Respiratory System
The respiratory system’s major function is gas exchange, in which air enters the body on inhalation (inspiration); travels throughout the respiratory passages, exchanging oxygen for carbon dioxide at the tissue level; and expels carbon dioxide on exhalation (expiration).
The upper airway—composed of the nose, mouth, pharynx, and larynx—allows airflow into the lungs. This area is responsible for warming, humidifying, and filtering the air, thereby protecting the lower airway from foreign matter.
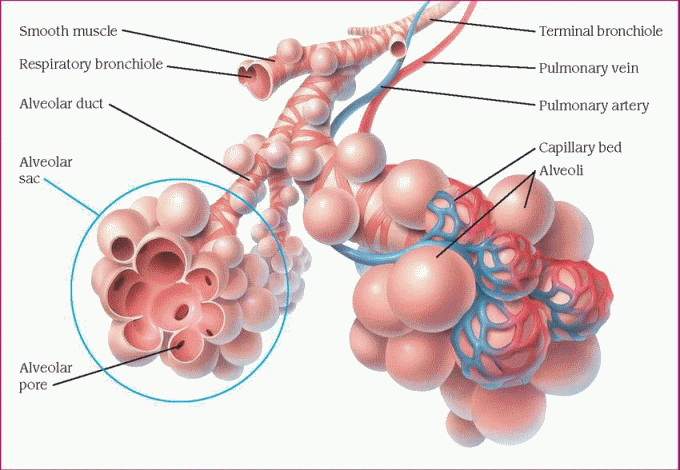
The lower airway consists of the trachea, mainstem bronchi, secondary bronchi, bronchioles, and terminal bronchioles. These structures are anatomic dead spaces and function only as passageways for moving air into and out of the lungs. Distal to each terminal bronchiole is the acinus, which consists of respiratory bronchioles, alveolar ducts, and alveolar sacs. The bronchioles and ducts function as conduits, and the alveoli are the chief units of gas exchange. These final subdivisions of the bronchial tree make up the lobules—the functional units of the lungs. (See Structure of the lobule.)
In addition to warming, humidifying, and filtering inspired air, the lower airway protects the lungs with several defense mechanisms. Clearance mechanisms include the cough reflex and mucociliary system. The mucociliary system produces mucus, which traps foreign particles. Foreign matter is then swept to the upper airway for expectoration by specialized fingerlike projections called cilia. A breakdown in the epithelium of the lungs or the mucociliary system can cause the defense mechanisms to malfunction, and pollutants and irritants then enter and inflame the lungs. The lower airway also provides immunologic protection and initiates pulmonary injury responses.
The external component of respiration (ventilation or breathing) delivers inspired air to the lower respiratory tract and alveoli. Contraction and relaxation of the respiratory muscles moves air into and out of the lungs.
Normal expiration is passive; the inspiratory muscles cease to contract, and the elastic recoil of the lungs and the chest wall causes them to contract again. These actions raise the pressure within the lungs to above atmospheric pressure, moving air from the lungs to the atmosphere.
An adult lung contains an estimated 300 million alveoli; each alveolus is supplied by many capillaries. To reach the capillary lumen, oxygen must cross the alveolar capillary membrane.
The pulmonary alveoli promote gas exchange by diffusion—the passage of gas molecules through respiratory membranes. In diffusion, oxygen passes to the blood, and carbon dioxide, a by-product of cellular metabolism, passes out of the blood and is channeled away.
Circulating blood delivers oxygen to the cells of the body for metabolism and transports metabolic wastes and carbon dioxide from the tissues back to the lungs. When oxygenated arterial blood reaches tissue capillaries, the oxygen diffuses from the blood into the cells because of an oxygen tension gradient. The amount of oxygen available to cells depends on the concentration of hemoglobin (the principal carrier of oxygen) in the blood, the regional blood flow, the arterial oxygen content, and cardiac output.
Because circulation is continuous, carbon dioxide doesn’t normally accumulate in tissues. Carbon dioxide produced during cellular respiration diffuses from tissues to regional capillaries and is transported by the systemic venous circulation. When carbon dioxide reaches the alveolar capillaries, it diffuses into the alveoli, where the partial pressure of carbon dioxide (PaCO2) is lower. Carbon dioxide is removed from the alveoli during exhalation.
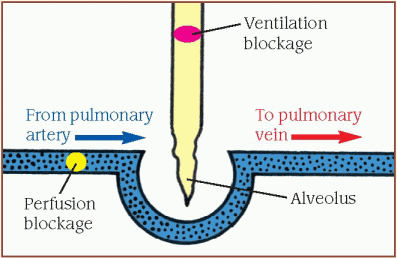
For effective gas exchange, ventilation and perfusion at the alveolar level must match closely. (See Understanding ventilation and perfusion, page 206.)
The ratio of ventilation to perfusion is called the [V with dot above]/[Q with dot above] ratio. A [V with dot above]/[Q with dot above] mismatch can result from ventilation-perfusion dysfunction or altered lung mechanics.
The amount of air carrying oxygen that reaches the lungs depends on lung volume and capacity, compliance, and resistance to airflow. Changes in compliance can occur in either the lung or the chest wall. Destruction of the lung’s elastic fibers, which occurs in acute respiratory distress syndrome, decreases lung compliance. The lungs become stiff, making breathing difficult. The alveolar capillary membrane may also be affected, causing hypoxia. Chest wall compliance is affected by disorders causing thoracic deformity, muscle spasm, and abdominal distention.
Respiration is also controlled neurologically by the lateral medulla oblongata of the brain stem. Impulses travel down the phrenic nerves to the diaphragm and then down the intercostal nerves to the intercostal muscles between the ribs. The rate and depth of respiration are controlled similarly.
Apneustic and pneumotaxic centers in the pons of the midbrain influence the pattern of breathing. Stimulation of the lower pontine apneustic center (by trauma, tumor, or stroke) produces forceful inspiratory gasps alternating with weak expiration. This pattern doesn’t occur if the vagi are intact. The apneustic center continually excites the medullary inspiratory center and thus facilitates inspiration. Signals from the pneumotaxic center and afferent impulses from the vagus nerve inhibit the apneustic center and “turn off” inspiration.
In addition, chemoreceptors respond to the hydrogen ion concentration of arterial blood
(pH), PaCO2, and the partial pressure of arterial oxygen (PaO2). Central chemoreceptors respond indirectly to arterial blood by sensing changes in the pH of the cerebrospinal fluid (CSF). PaCO2 also helps regulate ventilation by impacting the pH of CSF. If PaCO2 is high, the respiratory rate increases; if PaCO2 is low, the respiratory rate decreases. Information from peripheral chemoreceptors in the carotid and aortic bodies also responds to decreased PaO2 and pH. Either change results in increased respiratory drive within minutes.
