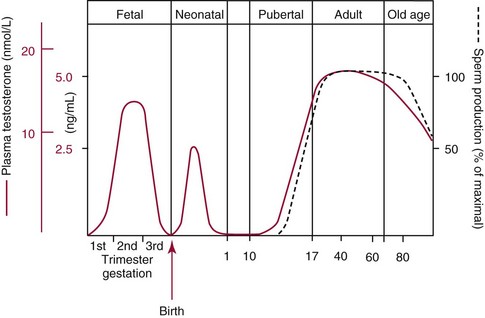
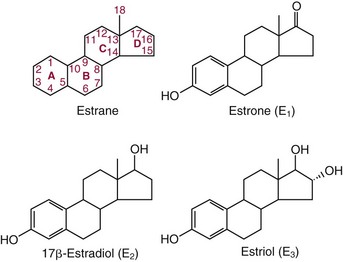
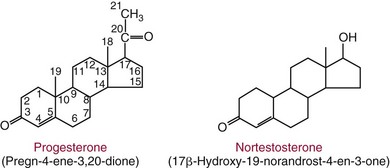
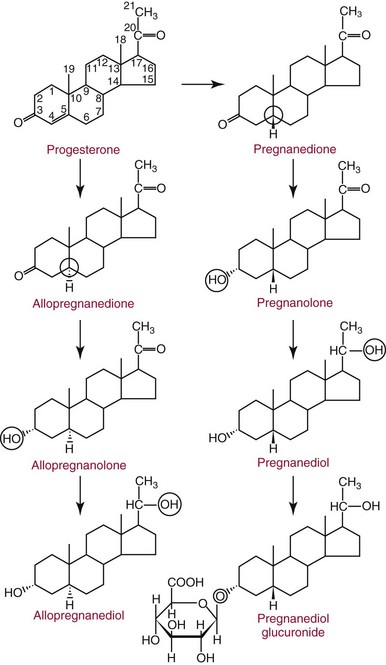
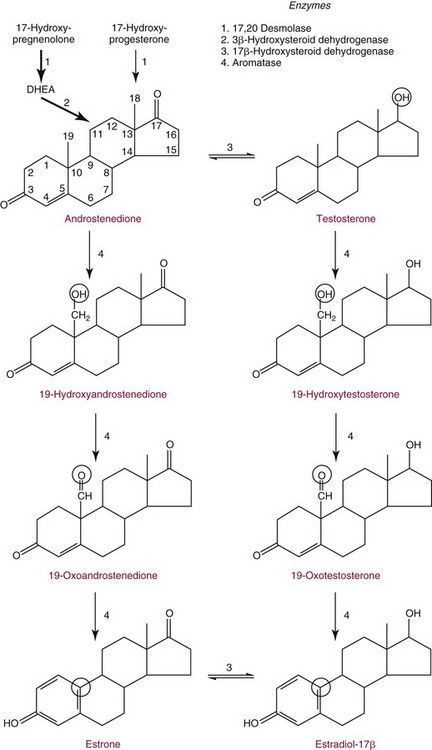
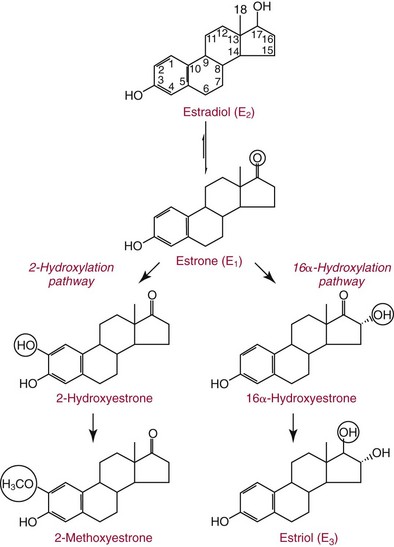
Chapter 56 Reproductive endocrinology encompasses the hormones of the hypothalamic-pituitary-gonadal axis, as well as the adrenal glands (see Chapter 54). These hormones are crucial for reproductive function and include gonadotropin-releasing hormone (GnRH), luteinizing hormone (LH), follicle-stimulating hormone (FSH), and a multitude of sex steroids. The sex steroids are synthesized by the ovaries, testes, and adrenal glands and are responsible for the manifestation of primary and secondary sex characteristics. This chapter is divided into four sections: Male Reproductive Biology, Female Reproductive Biology, Infertility, and Methods. Gonadotropin-releasing hormone (GnRH) is a decapeptide synthesized in the hypothalamus and transported to the anterior pituitary gland, where it stimulates the release of both FSH and LH (see also Chapter 53). In adult men, GnRH and thus LH and FSH are secreted in pulsatile patterns. A circadian rhythm is present, with higher concentrations found in the early morning hours and lower concentrations in the late evening.302 LH acts on Leydig cells to stimulate the conversion of cholesterol to pregnenolone. FSH acts on Sertoli cells and spermatocytes and is central to the initiation (in puberty) and maintenance (in adulthood) of spermatogenesis.297 Sex steroids and inhibin (a 32 kDa protein secreted by the Sertoli cells) provide negative feedback control of LH and FSH secretion, respectively. LH secretion is inhibited by testosterone and by its metabolites, estradiol and DHT. FSH may be elevated in disorders in which Sertoli cell numbers (and hence inhibin concentrations) are reduced. Likewise, a reduction in the number of Leydig cells (and hence testosterone secretion) leads to increased LH concentrations. Androgens are a group of C-19 steroids (Figure 56-1) responsible for masculinization of the genital tract and development and maintenance of male secondary sex characteristics. Testosterone is the principle androgen secreted in men. Testosterone is synthesized primarily by the Leydig cells of the testes (95%) and, to a lesser extent (≈5%) via peripheral conversion from the precursors dehydroepiandrosterone (DHEA) and androstenedione (which are synthesized in the zona reticularis of the adrenal glands). Synthesis of androgens begins with mobilization of cholesterol derived from lipoprotein cholesterol or by de novo synthesis.84,124 Cholesterol released from the lipid droplets migrates to the inner mitochondrial membrane, where pregnenolone formation is catalyzed by the cholesterol sidechain cleavage enzyme, CYP11A1. Conversion of cholesterol to pregnenolone is the rate-limiting step in testosterone synthesis113,281; however, it is thought that the rate of steroidogenesis is determined not by the activity of CYP11A1, but rather by delivery of cholesterol to the enzyme in the inner mitochondrial membrane by the steroidogenic regulatory protein (StAR)—a process thought to be regulated by LH.282 Following the formation of pregnenolone, four additional enzymatic steps are required to convert cholesterol to testosterone. The pathway for testosterone formation is shown in Figure 56-1, with the preferred pathway defined by heavy red arrows. Testosterone and DHT circulate in plasma freely (≈2 to 3%) or bound to plasma proteins. Binding proteins include the specific sex hormone–binding globulin (SHBG) and nonspecific proteins such as albumin. SHBG is an α-globulin that has low capacity for steroids but binds with very high affinity (Ka = 1 × 108 to 1 × 109), whereas albumin has high capacity but low affinity (Ka = 1 × 104 to 1 × 106).281 SHBG has the highest affinity for DHT and the lowest for estradiol. In men, testosterone circulates bound 44 to 65% to SHBG and 33 to 50% to albumin, whereas in women, testosterone is bound 66 to 78% to SHBG and 20 to 30% to albumin.222,230 