunohistochemistry
HRV infections produce relatively little cytopathology. Findings on biopsy during the peak of the illness include tissue edema and a sparse infiltration of neutrophils and mononuclear cells.253,254 The relative lack of pathology together with the small number of infected cells suggests that the immune response to HRV rather than direct virus-induced injury contributes most to respiratory symptoms.97
Mediators of Inflammation
HRV infection induced a variety of proinflammatory cytokines and mediators (Table 18.1). Most of these factors are present in peak concentrations during the acute cold, and correlate with illness severity and viral shedding.16 There is only circumstantial evidence that these immune mediators contribute to the pathogenesis of cold signs and symptoms, and the specific roles for most of these factors have yet to be defined. Kinins, leukotrienes, and histamine are of special interest due to their effects on vasodilation and edema, which are prominent findings during acute colds.110,262 HRV infection of volunteers with asthma induced increased mRNA and activity of human kallikrein, a critical enzyme for the release of kinins, in the lower airways.33 These mediators can also cause smooth muscle contraction as well as edema, and thus could promote airway obstruction in patients with asthma. Notably, common colds have little effect on concentrations of histamine in the nose,61 and second-generation antihistamines (which are specific H1-receptor antagonists), have no effect on cold symptoms.176 Leukotrienes (LTs) are overproduced during viral respiratory infections,70 but the cystinyl leukotriene receptor antagonist montelukast did not affect cold symptoms in experimentally infected volunteers.134 In addition, administration of LTB4 to the nose before experimental inoculation had no significant effects on either viral replication or symptoms.251 Kinins are increased early in the course of a cold,200 but testing of their role in pathogenesis awaits the development of safe and effective kinin inhibitors. HRV-induced cytokines may also promote vascular permeability; experiments conducted in vitro demonstrate that tumor necrosis factor α (TNF-α), IL-1β, and IFN-γ can each increase vascular permeability.212 Analysis of nasal secretions after experimental inoculation demonstrates that the thin rhinorrhea during the early stages of a cold contains serum proteins (e.g., immunoglobulin [IgG], albumin), and as the cold progresses the composition shifts to mucin proteins.110,262 This progression suggests that the pathophysiology of rhinorrhea is time-dependent, and shifts from vascular leakage initially to local secretion of mucus during the mid to latter stages of a cold.
Epithelium
The airway epithelium is the first line of defense against HRV infections. Mucins, antimicrobial peptides, and surfactant proteins in the mucous layer nonspecifically deter infection.103 In addition, well-differentiated epithelial cells are relatively
resistant to HRV infection.154 The epithelium may serve as a barrier against HRV infection; HRV replication is enhanced when apical cells of well-differentiated epithelial cell cultures are either damaged or stripped away.116 This property may help to explain how exposure to factors such as pollutants and allergic inflammation that can damage epithelium also increase the risk and/or severity of HRV illnesses. HRV infection itself can disrupt the barrier function of the epithelium,207 which may contribute to synergistic effects with bacteria, allergens, or irritants.
resistant to HRV infection.154 The epithelium may serve as a barrier against HRV infection; HRV replication is enhanced when apical cells of well-differentiated epithelial cell cultures are either damaged or stripped away.116 This property may help to explain how exposure to factors such as pollutants and allergic inflammation that can damage epithelium also increase the risk and/or severity of HRV illnesses. HRV infection itself can disrupt the barrier function of the epithelium,207 which may contribute to synergistic effects with bacteria, allergens, or irritants.
Table 18.1 HRV-Induced Mediators and Cytokines | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In addition to being the principal host cells for HRV infections, airway epithelial cells initiate antiviral and proinflammatory immune responses (Fig. 18.6). Attachment of HRV to ICAM-1 activates signaling cascades that lead to the activation of chemokine genes such as CXCL10.69,136 Once HRV particles uncoat in the endosome, the release of single-stranded RNA activates additional innate immune sensors. These include toll-like receptors (TLRs) (e.g., TLR-3, TLR-7) that are expressed on endosomal membranes, and the double-stranded RNA-dependent protein kinase (PKR) and RNA helicases (retinoic acid-induced gene I [RIG-I] and melanoma-differentiation-associated gene-5 [MDA-5]). MDA-5 binds to double-stranded RNA that is formed during the replication process, and appears to be an important contributor to anti-HRV IFN responses.220,244 The role of RIG-I, which binds to the 5′ triphosphate motif and longer stretches of double-stranded RNA,19 is more controversial. One recent in vitro study found that TLR-3 was constitutively expressed on airway
epithelial cells, and activation of TLR-3 led to induction of RIG-I and MDA-5.220 Subsequently, all three of these molecules contributed to upregulation of innate IFN responses to HRV infection.
epithelial cells, and activation of TLR-3 led to induction of RIG-I and MDA-5.220 Subsequently, all three of these molecules contributed to upregulation of innate IFN responses to HRV infection.