(pH), PaCO2, and the partial pressure of arterial oxygen (PaO2). Central chemoreceptors respond indirectly to arterial blood by sensing changes in the pH of the cerebrospinal fluid (CSF). PaCO2 also helps regulate ventilation by impacting the pH of CSF. If PaCO2 is high, the respiratory rate increases; if PaCO2 is low, the respiratory rate decreases. Information from peripheral chemoreceptors in the carotid and aortic bodies also responds to decreased PaO2 and pH. Either change results in increased respiratory drive within minutes.
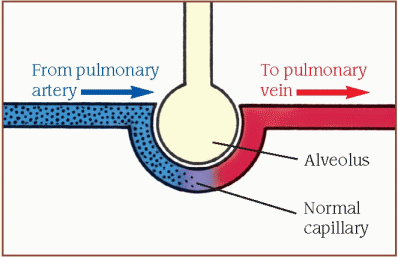
Effective gas exchange depends on the relationship between ventilation and perfusion, expressed as the [V with dot above]/[Q with dot above] ratio. The diagrams below show what happens when the [V with dot above]/[Q with dot above] ratio is normal and abnormal.
Normal ventilation and perfusion
When the [V with dot above]/[Q with dot above] ratio is matched, unoxygenated blood from the venous system returns to the right ventricle through the pulmonary artery to the lungs, carrying carbon dioxide. The arteries branch into the alveolar capillaries, where gas exchange occurs.
 |
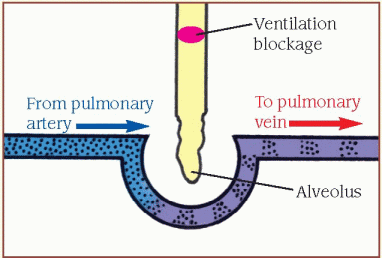
Inadequate ventilation (shunt)
When the [V with dot above]/[Q with dot above] ratio is low, pulmonary circulation is adequate, but oxygen is inadequate for normal diffusion (illustrated by the ventilation blockage). A portion of the blood flowing through the pulmonary vessels doesn’t become oxygenated.
 |
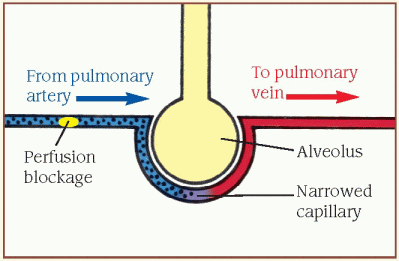
Inadequate perfusion (dead-space ventilation)
When the [V with dot above]/[Q with dot above] ratio is high, ventilation is normal, but alveolar perfusion is reduced or absent (illustrated by the perfusion blockage). This results from a perfusion defect, such as pulmonary embolism or a disorder that decreases cardiac output.
 |
Inadequate ventilation and perfusion (silent unit)
The silent unit indicates an absence of ventilation and perfusion to the lung area (illustrated by blockages in perfusion and ventilation). The silent unit may try to compensate for this [V with dot above]/[Q with dot above] imbalance by delivering blood flow to betterventilated lung areas.
 |
 |
Pathophysiologic changes
Pathophysiologic manifestations of respiratory disease may stem from atelectasis, bronchiectasis, cyanosis, and hypoxemia.
ATELECTASIS
Atelectasis occurs when the alveolar sacs or entire lung segments expand incompletely, producing a partial or complete lung collapse. This phenomenon removes certain regions of the lung from gas exchange, allowing unoxygenated blood to pass unchanged through these regions and resulting in hypoxia. Atelectasis may be chronic or acute, and commonly occurs in patients undergoing upper-abdominal or thoracic surgery. There are two major causes of collapse due to atelectasis: absorption atelectasis, secondary to bronchial or bronchiolar obstruction, and compression atelectasis.
Absorption atelectasis
Bronchial occlusion, which prevents air from entering the alveoli distal to the obstruction, can cause absorption atelectasis—the air present in the alveoli is absorbed gradually into the bloodstream and eventually the alveoli collapse. This may result from intrinsic or extrinsic bronchial obstruction. The most common intrinsic cause is retained secretions or exudate forming mucus plugs. Such disorders as cystic fibrosis, chronic bronchitis, and pneumonia increase the risk of absorption atelectasis. Extrinsic bronchial atelectasis usually results from occlusion caused by foreign bodies, bronchogenic carcinoma, and scar tissue.
Impaired production of surfactant can also cause absorption atelectasis. Increasing surface tension of the alveolus due to reduced surfactant leads to collapse.
Compression atelectasis
Compression atelectasis results from external compression, which drives the air out and causes the lung to collapse. This may result from upper-abdominal surgical incisions, rib fractures, pleuritic chest pain, tight chest dressings, and obesity (which elevates the diaphragm and reduces tidal volume). These situations inhibit full lung expansion or make deep breathing painful, thus resulting in this disorder.
Causes of bronchiectasis
Bronchiectasis results from conditions associated with repeated damage to bronchial walls and with abnormal mucociliary clearance, leading to a breakdown in the supporting tissue adjacent to the airways. Such conditions include:
♦ complications of measles, pneumonia, pertussis, or influenza
♦ congenital anomalies, such as bronchomalacia, congenital bronchiectasis, and Kartagener’s syndrome (bronchiectasis, sinusitis, and dextrocardia)
♦ cystic fibrosis
♦ immune disorders (agammaglobulinemia)
♦ inhalation of corrosive gas or repeated aspiration of gastric juices into the lungs
♦ obstruction (from a foreign body, tumor, or stenosis) with recurrent infection
♦ rare disorders, such as immotile cilia syndrome
♦ recurrent bacterial respiratory tract infections that were inadequately treated (tuberculosis).
BRONCHIECTASIS
Bronchiectasis is marked by chronic abnormal dilation of the bronchi and destruction of the bronchial walls and can occur throughout the tracheobronchial tree. It may also be confined to a single segment or lobe. This disorder is usually bilateral in nature and involves the basilar segments of the lower lobes.
There are three forms of bronchiectasis: cylindrical, fusiform (varicose), and saccular (cystic). It results from conditions associated with repeated damage to bronchial walls with abnormal mucociliary clearance, which causes a breakdown of supporting tissue adjacent to the airways. (See Causes of bronchiectasis.)
In patients with bronchiectasis, sputum stagnates in the dilated bronchi and leads to secondary infection, characterized by inflammation and leukocytic accumulations. Additional debris collects within and occludes the bronchi. Increasing pressure from the retained secretions induces mucosal injury.
Major causes of hypoxemia
This chart lists the major causes of hypoxemia and contributing factors.
Major cause | Contributing factors |
Alveolar capillary diffusion abnormality | Emphysema, conditions resulting in fibrosis, or pulmonary edema |
Decrease in inspired oxygen | High altitudes, inhaling poorly oxygenated gases, or breathing in an enclosed space |
Hypoventilation | Respiratory center inappropriately stimulated (such as by oversedation, overdosage, or neurologic damage), chronic obstructive pulmonary disease |
Shunting | Acute respiratory distress syndrome, idiopathic respiratory distress syndrome of the newborn, or atelectasis |
Ventilation-perfusion mismatch | Asthma, chronic bronchitis, or pneumonia |
CYANOSIS
Cyanosis is a bluish discoloration of the skin and mucous membranes. In most populations, it’s readily detectable by a visible blue tinge on the nail beds and lips. Central cyanosis indicates decreased oxygen saturation of hemoglobin in arterial blood, which is best observed in the buccal mucous membranes and the lips. Peripheral cyanosis is a slowed blood circulation of the fingers and toes that’s best visualized by examining the nail bed area.