The biologically active fraction includes free testosterone; some have suggested that albumin-bound testosterone may also be available for tissue uptake.63,229 Therefore, the bioavailable testosterone is equal to ≈35% of the total quantity (free + albumin-bound). Whether albumin-bound testosterone dissociates sufficiently fast to enter tissues is controversial.192,200 However, concentrations of bioavailable testosterone correlate with those of free testosterone.209,306 Testosterone and SHBG exhibit rhythmic variation in their circulating concentrations. Testosterone concentrations peak at approximately 0400 to 0800 hours, and nadir concentrations occur at between 1600 and 2000 hours.262,298 Daily variations in SHBG concentrations are similar to those of other proteins and albumin in serum, with major changes related to posture.298 Concentrations of SHBG are elevated with hyperthyroidism and in hypogonadal men. Circulating testosterone serves as a precursor for the formation of two additional active metabolites: DHT and estradiol (Figure 56-2). In one pathway, 5α-reductase converts 6 to 8% of testosterone to DHT. Both testosterone and DHT bind the androgen receptor, but DHT binds with higher affinity. In an alternative pathway, testosterone and androstenedione are converted to estrogens (≈0.3%) through aromatase (CYP19). DHT is formed in androgen target tissues such as the skin and prostate, whereas aromatization occurs in many tissues, especially the liver and adipose tissue. Peripheral aromatization occurs primarily in adipose tissue (of both men and women) because of the high concentration of aromatase in this tissue. The rate of extraglandular aromatization therefore increases with body fat. Dihydrotestosterone is metabolized to 3α-androstanediol (see Figure 56-1) and then is conjugated to form 3α-androstanediol glucuronide. These metabolites have been used as markers of DHT production in peripheral tissues. Serum concentrations of 3α-androstanediol glucuronide or 3α-androstanediol reflect the production of DHT in peripheral tissues such as skin.138,183 However, DHT may also arise from precursors other than testosterone. The reduction in serum 3α-androstanediol glucuronide concentrations noted in patients treated with glucocorticoids that suppress adrenal glucocorticoid and androgen production supports this conclusion.199 The main excretory metabolites of androstenedione, testosterone, and DHEA are shown in Figure 56-3. Except for epitestosterone, these catabolites constitute a group of steroids known as 17-ketosteroids (17-KS); they are excreted primarily in the urine. Testosterone is required for proper sexual development and function throughout all stages of life—fetal, pubertal, and adult (Figure 56-4). Fetal testes produce testosterone around the seventh week of gestation with peak serum concentrations of ≈250 ng/dL observed at the beginning of the second trimester, and with concentrations gradually returning to baseline by birth. Shortly after birth, the concentration of testosterone begins to increase, peaking again at ≈250 ng/dL at 2 to 3 months of age, and then falls to baseline again by 6 to 12 months. The function of this neonatal testosterone surge is not entirely clear, but it is thought to be important for bone growth and remodeling272 and development of external male genitalia.32 The concentration of testosterone remains low (<50 ng/dL) until puberty, when the concentration of testosterone rises to 500 to 700 ng/dL. Testosterone remains elevated through adulthood until around the third to fourth decade. Men beyond the age of 30 to 40 years experience an age-dependent decrease in circulating testosterone concentration. This has been demonstrated consistently in both cross-sectional and longitudinal analyses. Collectively these studies have shown a 0.5 to 2% decrease per year in total serum testosterone from about the fourth decade onward.93,126,208,304 This decline in testosterone is thought to be due to (1) a decrease in Leydig cell numbers, (2) decreased GnRH pulse amplitude, and (3) increases in SHBG.208 