Rhabdoviridae
Douglas S. Lyles
Ivan V. Kuzmin
Charles E. Rupprecht
The family Rhabdoviridae consists of more than 185 different viruses isolated from both plants and animals. They are enveloped viruses that have helical nucleocapsids containing single-stranded, negative-sense RNA and share a common elongated, rod-like or bullet-like shape. This distinctive morphology separates rhabdoviruses from other taxa in the order Mononegavirales, the Bornaviridae, the Filoviridae, and the Paramyxoviridae. Rhabdoviruses can replicate in plants, invertebrates, or vertebrates. The family Rhabdoviridae contains many members that are significant medical, veterinary, and agricultural pathogens. Currently, animal rhabdoviruses include four genera: Lyssavirus, Vesiculovirus, Ephemerovirus, and Novirhabdovirus (Table 31.1). Many other rhabdoviruses have not received adequate study and are assigned to the family solely on the basis of morphology.
History
Lyssaviruses
Rabies is an archaic entity, one of the oldest recognized infectious diseases. The continuing biomedical preoccupation with rabies is understandable because of its “alarming manifestations in man and dog alike … and its almost inevitable progression to a fatal outcome have ensured unparalleled notoriety”.772 These concerns extend beyond the material to the spiritual plane, as revealed by the following prayer: “San Roque, San Roque, que este perro no me toque!” This supplication for protection to the patron saint against pestilence, taught to children in both the Old and the New World for invocation whenever they encountered a dog on the street, literally translates to “St. Roque, St. Roque, do not allow this dog to touch me!”—classically linking dogs, bites, and resulting misfortune.
Ancient civilizations were familiar with rabies. An early passage mentions the dangers of dog bites, in the pre-Mosaic
Eshnunna Code of Mesopotamia, circa the 23rd century BC: “If a dog is mad and the authorities have brought the fact to the knowledge of its owner; if he does not keep it in, and it bites a man and causes his death, then the owner shall pay two-thirds of a mina [40 shekels] of silver”.41 In The Iliad (700 BC), Hector is compared to a rabid dog. Chinese scholars warned of the dangers of rabid dogs in 500 BC, and Aristotle (4th century BC) correctly associated the disease with animals but erroneously exempted humans from contracting it from a mad dog’s bite. In Rome, Cordamus guessed that a poison (i.e., a “virus”) was present in saliva. Similarly, in the 1st century AD, another Roman, Celsus, described clinical aspects of human infection: “The patient is tortured at the same time by thirst and by invincible repulsion toward water.” For prevention, he recommended immediate excision of the bitten tissue, cauterization of the wound by a hot iron, and dunking the victim into a pool. The Hebrew Talmud, also dating from the 1st century, makes several references to the disease. Throughout the ages, ingestion of a wide variety of substances (e.g., the liver from a mad dog, crayfish eyes, a cock’s brain or comb, or the cast slough of snakes pounded in wine with a male crab) and carrying sacred talismans or “madstones” were believed to be cures for rabies.41
Eshnunna Code of Mesopotamia, circa the 23rd century BC: “If a dog is mad and the authorities have brought the fact to the knowledge of its owner; if he does not keep it in, and it bites a man and causes his death, then the owner shall pay two-thirds of a mina [40 shekels] of silver”.41 In The Iliad (700 BC), Hector is compared to a rabid dog. Chinese scholars warned of the dangers of rabid dogs in 500 BC, and Aristotle (4th century BC) correctly associated the disease with animals but erroneously exempted humans from contracting it from a mad dog’s bite. In Rome, Cordamus guessed that a poison (i.e., a “virus”) was present in saliva. Similarly, in the 1st century AD, another Roman, Celsus, described clinical aspects of human infection: “The patient is tortured at the same time by thirst and by invincible repulsion toward water.” For prevention, he recommended immediate excision of the bitten tissue, cauterization of the wound by a hot iron, and dunking the victim into a pool. The Hebrew Talmud, also dating from the 1st century, makes several references to the disease. Throughout the ages, ingestion of a wide variety of substances (e.g., the liver from a mad dog, crayfish eyes, a cock’s brain or comb, or the cast slough of snakes pounded in wine with a male crab) and carrying sacred talismans or “madstones” were believed to be cures for rabies.41
 |
The transition from the medieval era to the Renaissance period of pragmatism and experimentation resulted in a remarkable treatise in 1546, entitled “The Incurable Wound,” by Fracastoro. This Italian physician clearly stated that human beings are susceptible to rabies, and he vividly described a clinical case:
Its incubation [following a bite by a rabid animal] is so stealthy, slow and gradual that the infection is very rarely manifest before the 20th day, in most cases after the 30th, and in many cases not until four or six months have elapsed. There are cases recorded in which it became manifest a year after the bite. [Once the disease takes hold,] the patient can neither stand nor lie down; like a madman he flings himself hither and thither, tears his flesh with his hands, and feels intolerable thirst. This is the most distressing symptom, for he so shrinks from water and all liquids that he would rather die than drink or be brought near to water; it is then that they bite other persons, foam at the mouth, their eyes look twisted, and finally they are exhausted and painfully breathe their last.360
His portrayal of human rabies is accurate in that the incubation periods can extend from months to years after initial exposure,662 but a biting attack on others by a rabid patient with resultant disease is an uncommon event.218
Although rabies is known to have been widespread in the Old World for thousands of years, its occurrence in the New World is less understood because of a dearth of records before European arrival. Rabies in the Americas was reported by the Reverend Marmolejo in Mexico as early as 1709, but some suspect that it was present before Columbus’s arrival in the 15th century. For example, not long after the discovery of the Americas, the bishop Petrus Martyr-Anglerius wrote in his De Rebus Oceanicis et de Orbi Novi Decades Octo, “In several places bats not much smaller than turtle doves used to fly at them [the Spanish sailors and soldiers] in the early evening with brutal fury and with their venomous bites brought those injured to madness … [and] bats … come in from the marshes on the river and attack our men with deadly bite”.388 This may have been one of the first descriptions of rabies transmission by vampire bats.
The bite of a rabid animal was considered a likely source of rabies infection by many, but it was only in 1804 that Zinke used dog saliva for transmission.386 Later in 1879, Galtier is credited with experimental rabies transmission and serial passage in rabbits.386 Clinical descriptions formed the basis for diagnosis until the advent of light microscopy. A clear description of viral and neuronal interactions was made by Negri in 1903, with the detection of cytoplasmic inclusions (Negri bodies) in neurons of rabid animals.393 Although the diagnostic value of Negri bodies was established by 1913, their viral composition had to wait until the later development of electron microscopy.
Pasteur’s research on rabies is perhaps the most well-known historical achievement in the field. First, through adaptation of “street” (wild-type) virus to laboratory animals, he was able to change its properties. Today, one could apply the term attenuated to his “fixed” virus strains. Second, Pasteur and his team developed concepts and experimental approaches to the first protective vaccination against rabies.388 Desiccated spinal cords from rabies virus–infected rabbits became the first rabies vaccine, and they were supposedly safe, although now it is known that the fixed viruses from which these vaccines were derived were not apathogenic but could actually cause the disease. July 6, 1885, is a milestone in the history of rabies. On that day, 9-year-old Joseph Meister was bitten at multiple sites by a rabid dog and received the first postexposure prophylaxis with Pasteur’s vaccine. Remarkably, Joseph survived.386 Pasteur’s vaccine, with all its modifications, became the accepted rabies prophylactic throughout the world in the early 20th century. Problems remained, however, because improperly inactivated virus caused rabies, and animal brain tissue induced allergic reactions leading to neuroparalytic accidents. Moreover, the vaccine was not very effective in cases of severe bites, such as those inflicted on the face and neck by rabid wolves and dogs.
Postexposure prophylaxis against rabies through simultaneous administration of antirabies serum and vaccine was introduced in 1889 by Babes.29 This approach found few adherents and languished until about 1940, when interest in the use of serum-containing rabies virus (RABV) antibodies was revived. In a trial organized by the World Health Organization in 1954, the combined use of serum and vaccine was found to be more protective than vaccine alone,288 an observation later corroborated by Chinese findings.212 Today, the combination of immune globulin and vaccine is the recommended standard for prophylaxis in human rabies exposure.
In the 1960s, an RABV grown in human diploid cells was used to produce a safe and efficacious inactivated vaccine,385,386 eliminating many of the problems connected with vaccines produced in brain tissue. This vaccine and others derived from cell culture are used widely throughout the world, although for economic reasons, several developing countries still use nervous tissue vaccines. Other RABV strains are used for vaccine production for human and animal use in addition to the original Pasteur virus (PV) strain. Given the progress in biotechnology, improved versions of rabies vaccines are currently under development.
Vesiculoviruses
Vesicular stomatitis virus (VSV) is the best-studied member of the genus Vesiculovirus. The extensive body of knowledge about
the replication of VSV reflects its status as a widely studied prototype for the nonsegmented, negative-strand RNA viruses. VSV produces an acute disease in cattle, horses, and pigs characterized by fever and vesicles in the mucosa of the oral cavity and in the skin of the coronary band and teat. Clinically, VS is very similar to foot-and-mouth disease (FMD). VSV can also cause an acute febrile disease in humans. Laboratory-adapted strains, however, are rarely pathogenic for humans.
the replication of VSV reflects its status as a widely studied prototype for the nonsegmented, negative-strand RNA viruses. VSV produces an acute disease in cattle, horses, and pigs characterized by fever and vesicles in the mucosa of the oral cavity and in the skin of the coronary band and teat. Clinically, VS is very similar to foot-and-mouth disease (FMD). VSV can also cause an acute febrile disease in humans. Laboratory-adapted strains, however, are rarely pathogenic for humans.
Although VS was first reported in the United States in 1916 during an epidemic in cattle and horses,695 a clinically similar disease was previously described in army horses in 1862, during the U.S. Civil War.484 In 1915, French veterinarians described a disease clinically similar to VS in horses imported to Europe from the United States and Canada during World War I. At that time, the etiology of this disease could not be determined with certainty, but it could be transmitted from horse to horse by rubbing the saliva of a sick animal on the tongue of a healthy one, establishing the infectious nature of the disease.295 In 1925, cattle transported from Kansas City, Missouri, to Richmond, Indiana, initiated an outbreak of VS in the area. The disease was experimentally transmitted to horses and the infectious agent was maintained by serial passages in animals. This strain became the VS-Indiana virus (VSIV) strain.149 In 1926, an outbreak of VS in cattle occurred in New Jersey. The causative agent was found to be a filterable agent that could infect cattle, horses, and guinea pigs. This virus, serologically different from the VSIV strain, is currently known as the VS-New Jersey virus (VSNJV) strain.148,149
The VSIV and VSNJV viruses represent the two serotypes most commonly isolated in the Americas. Most of the commonly studied laboratory-adapted strains of VSV (e.g., Glasgow, Orsay, San Juan, Mudd-Summers) belong to the VSIV serotype. In the United States, the last reported outbreak of the VSIV serotype occurred in 1965.751 The VSNJV serotype was responsible for outbreaks in the United States in 1944, 1949, 1957, 1959, 1963, 1982–1983, 1985, and 1995.87 In 1997, isolated cases were diagnosed in several horses in New Mexico, but this did not initiate an outbreak.21 Between 1946 and 1954, during an outbreak of FMD in Mexico,270 the joint Mexico–American commission for the control of FMD developed techniques for the differential diagnosis of FMD and VS based on the isolation of the agent and complement fixation methods.295 The availability of a more efficient diagnostic methodology demonstrated that VS was prevalent throughout the year in the tropical areas of Mexico.295
In South America, the disease was reported in 1939 in La Plata, Argentina.295 Later, VSV was isolated in Barinas, Venezuela, in 1941, and in Colombia in 1943.295 Currently, VSV is endemic in many Latin American countries and is responsible for important economic losses in the livestock industry. Disease caused by VSV was reported in 11 countries of Latin America in 1996.788 Although the presence of VS was previously suggested in Africa in 1884 to 1887 and in Asia in 1944,295 presently the disease is considered enzootic only in the Americas.788
Other vesiculoviruses are endemic in the Americas, Asia, and Africa. Piry virus was isolated from an opossum (Philander opossum) in Brazil in 1960701 and caused a febrile disease in humans.474 Cocal virus (COCV, or Indiana 2) was isolated from mites of the genus Gigantolaelaps from rice rats (Oryzomys laticeps velutinus) trapped during 1961, on Bush Bush Island in the Nariva swamp in eastern Trinidad.354 The VS Alagoas virus (VSAV, or Indiana 3) was isolated from domestic animals in the state of Alagoas, Brazil, during a VS outbreak.700 Later it also was isolated from sand flies and seropositive (but otherwise healthy) livestock in Colombia.700 Maraba virus (MARAV) was isolated from sand flies (Lutzomyia sp.) collected in the state of Pará, Brazil. Although humans are infrequently infected based on serology, the actual public health significance of MARAV has not been assessed.709
Vesiculoviruses endemic in Asia include Chandipura virus (CHPV) and Isfahan virus (ISFV). ISFV was isolated from sand flies (Phlebotomus papatasi) collected in Dormian, Isfahan Province, Iran, in 1975.699 From serologic analyses, the presence of ISFV has been detected in India, Iran, Turkmenistan, and other Asian countries.474 CHPV was obtained from the sera of two patients with a febrile illness in Nagpur City, Maharashtra State, India, in 1965 during an epidemic of chikungunya and dengue.66 This virus was also isolated from phlebotomine sand flies in West Africa in 1991.243 CHPV is now known to be a cause of viral encephalitis in children, following its identification as the cause of two recent outbreaks. One outbreak in 2003 in Andhra Pradesh State, India,580 included 329 cases (183 fatalities), and another in 2004 in Gujarat State, India,114 included 26 cases (at least 18 fatalities).