 In patients with black or dark complexions, cyanosis may not be evident in the lip area or nail beds. A better indicator in these individuals is to assess the membranes of the oral mucosa (buccal mucous membranes) and of the conjunctivae of the eyes.
In patients with black or dark complexions, cyanosis may not be evident in the lip area or nail beds. A better indicator in these individuals is to assess the membranes of the oral mucosa (buccal mucous membranes) and of the conjunctivae of the eyes.Cyanosis is caused by desaturation of arterial blood with oxygen or reduced hemoglobin amounts. It develops when unsaturated hemoglobin reaches 5 g/ml, even if hemoglobin counts are adequate or reduced. Conditions that result in cyanosis include decreased arterial oxygenation (indicated by low PaO2), pulmonary or cardiac right-to-left shunts, decreased cardiac output, anxiety, and a cold environment.
An individual who isn’t cyanotic doesn’t necessarily have adequate oxygenation. Inadequate tissue oxygenation occurs in severe anemia, resulting in inadequate hemoglobin concentration. It also occurs in carbon monoxide poisoning, in which hemoglobin binds to carbon monoxide instead of to oxygen. Although assessment doesn’t reveal cyanosis, oxygenation is inadequate.
Another patient may appear cyanotic even though oxygenation is adequate—as in polycythemia, an abnormal increase in the red blood cell count. Because the hemoglobin level is increased and oxygenation occurs at a normal rate, the patient may still present with cyanosis.
Cyanosis as a presenting condition must be interpreted in relation to the patient’s underlying pathophysiology. Diagnosis of inadequate oxygenation may be confirmed by analyzing arterial blood gases and measuring PaO2.
HYPOXEMIA
Hypoxemia is reduced oxygenation of the arterial blood, evidenced by reduced PaO2 of arterial blood. It’s caused by respiratory alterations, whereas hypoxia is diminished tissue oxygenation at the cellular level that may be caused by conditions affecting other body systems that are unrelated to alterations of pulmonary function. Low cardiac output or cyanide poisoning can result in hypoxia, in addition to alterations in respiration. Hypoxia can occur anywhere in the body. If hypoxia occurs in the blood, it’s termed hypoxemia. Hypoxemia can lead to tissue hypoxia.
Hypoxemia can be caused by decreased oxygen content of inspired gas, hypoventilation, diffusion abnormalities, abnormal [V with dot above]/[Q with dot above] ratios, and pulmonary right-to-left shunts. The physiologic mechanism for each cause of hypoxemia varies. (See Major causes of hypoxemia.)
Disorders
Respiratory disorders can be acute or chronic. The disorders described here include examples from each type.
Acute respiratory distress syndrome (ARDS) is a form of pulmonary edema that can quickly lead to acute respiratory failure. Also known as shock lung, stiff lung, white lung, wet lung, or Da Nang lung, ARDS may follow direct or indirect injury to the lung. However, its diagnosis is difficult, and death can occur within 48 hours of onset if not promptly diagnosed and treated. A differential diagnosis needs to rule out cardiogenic pulmonary edema, pulmonary vasculitis, and diffuse pulmonary hemorrhage. Patients who recover may have little or no permanent lung damage.
Causes
♦ Acute miliary tuberculosis
♦ Anaphylaxis
♦ Aspiration of gastric contents
♦ Coronary artery bypass grafting
♦ Diffuse pneumonia, especially viral pneumonia
♦ Drug overdose, such as heroin, aspirin, or ethchlorvynol
♦ Hemodialysis
♦ Idiosyncratic drug reaction to ampicillin or hydrochlorothiazide
♦ Inhalation of noxious gases, such as nitrous oxide, ammonia, or chlorine
♦ Injury to the lung from trauma (most common cause) such as airway contusion
♦ Leukemia
♦ Near drowning
♦ Oxygen toxicity
♦ Pancreatitis
♦ Sepsis
♦ Thrombotic thrombocytopenic purpura
♦ Trauma-related factors, such as fat emboli, sepsis, shock, pulmonary contusions, and multiple transfusions, which increase the likelihood that microemboli will develop
♦ Uremia
♦ Venous air embolism
Pathophysiology
Signs and symptoms
♦ Rapid, shallow breathing and dyspnea, which occur hours to days after the initial injury in response to decreasing oxygen levels in the blood
♦ Increased rate of ventilation due to hypoxemia and its effects on the pneumotaxic center
♦ Intercostal and suprasternal retractions due to the increased effort required to expand the stiff lung
♦ Crackles and rhonchi, which are audible and result from fluid accumulation in the lungs
♦ Restlessness, apprehension, and mental sluggishness, which occur as the result of brain hypoxia
♦ Motor dysfunction, which occurs as hypoxia progresses
♦ Tachycardia, which signals the heart’s effort to deliver more oxygen to the cells and vital organs
♦ Respiratory acidosis, which occurs as carbon dioxide accumulates in the blood and oxygen levels decrease
♦ Metabolic acidosis, which eventually results from failure of compensatory mechanisms
Complications
♦ Hypotension
♦ Decreased urine output
♦ Metabolic acidosis
♦ Respiratory acidosis
♦ Multiple organ dysfunction syndrome
♦ Ventricular fibrillation
♦ Ventricular standstill
Diagnosis
♦ Arterial blood gas (ABG) analysis with the patient breathing room air initially reveals a decreased ratio of PaO2 to the fraction of inspired oxygen (less than or equal to 200 mm Hg), a long with a reduced PaO2 (less than 60 mm Hg) and a decreased PaCO2 (less than 35 mm Hg). Hypoxemia, despite increased supplemental oxygen, is the hallmark of ARDS; the resulting blood pH reflects respiratory alkalosis. As ARDS worsens, ABG values show respiratory acidosis evident by an increasing PaCO2 (over 45 mm Hg), metabolic acidosis evident by a decreasing HCO3 – less than 22 mEq/L, and a declining PaO2 despite oxygen therapy.
♦ Pulmonary artery catheterization helps identify the cause of pulmonary edema (cardiac versus noncardiac) by measuring pulmonary artery wedge pressure (PAWP); allows collection of pulmonary artery blood, which shows decreased
oxygen saturation, reflecting tissue hypoxia; measures pulmonary artery pressure; measures cardiac output by thermodilution techniques; and provides information to allow calculation of the percentages of blood shunted though the lungs.
oxygen saturation, reflecting tissue hypoxia; measures pulmonary artery pressure; measures cardiac output by thermodilution techniques; and provides information to allow calculation of the percentages of blood shunted though the lungs.