In the past, these decreases in circulating concentrations of testosterone were viewed as a normal part of the aging process. Now, however, these decreases, when accompanied by symptoms of decreased libido, sexual dysfunction, decreased energy levels, and decreased muscle mass, are regarded as a syndrome with a variety of names, such as (1) androgen deficiency in the aging male (ADAM), (2) partial androgen deficiency of the aging male (PADAM), (3) late-onset hypogonadism (LOH), and, erroneously, (4) andropause. The name andropause is inaccurate and misleading, given that in contrast to menopause in women, concentrations of sex steroids in men do not decrease sharply with secondary cessation of reproductive function. A name put forward more recently is testosterone deficiency syndrome (TDS), highlighting a specific deficit in testosterone as part of the clinical picture.207,294 Diagnosis of LOH (TDS) should be based on both clinical and laboratory assessment.40 Clinically, patients should exhibit symptoms suggestive of testosterone deficiency, such as (1) decreased libido, (2) erectile dysfunction, (3) decreased muscle mass and strength, (4) decreased bone mineral density, and (5) changes in mood. Patients should exhibit one to three of these symptoms with a concomitant low concentration of serum testosterone to fit various diagnostic criteria. Total serum testosterone is the most widely used biochemical parameter for assessment of hypogonadism; although there is no agreed upon lower limit of normal, recently published consensus recommendations drafted by five professional andrology/urology societies concur that total testosterone above 12 nmol/L (350 ng/dL) does not require testosterone supplementation, whereas patients whose concentrations fall below 8 nmol/L (230 ng/dL) may benefit from testosterone replacement. This value is similar to the 200 ng/dL intent-to-treat cutoff published in the 2002 practice guidelines of the American Association of Clinical Endocrinology.234 The joint societies further recommend, for those patients falling in the gray zone between 8 and 12 nmol/L, repeat measurement of serum total testosterone with measurement of SHBG to calculate free testosterone, or direct measurement of free testosterone by equilibrium dialysis (if available). Measurement of free or bioavailable testosterone should be considered when total testosterone is not diagnostic despite the clinical presentation of hypogonadism.101 This is particularly true in the setting of obesity and advanced age, where concentrations of SHBG have been shown to be elevated. High concentrations of SHBG may mask a true deficit in testosterone. Transient decreases in testosterone secondary to acute illness should be excluded during this assessment. Moreover, underlying chronic disease that lowers concentrations of testosterone should be taken into consideration and treated appropriately. To assess whether hypogonadism is primary or secondary, serum LH should be measured; a serum prolactin measurement is indicated when serum testosterone concentrations are lower than 5.2 nmol/L (150 ng/dL), or when secondary hypogonadism is suspected.312–314 In sum, no absolute cutoffs or specific tests (total vs. bioavailable vs. free testosterone) are available for the laboratory diagnosis of hypogonadism in the aging male. Each patient’s laboratory results should be interpreted on an individual basis, with particular attention given to those parameters of the biochemistry of testosterone (e.g., obesity, age, comorbidities, medications) that may affect the findings. Patients who meet these criteria may be candidates for treatment with testosterone replacement therapy (TRT). The goal of therapy is to improve the symptoms and signs associated with testosterone deficiency. For more information on TRT, readers may consult recently published guidelines and position statements.312–314 Considerable controversy surrounds TRT, primarily regarding potential adverse effects, specifically, the effects of TRT on prostate and cardiovascular health. TRT in younger patients diagnosed