Several vesiculoviruses infect fish, and at least one of these, Spring viremia of carp virus (SVCV), has been recognized as a species of the Vesiculovirus genus. Dating back possibly to the Middle Ages, common carp Cyprinus carpio in European pond culture have been plagued by a complex of infectious diseases variously known as infectious dropsy, rubella, infectious ascites, hemorrhagic septicemia, and red contagious disease.59,316,621,706 These diseases proved to be of great economic importance, causing serious losses in carp pond fisheries of the central and eastern parts of Europe.227,228 The proposed causes (nutrition, environment, parasites, bacteria, viruses) for the acute and chronic forms of the epizootics remained controversial for a long time. However, a viral etiology for the acute form of infectious dropsy became evident when a cytopathic agent was isolated,706 and River’s postulates were fulfilled using virus isolated from affected carp.229 In order to distinguish the viral disease from other etiologic entities within the infectious dropsy complex, the disease was renamed spring viremia of carp (SVC), and the causative virus was termed SVCV (or, initially, Rhabdovirus carpio).229
SVCV has been identified in different parts of Europe, Russia, and the Middle East, causing mortality of up to 70% of young carps.9,58,61,99,229,645,698,706 In 2002, SVCV was first reported in U.S. waters at a North Carolina koi hatchery. Unfortunately, there is evidence that koi had been distributed from this hatchery to most of the 48 contiguous states before being confirmed with SVC. The first common carp die-off of wild fish that tested positive for SVC occurred in 2002 at Cedar Lake, Wisconsin,183 and the virus has rapidly disseminated to other states.
Ephemeroviruses
The first reference to bovine ephemeral fever (BEF) can be found in the book the Heart of Africa.636 Not until the 20th century was the disease reported among ruminants in much of its natural range throughout the tropical and subtropical regions of Africa, Asia, Australia, and the Middle East.671 The apparent emergence and re-emergence of BEF over 125 years is likely due to the expansive growth of the cattle industry and
improved surveillance.735 Until 1966, when bovine ephemeral fever virus (BEFV) was grown in mice,723 research on the agent was restricted largely to transmission studies in cattle.134 Characterization of ephemeroviruses is an ongoing process. For example, viruses Obodhiang (Sudan, 1963) and Kotonkan (Nigeria, 1967), isolated from mosquitoes, were initially suggested as “rabies related,” based on a limited antigenic cross-reactivity with lyssaviruses.60 However, gene sequencing and phylogenetic reconstructions demonstrated that these viruses belong to ephemeroviruses.401 Likely, more members of the genus will be recovered among other rhabdoviruses, isolated decades ago and awaiting molecular characterization.
improved surveillance.735 Until 1966, when bovine ephemeral fever virus (BEFV) was grown in mice,723 research on the agent was restricted largely to transmission studies in cattle.134 Characterization of ephemeroviruses is an ongoing process. For example, viruses Obodhiang (Sudan, 1963) and Kotonkan (Nigeria, 1967), isolated from mosquitoes, were initially suggested as “rabies related,” based on a limited antigenic cross-reactivity with lyssaviruses.60 However, gene sequencing and phylogenetic reconstructions demonstrated that these viruses belong to ephemeroviruses.401 Likely, more members of the genus will be recovered among other rhabdoviruses, isolated decades ago and awaiting molecular characterization.
Novirhabdoviruses
Infectious hematopoietic necrosis virus (IHNV) was first discovered in sockeye salmon (Oncorhynchus nerka) dying at hatcheries in Washington in 1953.611 Similar outbreaks among hatchery-reared salmonid fish in California were reported in the following decades.285,777 It was thought that IHNV was confined to salmonid fish in the Pacific coast of North America.482 However, the virus spread during the 1970s to the eastern United States, Europe, Japan, Korea, Taiwan, and China by shipment of infected fish and eggs.85,418,617 Electron microscopy of IHNV particles along with physicochemical and serologic analysis demonstrated that IHNV is a member of the Rhabdoviridae.16,314,483 Gene sequencing demonstrated that IHNV has the five structural genes common to rhabdoviruses, with the addition of a nonstructural, nonvirion (NV) gene between the genes for the G and L proteins.399 Several more fish rhabdoviruses, which demonstrate similar pathobiology and have similar genome organization, have been described, including Hirame rhabdovirus, snakehead virus, and viral hemorrhagic septicemia virus. These viruses were identified not only in North America but also in eastern and southern Asia, where they appear to be endemic.363,374 These viruses were first assigned into the genus Novirhabdovirus, based on the presence of the NV gene, in the Seventh Report of the International Committee on Taxonomy of Viruses (ICTV).736
Sigma Virus
This virus, a natural pathogen of Drosophila spp. fruit flies, was described in 1937.407 Sigma virus appears to be distributed worldwide. This is the only arthropod-specific rhabdovirus described to date, with an unusual mode of transmission: it is only transmitted vertically through both eggs and sperm and does not move horizontally between hosts.145 Sigma virus was initially placed in the Rhabdoviridae based on its bullet-shaped viral particles,64,696 and this has subsequently been confirmed using sequence data.71 Initially it was believed that Sigma virus infects only D. melanogaster. However, additional surveillance identified recently that related variants of Sigma virus infect D. affinis and D. obscura.451
Taxonomy
The rhabdoviruses share a variety of gross morphologic and functional attributes with other members of the order Mononegavirales. For example, the virions are large structures that mature by budding, with membrane-bound spikes and a helical nucleocapsid. They possess single-stranded, nonsegmented, negative-polarity RNA, with a similar gene arrangement. Within the family, recent analyses support the concept of a unified phylogeny and suggest an evolutionary history influenced by host species and transmission dynamics.83,404 Currently, the ICTV recognizes four genera of animal rhabdoviruses and two genera of plant rhabdoviruses. Furthermore, several rhabdovirus species have been recognized without inclusion into any of the established genera.177 Figure 31.1 shows phylogenetic relationships among rhabdoviruses.
Within the genus Lyssavirus only one major serogroup had been established, although various serotypes were defined.613 Placement within the genus was determined by serologic cross-reactivity of viral antigens, primarily based on antigenic sites on the nucleoprotein (the N protein). Historically, placement of a species as a rabies or rabies-related virus was determined by recognition of antigenic sites of the glycoprotein (the G protein) via virus neutralization tests. As nucleotide sequence data became available for a number of other Lyssavirus species,44,84,173,377,400,402,405,663 a trend toward genetic classification was established. Currently ICTV recognizes 11 Lyssavirus species, and one more representative (Shimoni bat virus [SHIBV]) is included in the genus provisionally without established species status (Table 31.1).
In general, demarcation criteria for Lyssavirus species include the following: (1) Genetic distances, with the threshold of 80% to 82% nucleotide identity for the complete N gene or 80% to 81% nucleotide identity for concatenated coding regions of N+P+M+G+L genes. Globally, all isolates belonging to the same species have higher identity values than the threshold, except the viruses currently included into the Lagos bat virus (LBV) species. For that reason some authors suggested that LBV be subdivided into several genotypes.173,473 However, as these LBV representatives are segregated into a monophyletic cluster in the majority of phylogenetic reconstructions, in the absence of other sufficient demarcation characters there is currently no possibility to subdivide LBV into several viral species. (2) Topology and consistency of phylogenetic trees, obtained with various evolutionary models. (3) Antigenic patterns in reactions with antinucleocapsid monoclonal antibodies (preceded by serologic cross-reactivity and definition of Lyssavirus serotypes, using polyclonal antisera). (4) Whenever available, additional characteristics, such as ecological properties, host and geographic range, and pathologic features, are considered.177 Moreover, based on genetic distances and serologic cross-reactivity, the genus has been subdivided into two phylogroups. Phylogroup I includes RABV, European bat Lyssavirus type 1 (EBLV-1), EBLV-2, Duvenhage virus (DUVV), Australian bat Lyssavirus (ABLV), Aravan virus (ARAV), Khujand virus (KHUV), and Irkut virus (IRKV). Phylogroup II includes LBV, Mokola virus (MOKV), and SHIBV. The remaining species of the genus, West Caucasian bat virus (WCBV), cannot be included in either of these phylogroups and is suggested to be considered as a representative of independent phylogroup III.400,402
Based on the serologic cross-reactivity patterns and sequence analyses of the members of the genus Vesiculovirus, a unique VSV serogroup has been established. This serogroup includes VSIV (currently the type species of the genus), VSNJV, VSAV, Carajas virus, CHPV, COCV, ISFV, MARAV, and Piry virus.177 Also recognized as a Vesiculovirus species is SVCV. Furthermore, 19 viruses are provisionally included in the genus without established species status (Table 31.1).