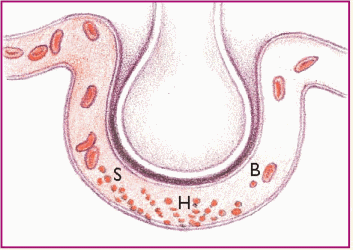
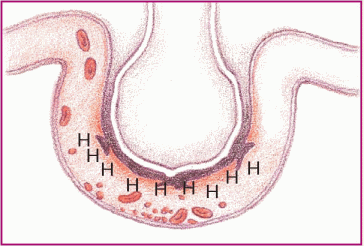
These diagrams show the process and progress of acute respiratory distress syndrome (ARDS).
Phase 1. Injury reduces normal blood flow to the lungs. Platelets aggregate and release histamine (H), serotonin (S), and bradykinin (B).
 |
Phase 2. The released substances inflame and damage the alveolar capillary membrane, increasing capillary permeability. Fluids then shift into the interstitial space.
 |
Phase 3. Capillary permeability increases and proteins and fluids leak out, increasing interstitial osmotic pressure and causing pulmonary edema.
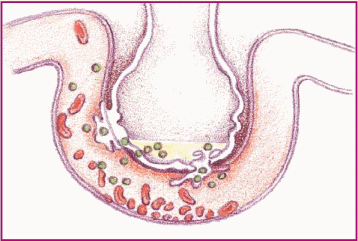 |
Phase 4. Decreased blood flow and fluids in the alveoli damage surfactant and impair the cell’s ability to produce more. The alveoli then collapse, thus impairing gas exchange.
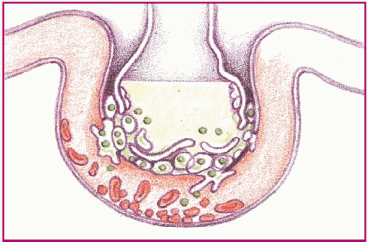 |
Phase 5. Oxygenation is impaired, but carbon dioxide (CO2) easily crosses the alveolar capillary membrane and is expired. Blood oxygen (O2) and CO2 levels are low.
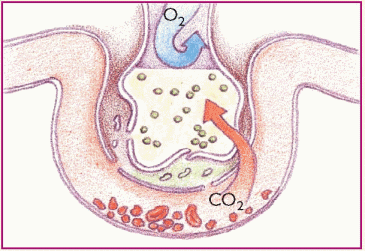 |
Phase 6. Pulmonary edema worsens and inflammation leads to fibrosis. Gas exchange is further impeded.
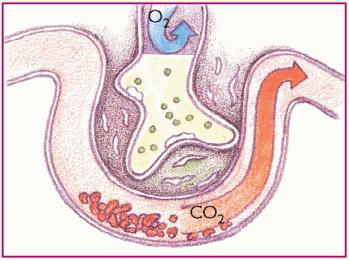 |
♦ Serial chest X-rays in early stages show bilateral infiltrates; in later stages, lung fields with a ground-glass appearance and “whiteouts” of both lung fields (with irreversible hypoxemia) may be observed. To differentiate ARDS from heart failure, note that the normal cardiac silhouette appears diffuse; bilateral infiltrates tend to be more peripheral and patchy, as opposed to the usual perihilar “bat wing” appearance of cardiogenic pulmonary edema; and there are fewer pleural effusions.
♦ Sputum analysis, including Gram stain and culture and sensitivity, identifies causative organisms.
♦ Blood cultures aid in identifying infectious organisms.
♦ Toxicology testing screens for drug ingestion.
♦ Serum amylase rules out pancreatitis.
Treatment
Therapy is focused on correcting the causes of ARDS and preventing progression of hypoxemia and respiratory acidosis; it may involve:
♦ administration of humidified oxygen by a tight-fitting mask, which allows for the use of continuous positive airway pressure
♦ for hypoxemia that doesn’t respond adequately to the above measures, ventilatory support with intubation, volume ventilation, and positive end-expiratory pressure (PEEP)
♦ pressure-controlled inverse ratio ventilation to reverse the conventional inspiration-toexpiration ratio and minimize the risk of barotrauma (Mechanical breaths are pressurelimited to prevent increased damage to the alveoli.)
♦ permissive hypercapnia to limit peak inspiratory pressure (Although carbon dioxide removal is compromised, treatment isn’t given for subsequent changes in blood hydrogen and oxygen concentration.)
♦ a sedative, an opioid, or a neuromuscular blocker, such as vecuronium, which may be given during mechanical ventilation to minimize restlessness, oxygen consumption, and carbon dioxide production and to facilitate ventilation
♦ sodium bicarbonate, which may reverse severe metabolic acidosis
♦ I.V. fluid administration to maintain blood pressure by treating hypovolemia
♦ a vasopressor to maintain blood pressure
♦ an antimicrobial to treat nonviral infections and to prevent ventilator-associated infections (based on culture results)
♦ a diuretic to reduce interstitial and pulmonary edema
♦ corticosteroids, as necessary to reduce inflammation
♦ correction of electrolyte and acid-base imbalances to maintain cellular integrity, particularly the sodium-potassium pump
♦ fluid restriction to prevent increase of interstitial and alveolar edema.
Special considerations
Caring for the patient with ARDS requires careful monitoring and supportive care.
♦ Frequently assess the patient’s respiratory status. Be alert for retractions on inspiration. Note the rate, rhythm, and depth of respirations; watch for dyspnea and the use of accessory muscles of respiration. On auscultation, listen for adventitious or diminished breath sounds. Check for clear, frothy sputum, which may indicate pulmonary edema.
♦ Observe and document the hypoxemic patient’s neurologic status (level of consciousness and mental sluggishness).
♦ Maintain a patent airway by suctioning, using sterile, nontraumatic technique. Ensure adequate humidification to help liquefy tenacious secretions.
♦ Closely monitor the patient’s heart rate and blood pressure. Watch for arrhythmias that may result from hypoxemia, acid-base disturbances, or electrolyte imbalances. With pulmonary artery catheterization, know the desired PAWP level. Check readings often, and watch for decreasing mixed venous oxygen saturation.
♦ Monitor serum electrolyte levels and correct imbalances. Measure intake and output; weigh the patient daily.
♦ Check ventilator settings frequently, and empty condensate from tubing promptly to ensure maximum oxygen delivery. Monitor ABG levels; check for metabolic and respiratory acidosis and PaO2 changes. The patient with severe hypoxemia may need controlled mechanical ventilation with positive pressure. Give a sedative, as needed, to reduce restlessness.
♦ Because PEEP may decrease cardiac output, check for hypotension, tachycardia, and decreased urine output. Suction only as needed to maintain PEEP or use an in-line suctioning apparatus. Reposition the patient often and record an increase in secretions, temperature, or hypotension that may indicate a deteriorating condition. Monitor peak pressures during ventilation. Because of stiff, noncompliant lungs, the
patient is at high risk for barotrauma (pneumothorax), evidenced by increased peak pressures, decreased breath sounds on one side, and restlessness.
patient is at high risk for barotrauma (pneumothorax), evidenced by increased peak pressures, decreased breath sounds on one side, and restlessness.