with hypogonadism has proved both safe and effective. However, data from prospective randomized controlled trials regarding the efficacy and safety of TRT in the aged population are lacking. Despite this lack of evidence, TRT prescriptions are on the rise; thus a sustained role for the laboratory in the diagnosis of LOH becomes evident, particularly given a growing aging population of males older than 65 years, projected to number some 31.3 million in the United States by the year 2030.137 A wide variety of abnormalities affect the male reproductive system before birth, in childhood, or in adulthood. For the purposes of this chapter, they have been divided into categories of (1) hypogonadotropic hypogonadism, (2) hypergonadotropic hypogonadism, (3) defects in androgen action (Box 56-1), (4) erectile dysfunction, and (5) gynecomastia. The effects of these abnormalities on infertility are discussed later in this chapter. Kallmann syndrome, the most common form of hypogonadotropic hypogonadism, results from a deficiency of GnRH in the hypothalamus during embryonic development.119 It is characterized by hypogonadism and anosmia (loss of the sense of smell) in male or female patients, and is inherited as an autosomal dominant trait with variable penetrance.50,262 This syndrome arises from a defect in the migration of GnRH neurons to the hypothalamus. The pituitary disorders are characterized by isolated gonadotropin deficiency with or without growth hormone deficiency. Patients with isolated gonadotropin deficiency display sexual infantilism and long arms and legs; those with combined deficiency do not have long arms and legs. These patients must be distinguished from those with growth delay. In all of these patients, LH, FSH, and testosterone concentrations are lower than normal. However, heterogeneity exists in the degree of gonadotropin deficiency; hence concentrations of LH, FSH, and testosterone have been shown to differ among affected patients. Hypergonadotropic hypogonadism results from a primary gonadal disorder. Patients with primary testicular failure have increased concentrations of LH and FSH and decreased concentrations of testosterone. Causes for primary hypogonadism are categorized as (1) acquired causes (irradiation, castration, mumps orchitis, or cytotoxic drugs), (2) chromosome defects (Klinefelter syndrome), (3) defective androgen synthesis (20α-hydroxylase deficiency), (4) testicular agenesis, (5) seminiferous tubular disease, and (6) other miscellaneous causes. Aging is associated with gonadal failure, specifically, decreased Leydig cell mass and reserve capacity with reduction in pulsatile secretion of GnRH by the hypothalamus, leading to decreased testosterone secretion.191 The most common and severe defect in androgen action is androgen insensitivity syndrome (AIS), a disorder arising from mutations in the androgen receptor gene (AR). AIS may be classified as complete (CAIS) or partial (PAIS), depending on the amount of residual receptor function. Individuals with complete AIS (formerly known as testicular feminization) have a male karyotype (46,XY) with female external genitalia (labia, clitoris, and vaginal opening). The testes are present intra-abdominally, and because they produce anti-Müllerian hormone (AMH) (also known as Müllerian inhibitory substance), no uterus, fallopian tubules, or proximal vagina is present. The circulating concentration of testosterone in these patients is greater than or equal to that of a healthy male.145 Concentrations of LH are increased, presumably because of resistance of the hypothalamic-pituitary system to androgen inhibition. Males with 5α-reductase deficiency (5-ARD) do not convert testosterone to the more potent DHT. Because DHT leads to masculinization of external genitalia in utero, males are born with ambiguous genitalia.325 High ratios of the circulating concentrations of testosterone to DHT are indicative of 5-ARD. Moreover, evidence indicates that DHT formation is deficient in the tissues of the urogenital tract in these patients.145 In patients with cryptorchidism