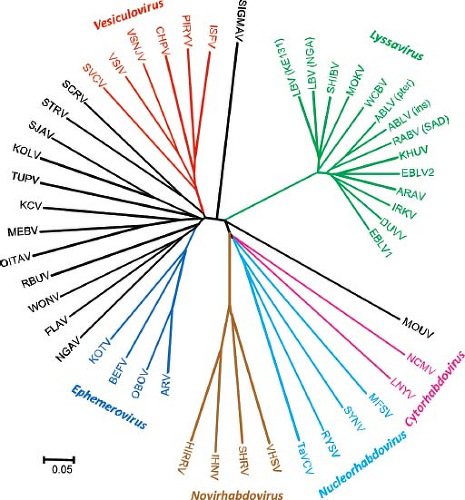 Figure 31.1. Phylogenetic relationships among rhabdoviruses. |
The members of the genus Ephemerovirus show limited cross-neutralization reactivity, but they are highly cross-reactive in complement fixation or indirect immunofluorescence tests. They exhibit similar but distinct genome organization with the common feature of a nonstructural glycoprotein (GNS) gene but variations in the number of accessory protein genes and the location of transcriptional control sequences. Different species may share up to 91% identity in N protein amino acid sequence. Currently the genus includes only three recognized species, but five more viruses are included provisionally177 (Table 31.1), based on antigenic reactivity and phylogenetic analysis of limited gene fragments.83,401 Phylogenetic relationships indicate that several intercontinental translocations of ephemeroviruses are likely to have occurred. Adelaide River virus (ARV) (Australia) and Obodhiang virus (OBOV) (Africa) demonstrate more genetic identity to each other than is observed between ARV and BEFV, both circulating in Australia. BEFV also circulates broadly in Africa, the Middle East, and southern areas of Asia, without significant genetic diversity.401,404,735
The genus Novirhabdovirus was established based on the presence a small NV protein of unknown function. The NV open reading frame (ORF) is located between the G and L genes and is preserved in diverse viruses and strains. The NV protein sequences are significantly less conserved between viruses in different species than sequences of the structural proteins.317,398 Species within the genus have been distinguished serologically on the basis of cross-neutralization with polyclonal rabbit antisera. Thus, IHNV and hirame rhabdovirus (HIRRV) each constitute single serotypes, and viral hemorrhagic septicemia virus (VHSV) has one major serotype with a small number of associated strains. Viruses from different species do not show cross-neutralization, but in some cases there is a low level of cross-reaction with specific proteins in western blot analyses. Nucleotide sequence data are available for most genes of these viruses and will undoubtedly contribute to the distinction of viral species in the future. For strains within a virus species, the nucleotide sequence divergence ranges up to a maximum of 8% for IHNV G and NV genes and 18% for the G genes of European and North American VHSV. N protein amino acid identity between IHNV and VHSV is approximately 34%.177
Members of two genera of the Rhabdoviridae infect plants and are transmitted via arthropod vectors, such as leafhoppers, planthoppers, and aphids.342 The cytorhabdoviruses and nucleorhabdoviruses are primarily distinguished based on their sites of virion maturation, in the cytoplasm and the nucleus, respectively. Genus classification based on sequence diversity has thus far correlated with classification by intracellular virus maturation. The genus Cytorhabdovirus currently includes nine recognized species and three provisional members, whereas genus Nucleorhabdovirus includes nine and four members, respectively
(Table 31.1). There is no significant sequence similarity (>50%) between analogous genes of the different species analyzed to date. However, nucleotide sequences are available for only a limited number of representatives and at the moment cannot be considered as sufficient to demarcate different species.177
(Table 31.1). There is no significant sequence similarity (>50%) between analogous genes of the different species analyzed to date. However, nucleotide sequences are available for only a limited number of representatives and at the moment cannot be considered as sufficient to demarcate different species.177
Recently, several rhabdoviruses, previously referred to as “unclassified”,707 were recognized by the ICTV as species, without assignment to any particular genus, based on their unique genome structure, phylogenetic and antigenic properties, and sufficient amount of knowledge on their ecology or pathobiology. These include Flanders, Tupaia, Sigma, Ngaingan, and Wongabel viruses.177 The recently described Moussa virus575 is another candidate for the establishment of a viral species without inclusion in any recognized genus.
Virion Structure
Rhabdoviruses are enveloped, rod- or cone-shaped particles (Fig. 31.2A, B), approximately 100 to 430 nm long and 45 to 100 nm in diameter. Animal rhabdoviruses are usually approximately 180 nm long and 80 nm wide, but those isolated from plants can be longer. The length of the virion is dictated by the length of the RNA genome, so that incorporation of additional genes into the viral genome results in correspondingly longer virions.629 Typically, mature virions appear either as bullet-shaped particles with one rounded and one flattened end or as bacilliform particles that appear hemispheric at both ends.
The genome RNAs of VSV and RABV, which are 11 to 12 kb, are encapsidated by approximately 1,200 copies of a single major nucleoprotein (N protein), with each molecule of N protein covering nine bases.277,702 Unlike the paramyxovirus rule of six, there is no requirement that the genome size be a multiple of this number. The nucleocapsid also contains 466 copies of the phosphoprotein (P protein, formerly called NS protein)702 and 50 copies of the large polymerase protein (L protein), which are responsible for the virion-associated RNA polymerase activity. The viral RNA polymerase cannot use naked RNA as a template but instead requires that the virion RNA template be encapsidated by N protein. P protein is responsible for binding L protein to the N protein–RNA template, and L protein is likely responsible for all of the enzymatic activities associated with RNA synthesis.
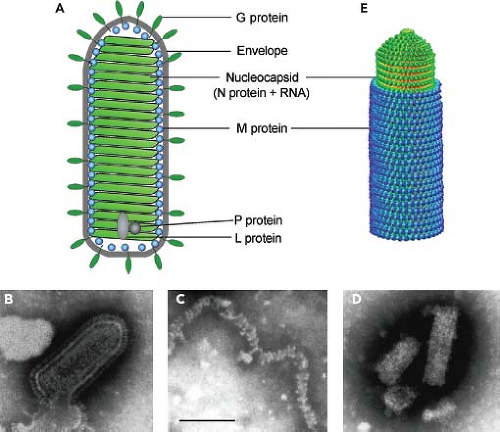 Figure 31.2. Structure of rhabdovirus virions. A: Diagram of virion. B: Negative stain electron micrograph of vesicular stomatitis virus (VSV) virion. C: VSV nucleocapsids prepared by solubilization of virion envelopes with triton X-100 in high-ionic-strength buffer. D: VSV nucleocapsid–M protein complexes prepared by solubilization of virion envelopes with triton X-100 in low-ionic-strength buffer. E: Model of the VSV nucleocapsid–M protein complex derived from cryoelectron microscopy data of Ge et al.264 Bar = 100 nm. (Negative stain electron micrographs by E. Alexander Flood, as described in [237].) |
The structures of N protein–RNA complexes from RABV and VSV have been determined by x-ray crystallography.11,276 The N protein molecule consists of two lobes, with the RNA inserted between the two lobes (Fig. 31.3). In the nucleocapsid, an amino-terminal extension from each N protein subunit interacts both with the adjacent subunit and with the subunit two positions away (Fig. 31.3, inset). Contacts between the C-terminal lobes also contribute to the stability of the nucleocapsid. N protein forms a stable nucleocapsid-like structure even in the absence of RNA.278,810 The C-terminal lobes of two N protein molecules in the nucleocapsid form a binding site for the P protein polymerase subunit, which is proposed to bind and dissociate in a processive manner during RNA synthesis.276
P protein consists of three domains, an acidic N-terminal domain, a central domain, and a C-terminal domain.185 P protein forms homo-oligomers, which are necessary for P protein to bind L protein to the nucleocapsid and for subsequent
transcriptase activity.256,257 The oligomerization is mediated by the P protein central domain, the structure of which has been determined by x-ray crystallography.184 Both the isolated central domain and the unphosphorylated full-length P protein form dimers.184,267 The phosphorylated transcriptionally active form of P protein was originally considered to be a trimer, based on epitope dilution experiments.256 Reanalysis of those data,693 however, suggests that P protein forms tetramers, similar to the P proteins of paramyxoviruses. Much of the N-terminal domain of P protein appears to be intrinsically disordered, although it probably adopts a well-defined structure upon binding ligands such as the L protein or soluble N protein (N0) involved in encapsidation of progeny genomes during genome replication.268,434 Two sites in the N-terminal domain must be phosphorylated by cellular casein kinase II for P protein to form oligomers and act in transcription.257,689 The C-terminal domain is responsible for binding the P protein to the nucleocapsid template, as described earlier. A basic region near the C-terminus of P protein is also necessary for interaction with L protein in viral transcription.164 The structures of the C-terminal domains of RV and VSV P proteins have been determined by x-ray crystallography and NMR spectroscopy.276,481,588
transcriptase activity.256,257 The oligomerization is mediated by the P protein central domain, the structure of which has been determined by x-ray crystallography.184 Both the isolated central domain and the unphosphorylated full-length P protein form dimers.184,267 The phosphorylated transcriptionally active form of P protein was originally considered to be a trimer, based on epitope dilution experiments.256 Reanalysis of those data,693 however, suggests that P protein forms tetramers, similar to the P proteins of paramyxoviruses. Much of the N-terminal domain of P protein appears to be intrinsically disordered, although it probably adopts a well-defined structure upon binding ligands such as the L protein or soluble N protein (N0) involved in encapsidation of progeny genomes during genome replication.268,434 Two sites in the N-terminal domain must be phosphorylated by cellular casein kinase II for P protein to form oligomers and act in transcription.257,689 The C-terminal domain is responsible for binding the P protein to the nucleocapsid template, as described earlier. A basic region near the C-terminus of P protein is also necessary for interaction with L protein in viral transcription.164 The structures of the C-terminal domains of RV and VSV P proteins have been determined by x-ray crystallography and NMR spectroscopy.276,481,588
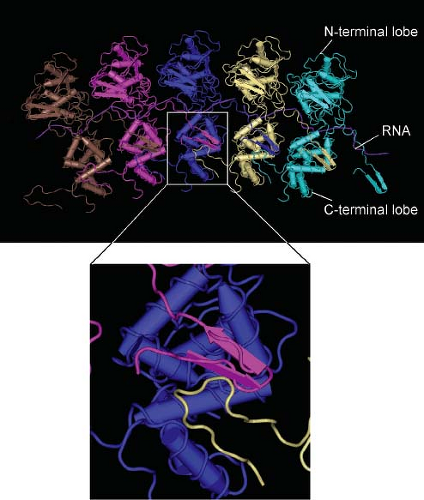 Figure 31.3. Structure of the vesicular stomatitis virus (VSV) nucleocapsid. Model of N protein and RNA in the VSV nucleocapsid derived from x-ray crystallography279 and cryoelectron microscopy.264 Inset shows interaction of the N-terminal extension from the pink N protein subunit with the C-terminal domain of the adjacent subunit (blue) as well as the subunit two positions away (white). (Assembled from PDB file 2WYY (MMDB ID: 80066) using Cn3D4.2 software.) |
The organization of the L protein has been deduced by analysis of sequence homology among members of the order Mononegavirales, which identified six conserved regions designated CRI through CRVI. The RNA polymerase activity has been mapped to CRIII.655 The L protein also has messenger RNA (mRNA) capping and methylation activity, which map to CRV437 and CRVI,435 respectively. High-resolution structures of L protein have not been published thus far, but its domain organization has been determined by negative stain electron microscopy combined with proteolytic digestion and deletion mutagenesis.577 The protein is organized into a ring-like structure that contains the RNA polymerase and an appendage of three globular domains. The capping activity maps to a globular domain attached directly to the ring, and the methylation activity maps to a more distal and flexibly connected domain.
When released from virions by treatment with detergents at high ionic strength, the nucleocapsid is loosely coiled and flexible (Fig. 31.2C), with a total length of 3.6 μm.702 In virions, however, the nucleocapsid is associated with the matrix (M) protein, which condenses the nucleocapsid into a tightly coiled helical nucleocapsid–M protein complex (Fig. 31.2D), sometimes referred to as the virus skeleton,54,514,515 which gives
the virion its bullet-like shape.54,463,514,515 The structure of the nucleocapsid–M protein complex has been determined to 10.6 Å resolution by analysis of cryoelectron micrographs of VSV virions (Fig. 31.2E).264 The N and M protein subunits in this structure were identified by fitting the electron density data from electron microscopy to that from x-ray crystallography. The N protein and RNA form an inner helical layer surrounded by an outer helical layer composed of M protein. The orientation of N protein subunits indicated that the 5′ end of the genome RNA is at the tip of the bullet, and the 3′ end is at the base. The conical tip of the bullet is formed by approximately seven successive turns of the N protein helix expanding gradually from 10 subunits per turn to 37.5 subunits per turn, with two turns forming a helical repeat. This pattern continues for approximately 29 turns to form the cylindrical trunk of the bullet. The M protein layer is formed by interaction of each M protein subunit with two successive turns of the N protein helix. The helical structure is held together by the M–N interactions as well as the interaction between M protein subunits in successive turns of the helix.
the virion its bullet-like shape.54,463,514,515 The structure of the nucleocapsid–M protein complex has been determined to 10.6 Å resolution by analysis of cryoelectron micrographs of VSV virions (Fig. 31.2E).264 The N and M protein subunits in this structure were identified by fitting the electron density data from electron microscopy to that from x-ray crystallography. The N protein and RNA form an inner helical layer surrounded by an outer helical layer composed of M protein. The orientation of N protein subunits indicated that the 5′ end of the genome RNA is at the tip of the bullet, and the 3′ end is at the base. The conical tip of the bullet is formed by approximately seven successive turns of the N protein helix expanding gradually from 10 subunits per turn to 37.5 subunits per turn, with two turns forming a helical repeat. This pattern continues for approximately 29 turns to form the cylindrical trunk of the bullet. The M protein layer is formed by interaction of each M protein subunit with two successive turns of the N protein helix. The helical structure is held together by the M–N interactions as well as the interaction between M protein subunits in successive turns of the helix.