The cascade of events that occurs in acute respiratory distress syndrome (ARDS) can eventually affect every body system. Here’s how the process occurs, step-by-step, and how multidiscipinary care can help.
Respiratory system
♦ Damage to alveolar and pulmonary capillary, membranes triggers neutrophils, macrophages, monocytes, and lymphocytes to produce various cytokines that promote cellular activation, chemotaxis, and adhesion.
♦ Damage can occur directly (by aspiration of gastric contents and inhalation of noxious gases) or indirectly (from chemical mediators released in response to systemic injury).
♦ The activated cells produce inflammatory mediators, including oxidants, proteases, kinins, growth factors, and neuropeptides, which initiate the complement cascade, intravascular coagulation, and fibrinolysis.
♦ Vascular permeability to proteins increases. Plasma and blood leak into the alveoli and interstitial space.
♦ Fluid accumulates in the lung interstitium, the alveolar spaces, and the small airways, causing the lungs to stiffen and thus impairing ventilation and reducing oxygenation of the pulmonary capillary blood.
♦ Pressure changes and decreased surfactant result in alveolar collapse and atelectasis.
♦ Interstitial inflammation develops, and epithelial cells proliferate.
♦ Fluid in the alveoli and alveolar cell damage reduce surfactant production. Without surfactant, surface tension in the alveoli increases.
♦ Lung surface area is decreased as the lungs become less compliant and the alveoli collapse.
♦ Gas exchange is impaired, and respirations increase to address hypoxia.
♦ Initially, oxygenation is affected and carbon dioxide (CO2) levels decrease because CO2 is more easily diffused across the impaired alveolar-capillary membrane. As gas exchange worsens, hypercapnia develops.
♦ Hyaline membranes form because of the lack of surfactant and the collection of tissue debris and white blood cells in the airway.
♦ Inflammation leads to fibrosis, further impeding gas exchange. Fibrosis progressively obliterates alveoli, respiratory bronchioles, and the interstitium. Functional residual capacity decreases, and shunting becomes more serious.
♦ Increasing partial pressure of arterial carbon dioxide leads to respiratory acidosis.
♦ Hypoxia further increases acidosis; pH decreases.
♦ Hypoxia and acidosis result in changes in mental status.
Immune system
♦ The lung injury causes an inflammatory response, which continues as ARDS progresses.
♦ Platelets aggregate at the lung injury site and release substances—such as serotonin, bradykinin, and histamine—that attract and activate neutrophils. These substances inflame and damage the alveolar membrane and increase capillary permeability.
♦ Additional chemotactic factors are released, including endotoxins (such as those present in septic states), tumor necrosis factor, and interleukin-1. The activated neutrophils also release several inflammatory mediators and platelet aggravating factors that damage the alveolar capillary membrane and increase capillary permeability.
♦ Histamines and other inflammatory substances increase capillary permeability, allowing fluids to move into the interstitial space. As capillary permeability increases, proteins, blood cells, and more fluid leak out, increasing interstitial osmotic pressure and causing pulmonary edema.
♦ Mediators released by neutrophils and macrophages cause varying degrees of pulmonary vasoconstriction, resulting in pulmonary hypertension and causing a ventilationperfusion mismatch.
♦ Systemically, neutrophils and inflammatory mediators cause generalized endothelial damage and increased capillary permeability throughout the body.
♦ Multiple organ dysfunction syndrome (MODS) occurs as the cascade of mediators affects each body system.
♦ Death may occur from the influence of ARDS and MODS.
Collaborative management
A pulmonary specialist can help evaluate and treat the patient’s respiratory system. If infectious agents are involved, an infectious disease specialist may be called in; if cardiac involvement is suspected, a cardiologist may be consulted. If the patient progresses through the later stages of ARDS and a prolonged course is expected, a nutritional consult for total parenteral nutrition may be needed as well as specialists in physical and occupational therapy to assist with rehabilitation. If this patient develops a prolonged dependency on mechanical ventilation, he may require an extended care facility that can accommodate this treatment (after his condition is stabilized). If a prolonged stay in the health care facility is expected and the patient requires long-term care, social services should be consulted. Alternatively, if ARDS is successfully treated in the initial stages, the patient may be discharged to home with instructions to follow up with his physician.
♦ Monitor nutrition, maintain joint mobility, and prevent skin breakdown. Accurately record caloric intake. Give tube feedings and parenteral nutrition, as ordered. Perform passive rangeof-motion exercises or help the patient perform active exercises, if possible. Provide meticulous skin care. Plan patient care to allow periods of uninterrupted sleep.
♦ Provide emotional support. Warn the patient who’s recovering from ARDS that recovery will take some time and that he’ll feel weak for a while.
♦ Watch for and immediately report all respiratory changes in the patient with injuries that may adversely affect the lungs (especially during the 2- to 3-day period after the injury, when the patient may appear to be improving).
When the lungs can’t adequately maintain arterial oxygenation or eliminate carbon dioxide, acute respiratory failure (ARF) results, which can lead to tissue hypoxia. In patients with essentially normal lung tissue, ARF usually means partial pressure of arterial carbon dioxide (PaCO2) above 60 mm Hg or partial pressure of arterial oxygen (PaO2) below 50 mm Hg. These limits, however, don’t apply to patients with chronic obstructive pulmonary disease (COPD), who often have a consistently high PaCO2 and low PaO2. In patients with COPD, only acute deterioration in arterial blood gas (ABG) values, with corresponding clinical deterioration, indicates ARF.
Causes
Conditions that can result in alveolar hypoventilation, ventilation-perfusion ([V with dot above]/[Q with dot above]) mismatch, or right-to-left shunting can lead to respiratory failure; these include:
♦ atelectasis
♦ bronchitis
♦ bronchospasm
♦ central nervous system (CNS) depression—head trauma or injudicious use of sedatives, opioids, tranquilizers, or oxygen
♦ CNS disease
♦ COPD
♦ cor pulmonale
♦ cystic fibrosis
♦ heart failure
♦ pneumonia
♦ pneumothorax
♦ pulmonary edema
♦ pulmonary emboli
♦ ventilatory failure.
Pathophysiology
Respiratory failure results from impaired gas exchange. Conditions associated with alveolar hypoventilation, [V with dot above]/[Q with dot above] mismatch, and intrapulmonary (right-to-left) shunting can cause ARF if left untreated. (See How ARF affects the body, page 214.)
Signs and symptoms
Specific signs and symptoms vary with the underlying cause of ARF, but may include these systems:
♦ Respiratory—Rate may be increased, decreased, or normal depending on the cause; respirations may be shallow, deep, or alternate between the two; air hunger may occur. Cyanosis may or may not be present, depending on the hemoglobin level and arterial oxygenation. Auscultation of the chest may reveal crackles, rhonchi, wheezing, or diminished breath sounds secondary to possible airway obstruction and subsequent hypoventilation.