or ambiguous genitalia, identification of abdominal gonads is essential for proper diagnosis and treatment. The presence of testicular tissue has traditionally been detected by measurement of Leydig cell testosterone production after stimulation with hCG. A growing appreciation of assessment of Sertoli cell function has been noted. Inhibin and AMH reflect Sertoli cell function and may offer a noninvasive evaluation of seminiferous tubular integrity.99,174,206,244 In one study, the mean plasma AMH concentration in anorchid patients was 0.8 ng/mL, compared with 48.2 ng/mL in patients with normal testes.174 AMH concentrations are also elevated in boys with delayed puberty and partial androgen insensitivity. Inhibin B may be used as a basal serum marker for the presence and function of testicular tissue in boys with nonpalpable testicles.12,118,164 Studies have shown that boys with anorchia have undetectable serum inhibin B concentrations.164 Boys with severe testicular damage or gonadal dysgenesis also have undetectable or very low concentrations of inhibin B, whereas normal serum inhibin B concentrations are observed among boys with abdominal “normal” testes.164 Erectile dysfunction (formerly referred to as impotence) is the persistent inability to develop or maintain a penile erection that is sufficient for intercourse and ejaculation in 50% or more of attempts.163,262 A wide variety of organic and psychological abnormalities may cause changes in sexual drive and in the ability to have an erection or to ejaculate. Psychogenic erectile dysfunction is the most common diagnosis. Other causes include vascular disease, diabetes mellitus, hypertension, uremia, neurologic disease, hypogonadism, hyperthyroidism and hypothyroidism, neoplasms, and drugs. The physician must pursue a careful evaluation of possible psychological factors, neuropathy, or vascular abnormalities that may be interfering with proper sexual function. If no obvious explanation for erectile dysfunction can be found, measurement of morning serum testosterone, LH, and thyroid-stimulating hormone (TSH) concentrations has been suggested.111,262 Elevated gonadotropin concentrations indicate primary hypogonadism. Total and even free testosterone concentrations may be within normal reference intervals, yet still may be subnormal for a given patient if found in the presence of elevated LH or FSH. Hyperprolactinemia is an infrequent cause of erectile dysfunction but should be considered in unusual situations. Sildenafil (sold under the trade names Viagra, Revatio, and others) was approved by the U.S. Food and Drug Administration (FDA) in April 1998 for use as an oral therapeutic agent for male erectile dysfunction.33,34,130,212,264 This agent and the drugs tadalafil (Cialis) and vardenafil (Levitra) are selective inhibitors of phosphodiesterase 5 (PDE5).34 By inhibiting PDE5 in the corpus cavernosum of the penis, these drugs block degradation of cyclic guanosine monophosphate (GMP), which is increased during sexual arousal. Increased cyclic GMP results in relaxation of vascular smooth muscle and increased inflow of blood. A high-performance liquid chromatography (HPLC) method for sildenafil has been developed.62 Gynecomastia, the benign growth of glandular breast tissue in men, is a common finding among males of varied ages.39,103,194 Gynecomastia, which is associated with an increase in the estrogen/androgen ratio, is commonly associated with three distinct periods of life. First, transient gynecomastia can be found in 60 to 90% of all newborns because of high estrogen concentrations that cross the placenta. The second peak occurs during puberty in 50 to 70% of normal boys. It is usually self-limited and may be due to low serum testosterone, low DHT, or a high estrogen/androgen ratio. The last peak is found in the adult population, most frequently among men aged 50 to 80 years. Gynecomastia may be due to testicular failure, resulting in an increased estrogen/androgen ratio, or to increased body fat, resulting in increased peripheral aromatization of testosterone to