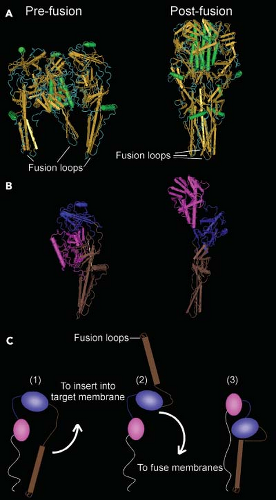 Figure 31.4. Structure of the vesicular stomatitis virus (VSV) G protein. A: G protein ectodomain trimers in the prefusion and postfusion state. G protein ectodomain (residues 1 to 422) was generated by limited proteolysis of virions with thermolysin.595,596 B: Comparison of domain organization of a single G protein subunit in the pre- and postfusion states. C: Diagram of domain rearrangements in transition from prefusion state (1) to proposed intermediate inserted into target membrane (2) to postfusion state (3). White line represents G stem leading to membrane anchor sequence that is missing from the crystal structure. (Assembled from PDB files 2CMZ and 2J6J using Cn3D4.2 software.) |
The amino-terminal 50 to 57 amino acids of M protein appear to be largely disordered in purified M protein.260,274,362 However, this sequence may form an ordered structure upon binding to the nucleocapsid.264 The remainder of the M protein sequence forms a compactly folded C-terminal domain, whose structure has been determined by x-ray crystallography.261,274 Sequences in the amino-terminal region are involved in interaction with N protein in the nucleocapsid–M protein complex, as well as interacting with sequences in the C-terminal domain in M–M interactions in the complex.144,160,264,274 The C-terminal domain of M protein also appears to interact with the virus envelope, perhaps with the cytoplasmic domain of the envelope glycoprotein.264
The structure derived from cryoelectron microscopy accounts for approximately 1,200 of the 1,800 copies of M protein in the virion. The remaining 600 M protein subunits are likely present in a nonhelical arrangement, thus rendering them undetectable in the analysis. One likely location is in association with the envelope lipid bilayer. M protein interacts with the lipid bilayer of the virus envelope, which was shown using lipophilic photoreactive probes.430,804 The M protein sequences involved in the interaction are present in the N-terminal region, partially overlapping the sequences involved in interaction with the nucleocapsid.160,430 This supports the idea that there are two populations of M protein in the virion, one involved in the nucleocapsid–M protein complex and the other involved in interaction with the envelope lipid bilayer.
The lipids of the envelope are derived from the host cell membrane during virus assembly by budding. The lipid composition of the envelope generally reflects that of the host membrane from which the virus buds, consisting primarily of phospholipids and cholesterol, although virus envelopes appear to be enriched in cholesterol and sphingomyelin compared with the host membranes from which they were derived.455 The virus envelope contains approximately 300 to 400 spike-like projections composed of a single species of viral glycoprotein (G protein). The individual spikes are trimers of G protein,188,262,765 which function in virus attachment and penetration by fusion of the virus envelope with endosome membranes. G protein is anchored in the envelope lipid bilayer by a 20–amino acid hydrophobic transmembrane domain near the C-terminus, which is followed by a 29–amino acid cytoplasmic domain, which is inside the virus envelope.606 The structure of the 446–amino acid external domain (ectodomain) of the VSV G protein has been determined by x-ray crystallography in both the neutral pH (“prefusion”) and low pH (“postfusion”) conformations (Fig. 31.4).595,596 Like other viral fusion proteins, the two conformations are dramatically different, indicating that major structural rearrangements must occur during
the fusion process. The surprising result is that the folding of G protein bears no resemblance to the fusion proteins of other negative-strand or positive-strand RNA viruses that have been determined. Instead, the structure of the VSV G protein is homologous to that of the gB glycoprotein of herpesviruses.305 This raises interesting questions about the evolutionary origin of these proteins.
the fusion process. The surprising result is that the folding of G protein bears no resemblance to the fusion proteins of other negative-strand or positive-strand RNA viruses that have been determined. Instead, the structure of the VSV G protein is homologous to that of the gB glycoprotein of herpesviruses.305 This raises interesting questions about the evolutionary origin of these proteins.
Genome Structures
Genomes of rhabdoviruses are single-stranded, nonsegmented RNA of negative polarity. They lack 5′ caps and 3′ poly A, consistent with their inability to function as mRNA. Genomes of three genera of Rhabdoviridae are shown diagrammatically in Figure 31.5. Genomes of lyssaviruses and vesiculoviruses are similar to each other. The approximately 50 nucleotides at the 3′ and 5′ ends (the leader and trailer sequences, respectively) are partially complementary. They contain important cis-acting sequences that serve as promoters for transcription and replication and as signals for encapsidation of genomes and antigenomes during replication, as described later. Although they do not encode proteins, short RNAs of unknown function are generated from these sequences. The five protein-encoding genes are in the order 3′–N–P/C–M–G–L–5′, which is the order of the analogous genes in other nonsegmented, negative-strand RNA viruses, regardless of the number of additional viral genes. Each gene junction contains a conserved sequence specifying the end (E) of the upstream gene, a two-nucleotide intergenic (I) sequence, and the start (S) sequence for the downstream gene. These sequences control the activities of the viral RNA polymerase, which transcribes these genes according to a stop–start mechanism described later. In general, the 5′ and 3′ untranslated regions of the viral mRNA are short (10 to 50 nucleotides) and lack cis-acting sequences that control translation or mRNA turnover. The one exception is the P/C gene of vesiculoviruses, which contains alternate start codons. The upstream start codon initiates translation of the P protein, whereas two downstream start codons initiate translation of an alternate reading frame that encodes two small basic proteins, C and C′.392,667 Analogous proteins encoded by paramyxoviruses often play a role in pathogenesis by altering viral gene expression and suppressing host responses to virus infection. Mutation of the VSV P gene to introduce a stop codon in the C and C′ open reading frame (without altering the sequence of the P protein), however, had no detectable effect on virus replication in cell culture or pathogenesis in mice.392 This still leaves open the possibility that the C and C′ proteins play a role in replication in other hosts.
The genomes of ephemeroviruses are larger than those of most other rhabdoviruses. The genome of BEFV is 14.8 kb and contains 10 genes (3′–N–P–M–G–GNS–α1–α2–β–γ–L-5′) separated by intergenic regions of 26 and 53 nucleotides.177,735 The genome of the related Adelaide River virus is 14.6 kb in length and contains nine genes (3′–N–P–M–G–GNS–α1–α2–β–L–5′) separated by intergenic regions of one to four nucleotides.742 The GNS gene encodes a glycoprotein, which is synthesized in approximately the same amount as G protein during virus infection,737 but it is not found in the mature virion. Intracellularly, GNS protein is localized in the endoplasmic reticulum–Golgi complex, and it is associated with amorphous structures in the cell surface but not with viruses in the budding process. It is highly glycosylated, with a molecular weight of 90 kD. GNS protein shares significant amino acid sequence homology with the G protein, but it does not induce protective neutralizing antibodies.312 The function of GNS protein is unknown. It has been proposed that the gene coding for this protein originated by gene duplication by a copy-choice mechanism involving relocation of the polymerase in an upstream position during viral replication.742
Genomes of novirhabdoviruses contain an NV gene between the G and L genes. The NV protein (12 to 14 kD) is expressed at variable levels in infected cells but is not detectable in purified virions. The NV protein sequences are significantly less conserved between viruses in different species than sequences of the other structural proteins, such that there is no significant amino acid sequence similarity between the NV proteins of IHNV and VHSV. The specific function of the NV protein is not yet defined, but it is required for efficient virus replication. Results of studies with NV gene deletion mutants generated by reverse genetics are inconsistent in that the NV appears to be required for pathogenicity in IHNV and VHSV but not snakehead virus (SHRV).317,398
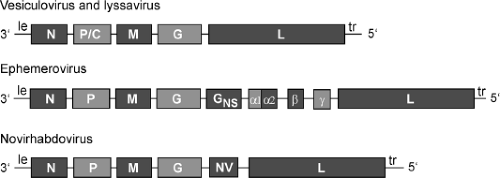 Figure 31.5. Diagram of rhabdovirus genomes. |
Rhabdoviruses that have not been assigned to a particular genus have a variety of additional transcription units. For example, the gene order of FLAV is 3′–N–P–pseudogene 1–19K–pseudogene 2–M–G–L–5′. The unique features include the gene encoding a 19-kD protein of unknown function, surrounded by two pseudogenes, about 500 nucleotides each, situated between the P and M genes.86,177 The gene order of TUPV is 3′–N–P/C–M–SH–G–L–5′. The unique small hydrophobic (SH) transcription unit between M and G genes encodes a protein with two hydrophobic amino acid stretches, including a potential signal sequence at the amino terminus
and a potential membrane-spanning sequence near the center. The C protein ORF that overlaps the TUPV P gene has the potential to encode a 221–amino acid basic protein that is more than three times larger than the VSV C protein.668
and a potential membrane-spanning sequence near the center. The C protein ORF that overlaps the TUPV P gene has the potential to encode a 221–amino acid basic protein that is more than three times larger than the VSV C protein.668
The genome of Ngaingan virus (NGAV) is over 15.7 kb, which is the largest genome yet described for any rhabdovirus, containing 13 ORFs in the order 3′–N–P–U1–U2–U3–M–U4–G–GNS–U5–U6–U7–L–5′. The NGAV P gene contains two alternative ORFs designated P1′ and P2′, analogous to alternative P ORFs referred to as either C or P′ in several other rhabdoviruses. The GNS gene encodes a nonstructural glycoprotein (568 amino acids [aa]) analogous to that of ephemeroviruses. NGAV contains seven additional genes (U1 through U7) with the potential to encode small proteins of unknown function. Although similar in size (81 to 153 aa) to proteins encoded by ORFs located in similar positions in several other rhabdoviruses, they lack significant sequence or structural similarity to any known protein. However, none of the small unique NGAV proteins has yet been detected in infected cells.283
The gene order in WONV is 3′–N–U4–P–U1–U2–U3–M–G–U5–L–5′. WONV lacks an alternative ORF in the P gene but contains five additional genes (U1 through U5), each of which encodes a protein that lacks significant amino acid sequence identity with other known proteins. The U1 protein (179 aa) is hydrophilic with numerous potential phosphorylation sites, an N-glycosylation site, an amidation site, and two N-myristoylation sites. The U2 protein (192 aa) contains two predicted N-myristoylation sites and a highly hydrophobic domain of 10 amino acids followed by a mitochondrial energy transfer signature that is characteristic of carrier and transport proteins. The U4 protein (49 aa) contains a single putative N-myristoylation site and shares overall 49% identity with guanosine triphosphate (GTP)-binding proteins of several bacteria. The U5 protein (127 aa) contains a predicted N-terminal extracellular domain, 22-aa transmembrane domain, and highly basic cytoplasmic tail and has overall structural similarity to the α1 proteins of ephemeroviruses, which have been suggested to be viroporins. Proteins of similar size to the U1, U2, U3, and U5 proteins have been detected in WONV-infected cells by immunoblot analysis using polyclonal mouse ascitic fluid.284
The genome of Moussa virus is similar to those of the genera Lyssavirus and Vesiculovirus. However, the ORFs located in the position of the P (ORF 2) and M (ORF 3) genes in other rhabdoviruses show no nucleotide or amino acid homology to sequences of other rhabdoviruses.575
The genomes of Sigma virus and members in the genera Cytorhabdovirus and Nucleorhabdovirus contain an additional gene between the P and M genes, referred to as “X” or “a.” The putative X protein is of unknown function but contains conserved domains found in reverse transcriptases. Another unusual feature is that M and G mRNAs overlap by 33 nucleotides.106,145
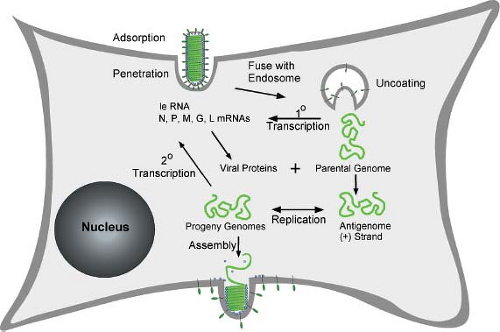 Figure 31.6. Diagram of rhabdovirus replication cycle. The steps illustrated are virus adsorption and penetration by endocytosis, envelope fusion with endosome membranes, release of nucleocapsids containing parental genomes into the cytoplasm, primary (1-degree) transcription, genome replication to produce nucleocapsids containing antigenomes and progeny genomes, secondary (2-degree) transcription, and assembly by budding from host plasma membrane. (Drawn by E. Alexander Flood.) |
Stages of Replication
The replication cycle of rhabdoviruses is typical of that of most nonsegmented, negative-strand RNA viruses (Fig. 31.6). The initial events of attachment, penetration, and uncoating result in release of the viral nucleocapsid into the cytoplasm of the host cell. The encapsidated parental genome RNA serves as a template for primary transcription by the virion RNA-dependent RNA polymerase, resulting in synthesis of leader (le) RNA and all five viral mRNAs. The accumulation of viral proteins synthesized from primary transcripts leads to replication of the genome, which involves synthesis of full-length positive-strand RNA, or antigenomes. The antigenomes, in turn, serve as templates for synthesis of progeny negative-strand genomes. Encapsidation of genomes and antigenomes occurs concomitantly with their synthesis and, indeed, is a key signal for the RNA polymerase to function as a replicase versus a transcriptase. Progeny nucleocapsids are used for three different purposes: (a) as templates for further rounds of replication; (b) as templates for secondary transcription, which is the major amplification step for viral gene expression; and (c) for assembly into progeny virions, which occurs by budding
from host membranes. In a single-cycle growth experiment, the early events including attachment, penetration, uncoating, and primary transcription occur within the first few hours postinfection. The processes of genome replication, secondary transcription, and virus assembly occur continuously throughout the remainder of the infectious cycle, which lasts for an additional 12 to 18 hours for VSV or several days for RABV.