♦ CNS—When hypoxemia and hypercapnia occur, the patient may show evidence of restlessness, confusion, loss of concentration, irritability, tremulousness, diminished tendon reflexes, papilledema, and coma.
Hypoxemia and hypercapnia that result from acute respiratory failure (ARF) stimulate strong compensatory responses by all body systems.
Rapid detection of the condition and a multidisciplinary approach to care allow for the best outcome.
Respiratory system
♦ Decreased oxygen saturation may result from alveolar hypoventilation, in which chronic airway obstruction reduces alveolar minute ventilation. Partial pressure of arterial oxygen (Pao2) levels fall and partial pressure of arterial carbon dioxide levels rise, resulting in hypoxemia. The most common cause of alveolar hypoventilation is airway obstruction, commonly seen with chronic obstructive pulmonary disease (emphysema or bronchitis).
♦ Most commonly, hypoxemia—ventilationperfusion ([V with dot above]/[Q with dot above]) imbalance—occurs when such conditions as pulmonary embolism or acute respiratory distress syndrome interrupt normal gas exchange in a specific lung region. Too little ventilation with normal blood flow or too little blood flow with normal ventilation may cause the imbalance, resulting in decreased PaO2 levels and, thus, hypoxemia.
♦ Although uncommon, a decreased fraction of inspired oxygen may lead to respiratory failure. Inspired air doesn’t contain adequate oxygen to establish an adequate gradient for diffusion into the blood—for example, at high altitudes or in confined, enclosed spaces. As a result, hypoxemia occurs.
♦ Tissue hypoxemia results in anaerobic metabolism and lactic acidosis. Respiratory acidosis occurs from hypercapnia. Cyanosis occurs because of increased amounts of unoxygenated blood. As respiratory failure worsens, intercostal, supraclavicular, and suprasternal retractions may also occur.
Cardiovascular system
♦ Untreated [V with dot above]/[Q with dot above] imbalances can lead to rightto-left shunting, in which blood passes from the heart’s right side to its left without being oxygenated. This results in unoxygenated blood reaching the arterial system to be distributed to the rest of the body.
♦ Heart rate and stroke volume increases; heart failure may occur.
♦ Hypoxemia deprives the myocardial tissue of oxygen and nutrients, possibly resulting in ischemia or a myocardial infarction.
Neurologic system
♦ In response to hypoxemia, the sympathetic nervous system triggers vasoconstriction, increases peripheral resistance, and increases the heart rate.
♦ Hypoxemia or hypercapnia (or both) causes the brain’s respiratory control center to increase respiratory depth (tidal volume) and then to increase the respiratory rate.
Hematologic system
♦ Hypoxia of the kidneys results in release of erythropoietin from renal cells, causing the bone marrow to increase production of red blood cells—an attempt by the body to increase the blood’s oxygen-carrying capacity.
Collaborative management
A pulmonary specialist can help evaluate and treat the patient’s respiratory conditions. A respiratory therapy team member can assist with oxygen therapy and ventilatory support. If infectious agents are involved, an infectious disease specialist may be required; if cardiac involvement is suspected, a cardiologist may be consulted. The patient may require nutritional support to maintain and improve overall nutrition, strengthen the immune system, and meet metabolic needs. Initially, the patient may require total parenteral nutrition, depending on the severity of the condition and the patient’s status. If the patient is able to eat, a registered dietitian can provide planning to meet the patient’s needs.
Physical and occupational therapy may be necessary to help with energy conservation and rehabilitation, depending on the patient’s condition and length of stay. If a prolonged stay in the health care facility is expected and the patient requires long-term care, social services should be contacted early.
♦ Cardiovascular—Tachycardia, with increased cardiac output and mildly elevated blood pressure secondary to adrenal release of catecholamine, occurs early in response to low PaO2. With myocardial hypoxia, arrhythmias may develop. Pulmonary hypertension, secondary to pulmonary capillary vasoconstriction, may cause increased pressures on the right side of the heart, neck vein distention, an enlarged liver, and peripheral edema.
Complications
♦ Tissue hypoxia
♦ Metabolic acidosis
♦ Multiple organ failure
♦ Cardiac arrest
Diagnosis
♦ ABG analysis indicates respiratory failure by deteriorating values and a pH below 7.3. Patients with COPD may have a lower than normal pH compared with previous levels.
♦ Chest X-rays identify pulmonary diseases or conditions, such as emphysema, atelectasis, lesions, pneumothorax, infiltrates, and effusions.
♦ Electrocardiography can demonstrate ventricular arrhythmias (indicating myocardial hypoxia) or right ventricular hypertrophy (indicating cor pulmonale).
♦ Pulse oximetry reveals decreasing arterial oxygen saturation.
♦ White blood cell count detects the underlying infection.
♦ Abnormally low hemoglobin levels and hematocrit signal blood loss, which indicates decreased oxygen-carrying capacity.
♦ Hypokalemia may result from compensatory hyperventilation, the body’s attempt to correct acidosis.
♦ Hypochloremia usually occurs in metabolic alkalosis.
♦ Blood cultures may aid in identifying pathogens.
♦ Pulmonary artery catheterization helps to distinguish pulmonary and cardiovascular causes of ARF and monitors hemodynamic pressures.
Treatment
♦ Oxygen therapy to promote oxygenation and raise PaO2
♦ Bidirectional positive-pressure airway mask over the oronasal region or mechanical ventilation with an endotracheal or a tracheostomy tube, if needed, to provide adequate oxygenation and reverse acidosis
♦ High-frequency ventilation, if the patient doesn’t respond to treatment, to force the airways open, promoting oxygenation and preventing alveoli collapse
♦ An antibiotic to treat infection
♦ A bronchodilator to maintain airway patency
♦ A corticosteroid to decrease inflammation
♦ Fluid restrictions in cor pulmonale to reduce volume and cardiac workload
♦ A positive inotropic agent to increase cardiac output
♦ A vasopressor to maintain blood pressure
♦ A diuretic to reduce edema and fluid overload
♦ Deep breathing with pursed lips if patient isn’t intubated and mechanically ventilated to help keep airway patent
♦ Incentive spirometry to increase lung volume
Special considerations
♦ Because the patient with ARF is usually treated in an intensive care unit (ICU), orient him to the environment, procedures, and routines to minimize his anxiety.
♦ To reverse hypoxemia, administer oxygen at appropriate concentrations to maintain PaO2 at a minimum of 50 to 60 mm Hg. Patients with COPD usually require only small amounts of supplemental oxygen. Watch for a positive response, such as improvement in the patient’s breathing, color, and ABG results.
♦ Maintain a patent airway. If the patient is retaining carbon dioxide, encourage him to cough and to breathe deeply. Teach him to use pursedlip and diaphragmatic breathing to control dyspnea. If the patient is alert, have him use an incentive spirometer; if he’s intubated and lethargic, turn him every 1 to 2 hours. Use postural drainage and chest physiotherapy to help clear secretions.