estradiol.39,103,194 Gynecomastia may also develop as the result of iatrogenic causes, hyperthyroidism, or liver disease. Liver disease impairs estrogen clearance and SHBG production, leading to increased bioavailable estrogen and subsequent gynecomastia. Finally, germinal cell or nonendocrine tumors that produce human chorionic gonadotropin (hCG), as well as estrogen-producing tumors of the adrenal glands, the testes, or the liver, will cause gynecomastia. hCG stimulates testicular aromatase activity and estrogen production, resulting in gynecomastia.103 In cases of striking gynecomastia in which history and physical examination point to no specific disorder, measurements of hCG, plasma estradiol, testosterone, and LH concentrations are appropriate.39 It is important to note that prolactin plays an important role in galactorrhea (milk production), but only an indirect role in gynecomastia. The ovaries produce ova and secrete the sex hormones progesterone and estrogen. Every healthy female neonate possesses approximately 400,000 primordial follicles, each containing an immature ovum. During the reproductive life span of an adult woman, 300 to 400 follicles will reach maturity.97,110 A single mature follicle is produced during each normal menstrual cycle at approximately day 14. Surrounding the oocyte of the mature follicle are three distinct cell layers: (1) theca externa, (2) theca interna, and (3) granulosa cells. The theca interna cells are the primary source of androgens, which are transported to adjacent granulosa cells, where they are aromatized to estrogens.50 Estrogens also have well-established effects on plasma proteins that influence endocrine testing. They increase concentrations of SHBG, corticosteroid-binding globulin, and thyroxine-binding globulin. Hence, boys and girls have comparable concentrations of SHBG, but adult men have SHBG concentrations that are about one half those of adult women. Concentrations of plasma proteins that bind copper and iron are also elevated in response to estrogen, as are those of high-density and very high-density lipoproteins. In addition, estrogens are believed to play a preventive role in coronary heart disease.114 The three most biologically active estrogens in order of potency are estrone (E1), estradiol (E2), and estriol (E3) (Figure 56-5). Structurally, estrogens are derivatives of the parent hydrocarbon estrane, which is an 18-carbon molecule with an aromatic ring A and a methyl group at C-13.50,333 All estrogens possess a phenolic hydroxyl group at C-3, which gives the compounds acidic properties, and lack a methyl group at C-10. In addition, estrogens may possess a ketone (estrone) or hydroxyl group (estradiol) at position C-17. The phenolic ring A and the hydroxyl group at C-17 are essential for biological activity. The biochemical pathway illustrating aromatization of testosterone to estradiol and androstenedione to estrone is shown in Figure 56-6. The role of estrogens in normal and abnormal menstrual cycles is described later in this chapter. Figure 56-6 Biosynthesis of estrogens. Heavy arrows indicate the Δ5-3β-hydroxy pathway. The circled area represents the site of chemical change. See Figure 56-1 for early synthetic steps. Estrogens are secreted primarily in healthy women by the ovarian follicles and the corpus luteum, and during pregnancy by the placenta. The adrenal glands and testes (in men) are also believed to secrete minute quantities of estrogens. The ovary synthesizes estrogens via aromatization of androgens. Synthesis of estrogens begins in the theca interna cells with the enzymatic synthesis of androstenedione from cholesterol. Androstenedione is then transported to the granulosa cells, where it is further metabolized directly to estrone (androstenedione → estrone), or first to testosterone and then to estradiol (androstenedione → testosterone → estradiol). These conversions are catalyzed by the enzyme aromatase. The healthy human ovary produces all three classes of sex steroids: estrogens, progestagens, and androgens; however, estradiol and progesterone are its