from host membranes. In a single-cycle growth experiment, the early events including attachment, penetration, uncoating, and primary transcription occur within the first few hours postinfection. The processes of genome replication, secondary transcription, and virus assembly occur continuously throughout the remainder of the infectious cycle, which lasts for an additional 12 to 18 hours for VSV or several days for RABV.
Mechanism of Attachment
Rhabdoviruses appear to use a variety of different receptors for attachment to different types of host cells. The RABV G protein binds most effectively to cells of neuronal origin,713 reflecting the neurotropism of RABV in vivo. Several different surface molecules expressed at high levels on neurons have been identified as potential receptors, including the nicotinic acetyl choline receptor,259,431 the neural cell adhesion molecule (CD56),704 and the low-affinity nerve-growth factor receptor p75NTR.714,715 Expression of CD56 and p75NTR has been shown to confer susceptibility to RABV on cells that are normally resistant to infection. Transgenic mice that lack CD56 show a delay in RABV spread through the central nervous system (CNS) and in RABV-induced mortality, but the mice still die following virus infection,704 indicating that other receptors are involved. RABV infection of transgenic mice that lack p75NTR was found to be similar to that of wild-type mice of the same strain,339 initially indicating that p75NTR was not an important receptor for RABV pathogenesis. However, RABV G protein binds to a region of p75NTR that is present on a splice variant of p75NTR that is still expressed in the transgenic mice.414 Thus, further experiments are required to fully evaluate the role of p75NTR in RABV pathogenesis.
In addition to the receptors that are enriched on cells of neuronal origin, RABV can also use receptors that are widely distributed among many cell types.582,685,793 These receptors appear to be of lower affinity than those on neuronal cell surfaces713 and have been difficult to identify. A similar difficulty exists in identifying receptors for VSV, which also binds to many different cell types in culture by interactions that appear to be of low affinity and often are not easily saturable. For both RABV and VSV, negatively charged lipids have been proposed to be cellular receptors for virus attachment. In the case of RABV, neuraminic acid-containing glycolipids (gangliosides) have been implicated686 and, in the case of VSV, phosphatidyl serine has been proposed as a cellular receptor,624 although later experiments have indicated that phosphatidyl serine is not the receptor for VSV.139 Instead, it seems likely that nonspecific electrostatic and hydrophobic interactions mediate attachment of VSV to cells. Treatment of cells with polycations and polyanions such as diethylaminoethyl-dextran (DEAE-dextran) and dextran sulfate can markedly enhance the efficiency of attachment and infection of cells by both VSV45 and RABV.793
An interesting feature of attachment by both viruses is that binding is markedly enhanced at lower pH in the range from pH 6.5 to 5.6.248,793 The pH dependence of attachment is similar to that of envelope fusion with cellular membranes (discussed in the next section), although fusion occurs most efficiently at slightly lower pH than does virus attachment. Furthermore, G protein mutations that shift the pH dependence of fusion also shift the pH dependence of attachment.248 This suggests that attachment to many cell types is mediated by G protein in a conformation that is similar to the fusion-active form of the viral G protein. This idea is supported by experiments with photoactivatable lipid probes, which indicate that the putative fusion peptide (described later) is inserted into target membranes under the optimal conditions for both attachment and fusion.197,542 This would also account for the affinity of the VSV G protein for phosphatidyl serine and other negatively charged phospholipids, because the presence of negatively charged lipids in the target membrane appears to be necessary for virus envelope fusion.105,199,796
Mechanism of Penetration
VSV was one of the early examples of a virus shown to penetrate into cells by clathrin-dependent endocytosis.478 Following attachment to host cell surfaces, virions can either migrate to preformed clathrin-coated pits or nucleate the formation of new coated pits,157,348 where they undergo endocytosis into coated vesicles (Fig. 31.7). Because VSV virions are longer than the typical diameter of a coated vesicle, the final closure of the endocytic vesicle requires participation of the actin cytoskeleton.157,158 The endocytic vesicles lose their clathrin coats to become early endosomes. The contents of early endosomes are transported to late endosomes and lysosomes for degradation. During this process, the endosomal vesicles often invaginate to form multiple intraluminal vesicles.282 Such membranes are referred to as multivesicular bodies (MVBs). As virions progress through the endocytic pathway, they are exposed to progressively lower pH. At a pH below 6.5, the G protein mediates fusion of the viral envelope with the endosome membrane. This fusion event releases the internal virion components into the cytoplasm (left side of Fig. 31.7). Most of the available evidence indicates that VSV virions fuse primarily with the membranes of early endosomes.348,498,648 Other evidence, however, suggests that many fusion events occur within MVBs (right side of Fig. 31.7), releasing the internal virion contents into the cytoplasmic contents trapped within the MVBs and requiring back-fusion of internal vesicles with the limiting membrane of the MVBs to release the viral nucleocapsid into the cytoplasm of the cell.422,460 Viral proteins that fail to be released into the cytoplasm are degraded by proteases and other enzymes in lysosomes.478
The mechanism by which rhabdovirus G proteins induce fusion of the virus envelope with cellular membranes shares many features with other viral envelope fusion proteins but is clearly distinct in several respects. The VSV G protein and the structurally similar fusion proteins of herpesviruses and baculoviruses are referred to as class III fusion proteins to distinguish them from class I proteins, which are structurally similar to the influenza virus hemagglutinin, and class II proteins, which include the envelope glycoproteins of the alphaviruses and flaviviruses.32 As with class I fusion proteins, rhabdovirus G proteins exist as a trimer of subunits held together by noncovalent bonds.188,262,765 Unlike most viral envelope proteins, however, the subunits of G protein are in a dynamic equilibrium between monomers and trimers because of the rapid dissociation and reassociation of subunits.464,801,802 As with most low pH-dependent fusion proteins, the effects of low pH are mediated by conformational changes in G protein. Unlike other viral fusion proteins, the conformational changes in G protein are reversible upon returning the pH to neutrality, whereas those of many other viral fusion proteins are not reversible.188,263,573
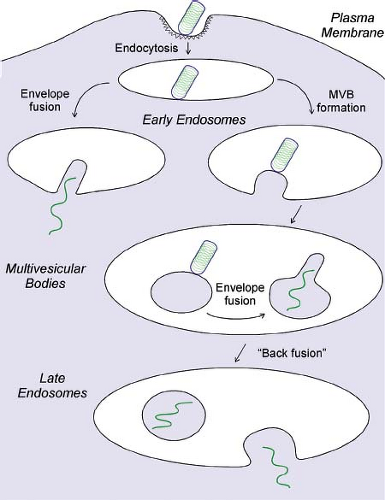 Figure 31.7. Diagram of rhabdovirus penetration by endocytosis. Pathway on the left shows virus envelope fusion with early endosomes. Pathway on the right shows virus envelope fusion with internal vesicles of multivesicular bodies (MVBs) and release of nucleocapsids into the cytoplasm by “back-fusion” with the MVB-limiting membrane. |
A general principle by which viral envelope proteins promote fusion is that they must insert into the target membrane through a region of their sequence referred to as the fusion peptide. In the class I fusion proteins, such as the influenza virus hemagglutinin (HA) and the paramyxovirus F proteins, the fusion peptide resides at the N-terminus of one of the subunits (HA2 or F1, respectively) generated by proteolysis of an inactive precursor. In contrast, proteolysis of rhabdovirus G proteins is not involved in activating fusion. This is similar to the case of the class II fusion proteins, in which the fusion peptide appears to be an internal region of the protein sequence. The regions of G protein that insert into target membranes at low pH have been mapped using photoactivatable lipid probes and mutagenesis studies in both the RABV and VSV G proteins.197,249,441,684,808 These sequences form two loops containing hydrophobic amino acids that extend from the protein structure (“fusion loops,” Fig. 31.4A). In the neutral pH “prefusion” state, the fusion loops are oriented toward the viral membrane (Fig. 31.4A, B). Upon lowering the pH, there is a proposed intermediate, in which the domain containing the fusion loops is reoriented to insert into the target membrane (Fig. 31.4C). Fusion of the viral and target membrane involves another domain rearrangement that brings the two membranes together in the “postfusion” state.
A second region of the G protein sequence functionally involved in fusion is the membrane-proximal ectodomain sequence immediately N-terminal to the membrane anchor sequence. Most of this sequence is not visible in the x-ray structures, because it was cleaved to solubilize the G protein. Mutations in this region dramatically inhibit fusion.347,647 G protein truncations containing part of this region (amino acids 421 to 461) together with the membrane anchor sequence and the cytoplasmic domain (G stems) enhance the fusion activity of other membrane fusion proteins and are able to cause hemifusion (mixing of the outer phospholipid leaflets of the two membranes) in the absence of other fusion proteins.347 The cooperation of the fusion loops and the membrane-proximal sequence may be analogous to similarly separate sequences in
other viral fusion proteins in bringing the viral and host membranes together for fusion.
other viral fusion proteins in bringing the viral and host membranes together for fusion.
Uncoating and Primary Transcription
Following fusion of the virus envelope with endosome membranes, which releases the internal virion components into the cytoplasm of the host cell, the viral M protein dissociates from the nucleocapsid.162,498,590 This step is necessary for viral RNA synthesis to occur, because M protein inhibits viral transcription.107,136,443,543,775 Binding of most of the M protein to nucleocapsids is readily reversible,461 and dissociation following envelope fusion is believed to occur spontaneously, although acidification of the virion interior appears to promote M protein dissociation from the nucleocapsid, similar to the M1 protein of influenza virus.497 Rhabdoviruses do not encode a separate ion channel protein analogous to the M2 protein of influenza viruses. Instead, the G protein is responsible for the permeability of the envelope to protons.497 Once the M protein has dissociated from the nucleocapsid, no further uncoating is necessary, because the encapsidated RNA is the template for the viral transcriptase complex.