♦ In an intubated patient, suction the trachea as needed after hyperoxygenation. Observe for change in quantity, consistency, and color of sputum. Provide humidification to liquefy secretions.
♦ Observe the patient closely for respiratory arrest. Auscultate for chest sounds. Monitor ABG levels and report changes immediately.
♦ Monitor and record serum electrolyte levels carefully, and correct imbalances; monitor fluid balance by recording intake and output or daily weight.
♦ Check the cardiac monitor for arrhythmias.
For mechanical ventilation
♦ Check ventilator settings, cuff pressures, and ABG values often because the fraction of inspired oxygen (FIO2) setting depends on ABG levels. Draw specimens for ABG analysis 20 to 30 minutes after every FIO2 change or check with oximetry.
♦ Prevent infection by using sterile technique while suctioning.
♦ Stress ulcers are common in intubated ICU patients. Check gastric secretions for evidence of bleeding if the patient has a nasogastric tube or complains of epigastric tenderness, nausea, or vomiting. Monitor hemoglobin level and hematocrit; check stool for occult blood. Administer an antacid, a histamine2-receptor antagonist, or sucralfate, as ordered.
♦ Prevent tracheal erosion, which can result from artificial airway cuff overinflation. Use the minimal leak technique and a cuffed tube with high residual volume (low-pressure cuff), a foam cuff, or a pressure-regulating valve on the cuff.
♦ To prevent oral or vocal cord trauma, make sure the endotracheal tube is positioned midline.
♦ To prevent nasal necrosis, keep the nasotracheal tube midline within the nostrils and provide good hygiene. Loosen the tape periodically to prevent skin breakdown. Avoid excessive movement of any tubes; make sure the ventilator tubing is adequately supported.
ASBESTOSIS
Considered a form of pneumoconiosis, asbestosis is characterized by diffuse interstitial pulmonary fibrosis. Prolonged exposure to airborne particles causes pleural plaques and tumors of the pleura and peritoneum. Asbestosis may develop 15 to 20 years after regular exposure to asbestos has ended. It’s a potent co-carcinogen and increases the smoker’s risk of lung cancer. An asbestos worker who smokes is 90 times more likely to develop lung cancer than a smoker who has never worked with asbestos.
Causes
♦ Exposure to asbestos used in paints, plastics, and brake and clutch linings
♦ Exposure to fibrous asbestos dust in deteriorating buildings or in waste piles from asbestos manufacturing plants
♦ Family members of asbestos workers, who may be exposed to stray fibers from the worker’s clothing
♦ Prolonged inhalation of asbestos fibers; people at high risk include workers in the mining, milling, construction, fireproofing, and textile industries
Pathophysiology
Asbestosis occurs when lung spaces become filled with asbestos fibers. The inhaled asbestos fibers (50 microns or more in length and 0.5 microns or less in diameter) travel down the airway and penetrate respiratory bronchioles and alveolar walls. Coughing attempts to expel the foreign matter. Mucus production and goblet cells are stimulated to protect the airway from the debris and aid in expectoration. Fibers then become encased in a brown, iron-rich proteinlike sheath in sputum or lung tissue, called asbestosis bodies. Chronic irritation by the fibers continues to affect the lower bronchioles and alveoli. The foreign material and inflammation swell airways, and fibrosis develops in response to the chronic irritation. Interstitial fibrosis may develop in lower lung zones, affecting lung parenchyma and the pleurae. Raised hyaline plaques may form in the parietal pleura, the diaphragm, and the pleura adjacent to the pericardium. Hypoxia develops as more alveoli and lower airways are affected.
Signs and symptoms
♦ Exertional dyspnea as a result of increased mucus production and airway narrowing
♦ Dyspnea at rest with extensive fibrosis
♦ Severe, nonproductive cough in nonsmokers or productive cough in smokers from chronic irritation of bronchial tree and mucus production
♦ Clubbed fingers due to chronic hypoxia
♦ Chest pain (commonly pleuritic) due to pleural irritation
♦ Recurrent respiratory tract infections as pulmonary defense mechanisms begin to fail
♦ Pleural friction rub due to fibrosis
♦ Crackles on auscultation attributed to air moving through thickened sputum
♦ Decreased lung inflation due to lung stiffness
♦ Recurrent pleural effusions due to fibrosis
♦ Decreased forced expiratory volume due to diminished alveoli
♦ Decreased vital capacity due to fibrotic changes
Complications
♦ Pulmonary fibrosis due to progression of asbestosis
♦ Respiratory failure
♦ Pulmonary hypertension
♦ Cor pulmonale
Diagnosis
♦ Chest X-rays may show fine, irregular, linear, and diffuse infiltrates. Extensive fibrosis is revealed by a honeycomb or ground-glass appearance. Chest X-rays may also show pleural thickening and calcification, bilateral obliteration of the costophrenic angles and, in later stages, an enlarged heart with a classic “shaggy” border.
♦ Pulmonary function studies may identify decreased vital capacity, forced vital capacity
(FVC), and total lung capacity; decreased or normal forced expiratory volume in 1 second (FEV1); a normal FEV1-to-FVC ratio; and reduced diffusing capacity for carbon monoxide when fibrosis destroys alveolar walls and thickens the alveolar capillary membrane.
(FVC), and total lung capacity; decreased or normal forced expiratory volume in 1 second (FEV1); a normal FEV1-to-FVC ratio; and reduced diffusing capacity for carbon monoxide when fibrosis destroys alveolar walls and thickens the alveolar capillary membrane.
♦ Arterial blood gas analysis may reveal decreased partial pressure of arterial oxygen and partial pressure of arterial carbon dioxide from hyperventilation.
Treatment
The goal of treatment is to relieve symptoms and control complications; it may involve:
♦ chest physiotherapy (controlled coughing and postural drainage with chest percussion and vibration) to help relieve respiratory signs and symptoms and manage hypoxia and cor pulmonale
♦ aerosol therapy to liquefy mucus
♦ an inhaled mucolytic to liquefy and mobilize secretions
♦ increased fluid intake to 3 qt (3 L) daily
♦ an antibiotic to treat respiratory tract infections
♦ oxygen administration to relieve hypoxia
♦ possibly a diuretic to decrease edema, digoxin to enhance cardiac output, and salt restriction to prevent fluid retention for patients with cor pulmonale.
Special considerations
♦ Teach the patient to prevent infections by avoiding crowds and persons with infections and by receiving influenza and pneumococcal vaccines.
♦ Improve the patient’s ventilatory efficiency by encouraging physical reconditioning, energy conservation in daily activities, and relaxation techniques.
ASTHMA
Asthma is a chronic inflammatory airway disorder characterized by airflow obstruction and airway hyperresponsiveness to a multiplicity of stimuli. This widespread but variable airflow obstruction is caused by bronchospasm, edema of the airway mucosa, and increased mucus production with plugging and airway remodeling. It’s a type of chronic obstructive pulmonary disease (COPD), a long-term pulmonary disease characterized by increased airflow resistance; other types of COPD include chronic bronchitis and emphysema.