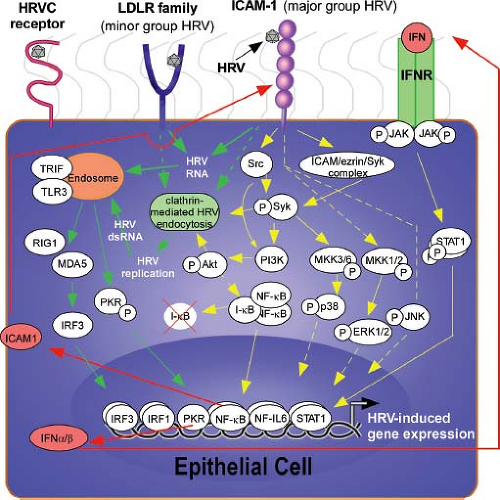 Figure 18.6. Airway epithelial cells initiate the immune response to human rhinovirus (HRV). Major group HRV binds to intercellular adhesion molecule 1 (ICAM-1) and minor group HRV binds to low-density lipoprotein receptors (LDL-Rs) on the surface of cells, such as epithelial cells and leukocytes, that would be abundant in the airway, to induce downstream signaling. The receptor and signaling pathways induced by HRV-C types has yet to be elucidated. Upon ligation of HRV to surface receptors on epithelial cells, direct uncoating of viral RNA or clathrin-mediated endocytosis leads to the release/replication of HRV RNA into the cytoplasm. This viral RNA is detected by endosomal receptors, such as toll-like receptor 3 (TLR-3), to propagate downstream signaling and induction of gene expression, including the production of interferons (IFNs) that can exert an autocrine/paracrine effect by binding to IFN receptors and triggering a janus kinase (JAK)-signal transducers and activators of transcription (STAT) signaling cascade. Replication-independent signaling is also induced upon HRV infection, and includes the activation of Src, Syk, and mitogen-activated protein kinase (MAPK) signaling cascades. Yellow solid lines, data to support link; dashed lines, indirectly connected (may have signaling molecules in between); red lines, signaling induced by product of HRV-induced gene transcription; green lines, replication-dependent signaling. (From Gavala ML, Bertics PJ, Gern JE. Rhinoviruses, allergic inflammation, and asthma. Immunol Rev 2011;242:69–90, with permission.) |
Engagement of cell surface receptors and RNA-sensing molecules leads to activation of several signaling cascades, including nuclear factor kappa B (NFκB), janus kinase (JAK)-signal transducers and activators of transcription (STAT) pathways, and mitogen-activated protein kinases (MAPKs).60,87,99,132,136,144,146,179,180,245,263 Viral proteases can also contribute to cell activation; the 3C protease cleaves the transcription factor organic cation transporter-1 (OCT-1),6 which negatively regulates CXCL8 (interleukin-8) transcription. The net result of these actions is activation of viral sensing and antiviral effector pathways, IFNs, and a variety of proinflammatory cytokines and chemokines.22,31,202 Type I IFNs inhibit HRV replication by inducing multiple antiviral pathways, and also prime epithelial cells to synthesize chemokines such as CXCL10.6,137 IFN-γ has several effects on viral replication and inflammatory responses. It is a potent inducer of ICAM-1135 and soluble ICAM-1,250 and effects on viral replication in cultured epithelial cells depend on experimental conditions. In addition to affecting viral receptor expression, IFN-γ also enhances the HRV-induced secretion of chemokines such as CCL5.135 HRV-induced chemokines from epithelial cells and other sources attract a variety of cells into the airway, including dendritic cells, monocytes, macrophages, epithelial cells, and lymphocytes (Table 18.1). The ensuing cellular response promotes antiviral effects and killing of infected cells, but also increases inflammation that contributes to the pathophysiology of illness.
Cellular Immune Responses
Macrophages are the principal cells in lower airway secretions, and it is likely that these cells are important contributors to the innate antiviral response. Considering that little to no replication of HRV has been observed in airway macrophages or blood monocytes,72,146 the signaling pathways could differ from those in epithelial cells, in which viral replication processes provide potent innate immune stimulation. As in epithelial cells, HRV activate NF-κB and STAT-1-dependent pathways. For example, HRV-induced secretion of CCL2 involves activation of NF-κB and phosphorylation of activating transcription factor-2 (ATF-2).87 Furthermore, HRV-A16 induces STAT-1 tyrosine phosphorylation and IFN-α release in monocytes.136 NF-κB activation, but not the p38 MAPK, has been reported to be involved in HRV-mediated TNF-α release by monocytes.146
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