The first biosynthetic step in the viral replicative cycle is primary transcription, mediated by the virion-associated, RNA-dependent RNA polymerase. The mechanism of primary transcription, defined as transcription from parental templates, appears to be identical to that of secondary transcription, or transcription from progeny templates following genome replication. The principal differences are in the much larger quantity of secondary transcripts, because of the larger number of progeny templates, and the brief time of primary transcription, compared with the prolonged period of secondary transcription throughout most of the viral infectious cycle.
The viral RNA polymerase is fully competent to synthesize all of the viral mRNA without new synthesis of viral proteins, as shown by the transcriptase activity of virion cores following solubilization of the envelope.51,506 Indeed, the first demonstration of a viral RNA-dependent RNA polymerase was made with virions of VSV.51 This cell-free transcriptase system has been a major tool in determining the mechanisms of viral transcription, establishing the requirement for both the L and P proteins for RNA polymerase activity203 and the requirement that the template RNA be encapsidated.70,202 An early insight was that a single entry point exists for the viral RNA polymerase near the 3′ end of the genome, and the viral mRNAs are transcribed sequentially in the order they appear in the genome: N–P–M–G–L. Thus, transcription of each gene depends on prior transcription of all upstream genes.1,50,201,336,337 This has since been found to be a general property of nonsegmented, negative-strand RNA viruses (see Chapter 30).
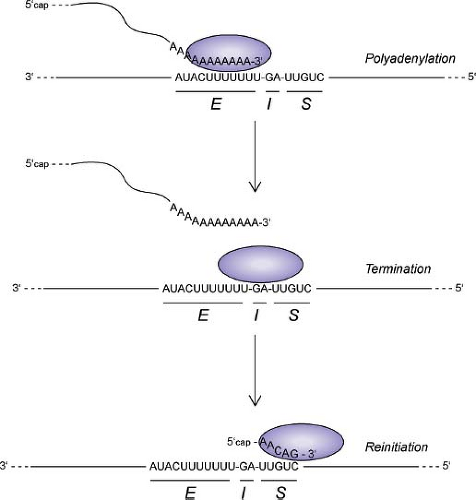 Figure 31.8. Diagram of rhabdovirus polymerase activities during transcription in response to gene end (E), intergenic (I), and gene start (S) sequences in the genomic RNA template. |
The mechanism of sequential transcription is generally considered to be a stop–start mechanism, in which cis-acting signals in the template RNA sequence govern the activities of the transcriptase complex at each gene junction (Fig. 31.8). With the exception of the junction between the leader and N
genes (discussed later), each of the VSV gene junctions contains a gene end sequence for the upstream gene (3′AUACUUUUUUU5′); an intergenic dinucleotide (G/CA), which is not transcribed; and a gene start sequence for the downstream gene (3′UUGUC5′).604 These sequences at each gene junction function as a signal for polyadenylation and termination of the upstream mRNA and also as a signal for the initiation, capping, and methylation of the downstream mRNA.55,56,57,315,630,677,678,679 Whereas the cis-acting signals in the template and the resulting modifications to the mRNA have been well defined, the mechanism by which these signals alter the activities of the transcriptase complex to accomplish these different tasks is a major question that remains to be addressed.
genes (discussed later), each of the VSV gene junctions contains a gene end sequence for the upstream gene (3′AUACUUUUUUU5′); an intergenic dinucleotide (G/CA), which is not transcribed; and a gene start sequence for the downstream gene (3′UUGUC5′).604 These sequences at each gene junction function as a signal for polyadenylation and termination of the upstream mRNA and also as a signal for the initiation, capping, and methylation of the downstream mRNA.55,56,57,315,630,677,678,679 Whereas the cis-acting signals in the template and the resulting modifications to the mRNA have been well defined, the mechanism by which these signals alter the activities of the transcriptase complex to accomplish these different tasks is a major question that remains to be addressed.
Transcript initiation requires both proper termination of the upstream gene and the gene start sequence 3′UUGUC5′.56,57,315,329,677,678,679 The requirements of the individual nucleotides in the start sequence for 5′ end modification are more rigorous than the requirements for correct initiation.679,741 Alterations of the capping and methylation of the transcripts affect the processivity of the polymerase and the extent of polyadenylation of the 3′ end, supporting a model in which the correct initiation and modification of the viral mRNAs play a regulatory role in the subsequent activities of the polymerase.255,436,741 The mechanism of 5′ end modification of VSV mRNAs and likely those of other negative-strand RNA viruses differs substantially from that of host mRNAs and mRNAs of other virus types. The viral mRNAs are capped by guanosine in a 5′–5′ triphosphate linkage, as are host mRNAs.2 The capping reaction differs, however, in that both the α- and β-phosphates are derived from the GTP donor, whereas for host capping enzymes, only the α-phosphate is derived from the GTP donor.2,533 This reaction occurs through an unusual covalent L protein–RNA intermediate.533 The host translation factor EF-1 is associated with the viral L and P proteins, and it has been proposed that the α-subunit of EF-1 plays a role in the capping reaction through its guanine nucleotide–binding activity.124,574
The mechanism of methylation of the viral mRNA cap is also unusual. S-adenosyl methionine is used as a methyl donor,2 as with host enzymes. However, instead of having separate enzymes that catalyze ribose 2′-O methylation versus guanine-N-7 methylation, both activities appear to reside in a single domain of L.254,275,435,438 This domain has a single binding site for S-adenosyl methionine and transfers the methyl groups in an unconventional order, in which 2′-O methylation precedes guanine-N-7 methylation.576
Following elongation of viral mRNA, the transcriptase complex encounters a termination signal at the end of each gene consisting of the sequence 3′AUACUUUUUUU. This signals the polymerase to “stutter” over the seven Us in the template, resulting in polyadenylation of the viral mRNA.55,56 Occasionally, the termination signal is ignored, resulting in read-through by the polymerase to give a dicistronic transcript.56,57,311 Some nonsegmented, negative-strand viruses have gene junctions with a high degree of read-through, which plays a substantial role in regulating the relative levels of the different viral proteins. Because read-through transcripts are rather uncommon for VSV, they are not thought to play a significant role in regulation of viral gene expression.
Following the polyadenylation reaction, which stops after addition of approximately 200 As, two possible fates exist for the transcriptase complex at each gene junction. The most common outcome is that the transcriptase complex traverses the two intergenic nucleotides and resumes transcription at the initiation signal of the downstream gene. Approximately 20% to 30% of transcriptase complexes fail to resume transcription of the downstream gene, however, and presumably dissociate from the template, leading to a 20% to 30% attenuation of expression of the downstream gene at each gene junction.336,730,757 This transcription attenuation results in a gradient of mRNA and protein expression, such that the abundance of each gene product depends on its distance from the 3′ end of the genome (i.e., N > P > M > G > L). The G–L gene junction is unusual in that the level of attenuation is much higher than that at the other gene junctions,49 resulting in much lower levels of L protein relative to the other viral proteins. The basis for this difference is not known, because the sequence of the G–L gene junction does not differ from that of the other gene junctions.
Transcription attenuation is a general feature of nonsegmented, negative-strand RNA viruses and is the major mechanism regulating abundance of the individual mRNA. The importance of the gene order in regulating the relative levels of viral proteins was dramatically illustrated by genetic engineering experiments to change the order of the genes of VSV. The resulting changes in relative abundance of the viral proteins resulted in substantial reductions in viral replication and pathogenesis.49,757 The similarity of the basic mechanisms in virus replication among nonsegmented, negative-strand RNA viruses and their dependence on the relative levels of each viral protein presumably accounts for the conservation of the basic gene order among these viruses.
The initiation and termination of transcription of the leader RNA differs from that of the viral mRNAs. The leader RNA is encoded by the 47 3′-terminal nucleotides of the genome. The leader gene differs from the other genes both in terms of the cis-acting signals in the template that initiate transcription760 and the nature of the product leader RNA, which is phosphorylated at the 5′ end and lacks a cap structure. In addition, the sequence at the leader–N gene junction is distinct from that of the other gene junctions and lacks the U7 sequence that governs polyadenylation.760 Correspondingly, leader RNA is not polyadenylated. Another unusual feature of transcriptional regulation at this gene junction is that its behavior is different in the cell-free transcription system versus transcription in infected cells. In the cell-free system, synthesis of leader RNA is required to transcribe the downstream N gene, consistent with the single polymerase entry site and stop–start model.201,762 In infected cells, however, the viral RNA polymerase can initiate synthesis at the first downstream gene without prior synthesis of leader RNA. This has been shown by inserting a small gene between the leader and N genes and determining the target size for UV inactivation of the inserted gene.762 This is the only gene junction that shows this behavior, because transcription initiation at all of the other genes requires prior transcription of the upstream gene both in the cell-free assay and in infected cells.
The difference in the site of initiation in infected cells versus that in the cell-free system indicates that host factors can influence the site of initiation. A viral transcriptase complex has been isolated from VSV-infected cells that contains, in addition to P and L proteins, the host proteins EF-1α, heat shock protein 60 (hsp60), and smaller amounts of the host mRNA capping enzyme guanyl transferase.574 Unlike the virion RNA polymerase, this complex initiates transcription at
the N gene and does not transcribe leader RNA.574 The ability of the polymerase to independently initiate at the first gene downstream of the leader gene appears to account for the phenotype of a VSV mutant (polR1) that synthesizes N mRNA in excess over leader RNA,128 which would be difficult to achieve if transcription of the N mRNA required prior transcription of the leader RNA. The mutation responsible for the polR phenotype is in the N protein associated with the template,127 indicating that the nature of the template can influence the site of initiation.
the N gene and does not transcribe leader RNA.574 The ability of the polymerase to independently initiate at the first gene downstream of the leader gene appears to account for the phenotype of a VSV mutant (polR1) that synthesizes N mRNA in excess over leader RNA,128 which would be difficult to achieve if transcription of the N mRNA required prior transcription of the leader RNA. The mutation responsible for the polR phenotype is in the N protein associated with the template,127 indicating that the nature of the template can influence the site of initiation.
Genome RNA Replication
Requirement for Encapsidation of Newly Synthesized RNA
A fundamental principle in replication of nonsegmented, negative-strand RNA viruses is that the ability of the RNA polymerase to replicate the viral genome depends on new viral protein synthesis to encapsidate the newly synthesized RNA. For example, treating infected cells with inhibitors of protein synthesis (e.g., cycloheximide) allows synthesis of viral mRNA but inhibits replication of genome RNA.324,756 The critical viral protein required for replication is the N protein, as shown by its ability to support synthesis of genome RNA in the cell-free system.550 In infected cells, however, a complex of N protein with P protein (often referred to as N0–P) is likely to be the active complex in promoting genome replication.555,556 The role of P protein in this complex appears to be to maintain the solubility and proper folding of N protein so that the nascent RNA synthesized by the RNA polymerase can be encapsidated.167,470,476,477 Analysis of an N0–P complex expressed in insect cells indicates that the complex contains one N protein and two P proteins.480
Encapsidation of nascent RNA appears to constitute a signal for the viral RNA polymerase to ignore the sequences in the genome template at each gene junction that govern the stop–start mechanism for transcription, thereby generating full-length, encapsidated RNA that is complementary to the genome (i.e., antigenomes). Use of antigenomes as templates results in synthesis of progeny genomes. The mechanism of RNA replication appears to be the same regardless of whether genomes or antigenomes are used as templates. In particular, replication of both templates requires that the nascent product RNA be encapsidated to generate full-length products. In addition to serving as a template for progeny genomes, the antigenome can be used as a template to generate a short, noncapped, nonpolyadenylated RNA complementary to the 3′ end of the antigenome that is analogous to the leader RNA. Variously called the minus strand leader or trailer RNA, this RNA is found in small amounts in infected cells and is the primary product produced in the cell-free system when antigenomes are used as templates in the absence of a source of new viral proteins.432,755,776
Cis-Acting Signals and RNA Polymerase Complexes that Govern Replication Versus Transcription
The critical cis-acting RNA sequences that govern replication are located at the 3′ ends of the genome and antigenome (Fig. 31.9). These sequences in the templates serve as promoters to initiate RNA synthesis, and their complementary sequences at the 5′ end of the product RNA serve as encapsidation signals, with the resulting encapsidation permitting elongation of the RNA into full-length products. The sequences required for encapsidation have been mapped using synthetic RNA in cell-free encapsidation assays and also in transfected cells using minigenomes (described later).163,507,548,760 The sequences in the templates that serve as promoters for replication and transcription have been defined by mutagenesis studies using minigenomes.439,440,548,758,760 The 3′ termini of the genome and antigenome of VSV are identical at 15 of 18 positions. These 18 nucleotides are essential elements of both the genomic and antigenomic promoters. The near identity of the 3′ termini of the genome and antigenome implies that both RNAs display terminal complementarity. This complementarity enhances the activity of these RNA as templates for replication,758 suggesting that base pairing of the termini is an important element of promoter recognition by the VSV RNA polymerase, similar to promoter recognition by the influenza virus RNA polymerase.