 Although asthma strikes at any age, about 50% of patients are younger than age 10; twice as many boys as girls are affected in this age-group. One-third of patients develop asthma between ages 10 and 30, and the incidence is the same in both sexes in this age-group. Moreover, about one-third of all patients share the disease with at least one immediate family member.
Although asthma strikes at any age, about 50% of patients are younger than age 10; twice as many boys as girls are affected in this age-group. One-third of patients develop asthma between ages 10 and 30, and the incidence is the same in both sexes in this age-group. Moreover, about one-third of all patients share the disease with at least one immediate family member.Asthma may result from sensitivity to extrinsic or intrinsic allergens. Extrinsic, or atopic, asthma begins in childhood; typically, patients are sensitive to specific external allergens.
 Extrinsic asthma is commonly accompanied by other hereditary allergies, such as eczema and allergic rhinitis, in children.
Extrinsic asthma is commonly accompanied by other hereditary allergies, such as eczema and allergic rhinitis, in children.Intrinsic, or nonatopic, patients with asthma react to internal, nonallergenic factors; external substances can’t be implicated in patients with intrinsic asthma. Most episodes occur after a severe respiratory tract infection, especially in adults. However, many patients with asthma, especially children, have intrinsic and extrinsic asthma.
 Asthma is a complex inheritable disease, meaning there are several genes that make a person susceptible to the disease, including genes on chromosomes 5, 6, 11, 12, and 14.
Asthma is a complex inheritable disease, meaning there are several genes that make a person susceptible to the disease, including genes on chromosomes 5, 6, 11, 12, and 14.The role of these genes in the development of asthma isn’t clear, but one of the most promising sites of study is chromosome 5. Even though researchers haven’t specifically identified a gene from this site, they do know that the area is rich in genes coding for molecules that play a key role in the inflammatory response seen in asthma. Scientists continue to search for specific asthma genes.
A significant number of adults acquire an allergic form of asthma or exacerbation of existing asthma from exposure to agents in the workplace. Irritants, such as chemicals in flour, acid anhydrides, toluene di-isocyanates, screw flies, river flies, and excreta of dust mites in carpet, have been identified as agents that trigger asthma.
Causes
Extrinsic allergens
♦ Animal dander
♦ Food additives containing sulfites
♦ House dust or mold
♦ Kapok or feather pillows
♦ Other sensitizing substances
♦ Pollen
Pathophysiology
There are two genetic influences identified with asthma, namely the ability of an individual to develop asthma (atopy) and the tendency to develop hyperresponsiveness of the airways independent of atopy. A locus of chromosome 11 associated with atopy contains an abnormal gene that encodes a part of the immunoglobulin (Ig) E receptor. Environmental factors interact with inherited factors to cause asthmatic reactions with associated bronchospasms.
In asthma, bronchial linings overreact to various stimuli, causing episodic smooth-muscle spasms that severely constrict the airways. (See Pathophysiology of asthma.) IgE antibodies, attached to histamine-containing mast cells and receptors on cell membranes, initiate intrinsic asthma attacks. When exposed to an antigen, such as pollen, the IgE antibody combines with the antigen.
On subsequent exposure to the antigen, mast cells degranulate and release mediators. Mast cells in the lung interstitium are stimulated to release histamine and leukotrienes. Histamine attaches to receptor sites in the larger bronchi, where it causes swelling in smooth muscles. Mucous membranes become inflamed, irritated, and swollen. The patient may experience dyspnea, prolonged expiration, and an increased respiratory rate.
Leukotrienes attach to receptor sites in the smaller bronchi and cause local swelling of the smooth muscle. Leukotrienes also cause prostaglandins to travel through the bloodstream to the lungs, where they enhance histamine’s effect. A wheeze may be audible during coughing—the higher the pitch, the narrower the bronchial lumen. Histamine stimulates the mucous membranes to secrete excessive mucus, further narrowing the bronchial lumen. Goblet cells secrete viscous mucus that’s difficult to cough up, resulting in coughing, rhonchi, increased-pitch wheezing, and increased respiratory distress. Mucosal edema and thickened secretions further block the airways. (See Looking at a bronchiole in asthma, page 220.)
On inhalation, the narrowed bronchial lumen can still expand slightly, allowing air to reach the alveoli. On exhalation, increased intrathoracic pressure closes the bronchial lumen completely. Air enters but can’t escape. The patient develops a barrel chest and hyperresonance to percussion.
Mucus fills the lung bases, inhibiting alveolar ventilation. Blood is shunted to alveoli in other lung parts, but still can’t compensate for diminished ventilation.
Hyperventilation is triggered by lung receptors to increase lung volume because of trapped air and obstructions. Intrapleural and alveolar gas pressures rise, causing a decreased perfusion of alveoli. Increased alveolar gas pressure, decreased ventilation, and decreased perfusion result in uneven ventilation-perfusion ratios and mismatching within different lung segments.
Hypoxia triggers hyperventilation by respiratory center stimulation, which in turn decreases partial pressure of arterial carbon dioxide (PaCO2) and increases pH, resulting in a respiratory alkalosis. As the airway obstruction increases in severity, more alveoli are affected. Ventilation and perfusion remain inadequate, and carbon dioxide retention develops. Respiratory acidosis results, and respiratory failure occurs.
If status asthmaticus occurs, hypoxia worsens and expiratory flows and volumes decrease even further. If treatment isn’t initiated, the patient begins to tire out. (See Averting an asthma attack, page 221.) Acidosis develops as arterial carbon dioxide increases. The situation becomes life-threatening as no air becomes audible upon auscultation (a silent chest) and PaCO2 rises to over 70 mm Hg.
Signs and symptoms
Extrinsic asthma is usually accompanied by signs and symptoms of atopy (type I IgEmediated allergy), such as eczema and allergic rhinitis. It commonly follows a severe respiratory tract infection, especially in adults.
An acute asthma attack begins dramatically, with simultaneous onset of severe multiple symptoms, or insidiously, with gradually increasing respiratory distress. Asthma that occurs with cyanosis, confusion, and lethargy indicates the onset of life-threatening status asthmaticus and respiratory failure.
Signs and symptoms of asthma include:
♦ sudden dyspnea, wheezing, and tightness in the chest from bronchoconstriction
♦ coughing that produces thick, clear, or yellow sputum resulting from excess mucus production
♦ tachypnea, along with use of accessory respiratory muscles due to increasing air trapping and respiratory distress
♦ rapid pulse from increased workload of the heart due to the effects of hypoxemia and hyperinflation on the pulmonary vasculature
In asthma, hyperresponsiveness of the airways and bronchospasms occur. These illustrations show the progression of an asthma attack.
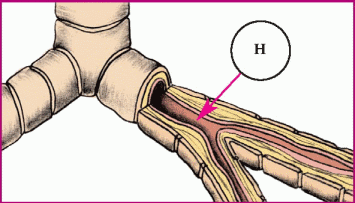 |
♦ Histamine (H) attaches to receptor sites in larger bronchi, causing swelling of the smooth muscles.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