 Figure 31.9. Diagram of activities of the rhabdovirus genomic and antigenomic promoters in transcription versus replication. |
The genomic and antigenomic promoters of VSV differ substantially at positions 19 to 29 and 34 to 46. These
sequences in the genomic promoter are required for mRNA synthesis, but not for replication.439,760 In contrast, these sequences in the antigenomic promoter serve as an enhancer of replication.440 As a result of this enhancer activity in the antigenomic promoter, replication of genomes versus antigenomes is asymmetric: many more genomes than antigenomes are synthesized in virus-infected cells.231,650,666,754 The functional differences between the genomic and antigenomic promoters were dramatically illustrated by engineering an ambisense RABV that contained the sequence of the genomic promoter at the 3′ ends of both the genome and antigenome.231 The promoter in the antigenome of this virus was engineered to drive the transcription of a foreign gene. As a result, both the genome and antigenome of this virus were used as templates for mRNA synthesis—the genome as a template for the five viral mRNAs and the antigenome as a template for the foreign mRNA. Furthermore, the normal asymmetry of replication was abolished, so that genomes and antigenomes were synthesized in approximately equal amounts.
sequences in the genomic promoter are required for mRNA synthesis, but not for replication.439,760 In contrast, these sequences in the antigenomic promoter serve as an enhancer of replication.440 As a result of this enhancer activity in the antigenomic promoter, replication of genomes versus antigenomes is asymmetric: many more genomes than antigenomes are synthesized in virus-infected cells.231,650,666,754 The functional differences between the genomic and antigenomic promoters were dramatically illustrated by engineering an ambisense RABV that contained the sequence of the genomic promoter at the 3′ ends of both the genome and antigenome.231 The promoter in the antigenome of this virus was engineered to drive the transcription of a foreign gene. As a result, both the genome and antigenome of this virus were used as templates for mRNA synthesis—the genome as a template for the five viral mRNAs and the antigenome as a template for the foreign mRNA. Furthermore, the normal asymmetry of replication was abolished, so that genomes and antigenomes were synthesized in approximately equal amounts.
In addition to differences in the cis-acting sequences that govern replication versus transcription, differences in the nature of the polymerase complex and the structural requirements of the P protein are also important for replication versus transcription. The C-terminal basic region of P protein that is involved in interaction with L protein is required for transcription, but not replication.164 Similarly, phosphorylation of serine and threonine residues in the N-terminal domain of P protein by cellular casein kinase II is required for transcription, but not for replication.550 In contrast, phosphorylation of sites in the C-terminal domain is required for replication, but not transcription.329 These differences in the structural requirements for P protein are consistent with the idea that the polymerase complex that carries out replication is distinct from that which carries out transcription. In particular, the transcriptase appears to be an L–(P4) complex, whereas the replicase appears to be an L–N–P4 complex.286 Establishing the structural basis for the differences in replication versus transcription, particularly the ability of the two polymerase complexes to respond to cis-acting signals in the template and the requirement for encapsidation of the nascent product RNA, is a key issue for understanding RNA synthesis by rhabdoviruses and other nonsegmented, negative-strand viruses.
Secondary Transcription
Once nucleocapsids containing progeny genomes begin to accumulate in infected cells, they are used as templates for secondary transcription, and they are assembled into progeny virions. In the case of VSV, most of the viral nucleocapsids that are made during the infectious cycle remain associated with infected cells and are not released in the form of progeny virions,379,666 suggesting that use of these nucleocapsids as templates predominates over their use for virion assembly. Although conceptually it is the last step in the virus replication cycle, virus assembly begins at approximately the same time as secondary transcription (for VSV, around 2 to 3 hours postinfection), reaches a maximum rate around 8 to 10 hours postinfection when viral protein synthesis is at its maximum, and declines concomitantly with a decline in viral protein synthesis toward the end of the infectious cycle around 16 to 20 hours postinfection.
Assembly of Progeny Virions
As with most viruses, the individual components of rhabdoviruses are assembled in separate cellular compartments and only come together in the final steps of virus assembly: the nucleocapsid is assembled during the process of RNA replication as described in the previous section, the G protein is assembled in the secretory pathway, and the M protein is synthesized as a soluble protein that then associates with the cytoplasmic surface of the host plasma membrane.
Assembly of G Protein
The assembly of the VSV G protein in the secretory pathway of host cells has been studied for many years by both virologists and cell biologists, not only for its importance for virus assembly, but also as a prototype for the assembly of other host and viral integral membrane proteins. G protein is synthesized by ribosomes bound to the rough endoplasmic reticulum (ER) and is inserted into the ER membrane in the typical type I orientation.605,606 An N-terminal signal sequence of 16 amino acids targets the protein for insertion and is cleaved from the nascent polypeptide.446 The new N-terminus and most of the protein sequence (446 amino acids) are transferred to the luminal side of the ER membrane to form the protein’s ectodomain.367 A hydrophobic sequence of 20 amino acids near the C-terminus serves as a stop–transfer sequence and becomes the membrane anchor.606 The 29 C-terminal amino acids remain on the cytoplasmic side of the ER membrane and form the protein’s cytoplasmic domain.367 Two asparagine residues in G protein are glycosylated during translation.368 The initial oligosaccharides added to G protein are of the high mannose type, which are later modified by enzymes in Golgi membranes to the complex type of oligosaccharides.581
Following insertion into the ER, G protein associates with two molecular chaperones, BiP (GRP78) and calnexin,290,467 which assist in the formation of the proper disulfide bonds and correct folding of the ectodomain. Mutations that prevent correct folding of the ectodomain or that prevent glycosylation, which is required for calnexin binding, result in the formation of aggregates of misfolded G protein together with BiP, which are not transported from the ER.189,467,468 Therefore, the ability of a mutant protein to be transported from the ER is a minimal criterion by which it can be said to be properly folded. Shortly after release of the properly folded G protein from the chaperones, G protein monomers associate into trimers189 and are transported to Golgi membranes by membrane vesicles that bud from the ER and subsequently fuse with Golgi membranes.394,705 G protein is one of the most rapidly transported integral membrane proteins, requiring approximately 15 minutes to be transported from ER to Golgi membranes.63 This rapid transport is dependent on a six–amino acid sequence in the cytoplasmic domain, which can function to concentrate G protein at the sites of vesicle budding.520,641
Once G protein is transported to Golgi membranes, it undergoes further posttranslational modifications, including conversion of its oligosaccharides from the high-mannose type to the complex type, containing additional N-acetyl glucosamine, galactose, and sialic acid.581 Although these modifications are not required for G protein function, they provide a convenient and widely used marker for transport of G protein through successive Golgi membranes.635 Another G protein
modification that occurs in Golgi membranes is the addition of the fatty acid palmitate to a cysteine residue in the cytoplasmic domain.606,625 Again, this modification does not appear to be critical, because some strains of VSV lack this modification, and mutation of the target cysteine residue, which abolishes palmitoylation, does not affect G protein function.766
modification that occurs in Golgi membranes is the addition of the fatty acid palmitate to a cysteine residue in the cytoplasmic domain.606,625 Again, this modification does not appear to be critical, because some strains of VSV lack this modification, and mutation of the target cysteine residue, which abolishes palmitoylation, does not affect G protein function.766
Transport of G protein from Golgi membranes to the plasma membrane requires approximately 15 minutes, so that the total time from synthesis in ER to appearance in the plasma membrane is about 30 minutes.63 In polarized epithelial cells, G protein is selectively transported to the basolateral surface.82,271,676 The same amino acid sequence in the cytoplasmic domain that promotes the rapid transport of G protein from ER to Golgi membranes is also necessary for the selective transport to the basolateral surface of polarized epithelial cells.703 Sorting of G protein and other basolaterally targeted proteins from those destined for the apical surface occurs first in Golgi membranes, and from Golgi membranes G protein is transported to the recycling endosome compartment prior to transport to the basolateral plasma membrane.20,104,155 This intermediate step presumably reflects additional sorting steps by which cells regulate the protein composition of their plasma membranes.
At the plasma membrane of infected cells, G protein is organized into clusters or microdomains that are approximately 100 to 150 nm in diameter.91,92 These G protein–containing microdomains are formed independently of other viral components92 and appear to be similar to cholesterol- and sphingolipid-rich lipid rafts that serve as sites of assembly for other viruses, such as influenza viruses.455,561 Lipid rafts have been defined in part by their resistance to solubilization with detergents at low temperatures.90 In contrast to envelope glycoproteins of influenza virus and other viruses that assemble at lipid rafts, G protein in host plasma membranes and in virion envelopes is detergent soluble.622 Nonetheless, the plasma membrane microdomains (and virus envelopes) that contain G protein resemble lipid rafts in that they are enriched in cholesterol and sphingolipids,455,561 but these lipids must not be in sufficiently high amounts to confer detergent resistance.93,622
The G protein–containing microdomains at the sites of virus budding are somewhat larger (300 to 400 nm) than the microdomains in the plasma membrane outside of virus budding sites (100 to 150 nm), implying that formation of virus budding sites involves clustering of membrane microdomains.91,93 An interesting feature of the budding process is that envelope glycoproteins from many unrelated viruses, as well as some host integral membrane proteins, can be incorporated into the envelopes of VSV or RABV in a process referred to as pseudotype formation or phenotypic mixing. Pseudotype formation was originally demonstrated by coinfection of cells with two different viruses.807 More recently the incorporation of heterologous glycoproteins into the envelopes of VSV and RABV has been demonstrated using recombinant viruses that express the foreign glycoprotein from the viral genome.101,240,358,391,479,629,690 Incorporation of heterologous glycoproteins into the virus envelope appears to result from clustering of microdomains containing the heterologous glycoprotein together with G protein–containing microdomains at the sites of virus assembly.93 It is not known what causes microdomains containing G protein or other glycoproteins to cluster at the sites of virus budding, but a model has been proposed in which clustering is driven by formation of the viral nucleocapsid–M protein complex.93,688
The efficiency with which heterologous glycoproteins are incorporated into the virus envelope varies over a considerable range, although thus far, none has been found to be incorporated as efficiently as G protein. In the case of RABV, interaction of the G protein cytoplasmic domain with the internal virion components may promote incorporation into the virus budding site, because appending the G protein cytoplasmic domain to foreign glycoproteins enhances their incorporation into RABV virions.489,490 In the case of VSV, incorporation of glycoproteins into the envelope does not depend on the sequence of the cytoplasmic domain, however, because substituting foreign sequences for the cytoplasmic domain of G protein or deleting the cytoplasmic domain does not alter the efficiency of G protein incorporation,92,628 and, with one exception (the human immunodeficiency virus [HIV] envelope glycoprotein),351,539 substituting the G protein cytoplasmic domain into foreign glycoproteins does not promote their incorporation into the virus envelope.358,594,629 Instead, the ability of a foreign glycoprotein to be incorporated into the VSV envelope may depend on the composition or physical properties of the microdomains containing the foreign glycoprotein. For example, the influenza virus hemagglutinin, which is in detergent-insoluble lipid rafts, is incorporated into the VSV envelope less efficiently than the T-cell antigen CD4, which is present in microdomains that are primarily detergent soluble.93,391,629
The presence of G protein in the plasma membrane is not essential for virus budding, as shown by studies with recombinant VSV and RABV in which the G gene has been mutated or deleted.378,491,594,632,690 In the absence of a complementing source of G protein, these viruses produce noninfectious particles that lack G protein but are otherwise indistinguishable from wild-type viruses. The efficiency of virus budding, however, is reduced by at least an order of magnitude in the absence of G protein, indicating that G protein plays a role in virus assembly to enhance the budding process. Thus, not only is it likely that internal virion components promote incorporation of G protein into the envelope as described earlier, but also it appears that G protein promotes assembly of internal virion components. As with the ability to be incorporated into the envelope, the ability of the VSV G protein to promote assembly is independent of the sequence of the cytoplasmic domain, although a minimal length of eight amino acids in the cytoplasmic domain does appear to be required.628 Instead of the cytoplasmic domain, the sequences in G protein that are responsible for promoting assembly appear to be in the membrane-proximal amino acids of the ectodomain.594 This has led to the suggestion that these sequences are responsible for introducing curvature in the membrane that promotes budding.594 This idea is supported by the observation that G protein expressed in the absence of other viral components is released from cells in membrane vesicles that may form by a process similar to virus budding.599
Role of M Protein in Virus Assembly
Unlike G protein, the viral M protein is synthesized as a soluble protein379,486 and associates with membranes in the manner of peripheral membrane proteins (i.e., through a combination of ionic and hydrophobic interactions and without spanning the membrane lipid bilayer).430,454,455,532,797,803,804 In virus-infected
cells, most of M protein is usually localized in the cytoplasm and is found in the soluble cytosolic fraction in subcellular fractionation experiments, with smaller amounts being membrane associated.238,379,535,537 This distribution is also observed in transfected cells that express M protein in the absence of other VSV components,123,124,797 indicating that association of M protein with membranes does not depend on other viral components. In addition, membrane association does not appear to require posttranslational modification, such as phosphorylation or covalent modification with lipids.361,430 Instead, M protein appears to spontaneously associate with membranes containing negatively charged phospholipids,454,455,532,797,803 which are enriched on the cytoplasmic surface of host plasma membranes. The N-terminal 20 amino acids of M protein, which are enriched in positively charged residues, appear to be responsible for membrane association, as shown using photoactivatable membrane probes.430 Mutational analysis has also implicated the N-terminal region of M protein in membrane binding.123,160,797 The membrane-bound M protein in infected cells is organized into membrane microdomains whose size is similar to G protein microdomains.688 However, M protein and G protein reside in separate microdomains except at the sites of virus budding.688
cells, most of M protein is usually localized in the cytoplasm and is found in the soluble cytosolic fraction in subcellular fractionation experiments, with smaller amounts being membrane associated.238,379,535,537 This distribution is also observed in transfected cells that express M protein in the absence of other VSV components,123,124,797 indicating that association of M protein with membranes does not depend on other viral components. In addition, membrane association does not appear to require posttranslational modification, such as phosphorylation or covalent modification with lipids.361,430 Instead, M protein appears to spontaneously associate with membranes containing negatively charged phospholipids,454,455,532,797,803 which are enriched on the cytoplasmic surface of host plasma membranes. The N-terminal 20 amino acids of M protein, which are enriched in positively charged residues, appear to be responsible for membrane association, as shown using photoactivatable membrane probes.430 Mutational analysis has also implicated the N-terminal region of M protein in membrane binding.123,160,797 The membrane-bound M protein in infected cells is organized into membrane microdomains whose size is similar to G protein microdomains.688 However, M protein and G protein reside in separate microdomains except at the sites of virus budding.688
Both the cytosolic and membrane-bound M protein are recruited into nucleocapsid–M protein complexes at the site of virus budding from host plasma membranes.237,528 Nucleocapsids clearly get selected for assembly with M protein, because most of the intracellular nucleocapsids, which are being used as templates for viral RNA synthesis, are not able to bind M protein.237,535,537 The only place in infected cells where co-localization of nucleocapsids and M protein is observed is in the nucleocapsid–M protein complexes in the process of budding from the plasma membrane.485,528,535,537 In the case of VSV, nucleocapsids containing genome RNA are incorporated into virus particles much more efficiently than those containing antigenomes.610,650,666,756 An RNA sequence near the 5′ end of the genome has been identified that is required for nucleocapsids containing this RNA to be incorporated into virus particles.761 If such a sequence is present in the RABV genome, it must also be present in the antigenome, because the recombinant ambisense RABV described earlier, which contains the genomic promoter in both the genome and antigenome, incorporates both genome and antigenome RNA into virions with equal efficiency.231 One of the important questions about virus assembly that needs to be addressed is how this RNA sequence promotes incorporation into virions, because selection of nucleocapsids for virion assembly is a critical step.
Once assembly of nucleocapsid–M protein complexes has begun, the recruitment of M protein into the complex appears to occur spontaneously. This process can be recreated in a cell-free system using purified M protein and virion nucleocapsids that have been stripped of most of their M protein by treatment with high-ionic-strength buffers.54,461,514,515 Nucleocapsids, from which M protein has been completely removed, however, cannot rebind M protein with the same high affinity observed in virion nucleocapsid–M protein complexes.237,461 This suggests that addition of the initial one or a few molecules of M protein to nucleocapsids (and, therefore, their removal) is fundamentally different from that of most M protein molecules in the nucleocapsid–M protein complex. Although the basis for this difference has yet to be discovered, it is tempting to think that it may be connected to the process of selection of intracellular nucleocapsids for virus assembly described in the previous paragraph.
Release of Assembled Virions
Following assembly of the nucleocapsid–M protein complex, the final step in virus assembly is release of the budding virion. This process is mediated by interaction of M protein with host proteins involved in MVB formation.298,299,331,332,345 The MVB machinery is also involved in release of retroviruses and filoviruses mediated by “late budding domains” in their Gag proteins or matrix proteins (VP40), respectively.564 In the formation of MVBs, vesicles derived from endosome membranes bud into the lumen of the endosome, carrying elements of the cytoplasm as their internal contents (Fig. 31.7). Modification of proteins on the cytoplasmic surface of the endosome membrane by covalent attachment of ubiquitin appears to be an important signal for incorporation of such cargo molecules into MVBs. The process of virus budding has the same membrane topology (cytoplasmic contents are internal), except that the process occurs at the plasma membrane rather than at the endosome membrane. A short sequence in M protein (PPPY in VSV, PPEY in RABV) appears to be responsible for redirecting this cellular machinery to the plasma membrane.298,299,331,332,345,785 No other viral components appear to be required, because expression of M protein in transfected cells in the absence of other viral components results in budding of membrane vesicles containing M protein.299,357,442 The PPPY sequence interacts with an E3 ubiquitin ligase called Nedd4, which is able to ubiquitinate M protein, in a cell-free assay.298 It has yet to be established whether Nedd4 itself, or one of its numerous family members, ubiquitinate M protein in infected cells, because this is likely to involve only a small proportion of the total M protein. Nonetheless, it is likely that ubiquitination of M protein is critical for release of budding virions, because mutation of the PPPY motif in M protein, or depletion of free ubiquitin in infected cells, dramatically reduces the release of virions and causes the accumulation of budding particles at the plasma membrane because of inhibition of their release.298,345 One of the questions that needs to be resolved is which cellular factors involved in MVB formation are involved in virus budding? Tsg101, a protein involved in recognizing ubiquitinated cargo proteins, and Vps4a, an adenosine triphosphatase (ATPase) involved in recycling the membrane trafficking machinery, have been implicated in budding of HIV but were reported not to be involved in budding of VSV or RABV.331,332 However, a more recent report indicated that Vps4a is involved in VSV budding.694
Molecular Genetics of Rhabdoviruses
Rapid Evolution and Existence of Quasispecies
Rhabdoviruses are classic examples of RNA viruses capable of undergoing rapid evolution. This is because of the high error rates of their RNA polymerases and their lack of proofreading activity. As a result, the rate of base substitutions during replication of genome RNA is approximately 1 in 104.190 Because their genomes are only slightly larger than 104 bases, this implies that nearly every genome contains at least one base substitution. Thus, even clonal populations of these viruses are actually collections of viruses with closely related sequences
(i.e., they are quasispecies). Because of their diversity of genome sequences, these viruses are capable of rapid genetic adaptation when placed under selective pressure of replication under different conditions. These viruses are genetically reasonably stable, however, when replication is maintained under a constant set of conditions.597 This is because the collection of genome sequences quickly reaches a consensus sequence representing the sequence with the highest level of fitness within a few replication cycles in a new host. This rapid adaptability may be advantageous in nature for viruses that alternate replication among different hosts. For example, VSV replicates both in arthropod hosts, where it establishes persistent infection, and in mammalian hosts, where it causes an acute infection. Transfer from one type of host to the other requires substantial increases in viral fitness to maintain optimal replication in the new host.806 In principle, this adaptation can occur through random mutation of the consensus sequence from the original host to one with greater fitness in the new host. However, in a virus population undergoing periodic cycling between insect and mammalian hosts, rapid adaptation to the mammalian host likely involves maintenance of a minority population of genomes in the insect host that quickly became dominant during mammalian infection.524 Replication of RABV is restricted to mammalian hosts, but similar though less drastic adaptation can also occur when RABV is transferred between different hosts.
(i.e., they are quasispecies). Because of their diversity of genome sequences, these viruses are capable of rapid genetic adaptation when placed under selective pressure of replication under different conditions. These viruses are genetically reasonably stable, however, when replication is maintained under a constant set of conditions.597 This is because the collection of genome sequences quickly reaches a consensus sequence representing the sequence with the highest level of fitness within a few replication cycles in a new host. This rapid adaptability may be advantageous in nature for viruses that alternate replication among different hosts. For example, VSV replicates both in arthropod hosts, where it establishes persistent infection, and in mammalian hosts, where it causes an acute infection. Transfer from one type of host to the other requires substantial increases in viral fitness to maintain optimal replication in the new host.806 In principle, this adaptation can occur through random mutation of the consensus sequence from the original host to one with greater fitness in the new host. However, in a virus population undergoing periodic cycling between insect and mammalian hosts, rapid adaptation to the mammalian host likely involves maintenance of a minority population of genomes in the insect host that quickly became dominant during mammalian infection.524 Replication of RABV is restricted to mammalian hosts, but similar though less drastic adaptation can also occur when RABV is transferred between different hosts.
The high rate of spontaneous mutation makes it feasible to isolate a variety of different types of viral mutants in the laboratory. For example, several large collections of temperature-sensitive mutants of VSV have been isolated, which fall into five or more complementation groups, corresponding to mutants with defects in each of the five viral genes.234,320,571 Similarly, antigenic variants that escape neutralization with monoclonal antibodies are readily isolated in the laboratory.410,426,459 Unlike influenza viruses and HIV, however, relatively little antigenic drift is found in rhabdoviruses during outbreaks in nature. This may reflect the harmful effects on G protein function of accumulating the multiple mutations necessary to escape neutralization by a polyclonal antibody response in intact animal hosts.525
Despite the advantages for rapid adaptation, the high rate of mutation makes these viruses susceptible to the harmful effects of genetic bottlenecks, in which only one or a few genomes are selected for further replication. In a process known as Muller’s ratchet, successive passage under conditions of limited genetic diversity, such as sequential passage by isolation of individual virus plaques, leads to progressive accumulation of base substitutions, most of which decrease virus replication, thus leading to progressively lower viral fitness192 (see Chapter 11).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