primary secretory products. Because the ovary lacks both the 21-hydroxylase and 11β–hydroxylase enzymes, glucocorticoids and mineralocorticoids are not produced in the ovary.50,333 More than 20 estrogens have been identified, but only 17β-estradiol (E2) and estriol (E3) are routinely measured clinically. The most potent estrogen secreted by the ovary is 17β-estradiol. Because it is derived almost exclusively from the ovaries, its measurement is often considered sufficient for evaluation of ovarian function. Estrogens are also produced by peripheral aromatization of androgens, primarily androstenedione. In healthy men and women, ≈1% of secreted androstenedione is converted to estrone.50,333 Although the ovaries of postmenopausal women do not secrete estrogens, these women have significant blood concentrations of estrone originating from the peripheral conversion of adrenal androstenedione. Because a major site of this conversion is adipose tissue, estrone is increased in obese postmenopausal women, sometimes yielding enough estrogen to produce bleeding.50,333,336 Research has shown that biosynthesis of estrogens differs qualitatively and quantitatively in pregnant women compared with nonpregnant ones. In pregnant women, the major source of estrogens is the placenta, whereas in nonpregnant women, the ovaries are the main site of synthesis.50,333,336 In contrast to the microgram quantities secreted by nonpregnant women, the quantity of estrogens secreted during pregnancy increases to milligram amounts. The major estrogen secreted by the ovary is estradiol (E2), whereas the major product secreted by the placenta is estriol (E3). E3 is formed in the placenta by sequential desulfation and aromatization of plasma dehydroepiandrosterone sulfate (DHEA-S). Except during pregnancy, measurements of E3 have little clinical value because in nonpregnant women, E3 is derived almost exclusively from E2 (see also Chapter 57). E3 is the predominant hormone of late pregnancy. Maternal E3 is almost entirely (≈90%) derived from fetal and placental sources. It is first detected during the ninth gestational week and gradually increases during the first and second trimesters. Plasma and salivary E3 concentrations peak approximately 3 to 5 weeks before labor and delivery.108 This characteristic surge in E3 has been observed in term, preterm, and post-term pregnancies. Some reports have suggested utility in the measurement of salivary E3 in the prediction of risk for spontaneous preterm birth.107,132–134,167,195 This test has a high negative predictive value but a low positive predictive value. Consequently, the American College of Obstetricians and Gynecologists does not now suggest measuring salivary E3 concentrations, except for research purposes.2 Details regarding techniques used to determine serum and salivary E3 concentrations are discussed later in the section on analytical methods. Serum unconjugated E3 measurements, along with alpha fetoprotein, hCG, and inhibin A, are commonly used as part of the “quad” maternal screens for Down syndrome–affected fetuses. On average, unconjugated E3 is 0.72 times less than normal (median value at 16 weeks: 0.30 to 1.50 µg/L) when fetal Down syndrome is present.47,198,287,309 For more on maternal serum screening, see Chapter 57. More than 97% of circulating E2 is bound to plasma proteins. It is bound specifically and with high affinity to SHBG, and nonspecifically to albumin.222,281 SHBG concentrations are increased by estrogens and therefore are higher in women than in men. They are also increased during (1) pregnancy, (2) oral contraceptive use, (3) hyperthyroidism, and (4) administration of certain antiepileptic drugs such as dilantoin. SHBG concentrations may decrease in hypothyroidism, obesity, or androgen excess. In women, E2 circulates bound 40 to 60% to SHBG, and 40 to 60% to albumin. SHBG has a higher affinity for testosterone than E2; therefore, in men, E2 circulates 20 to 30% bound to SHBG, and 70 to 80% bound to albumin.222,230 Only 2 to 3% of total E2 circulates in free form. In contrast, estrone and estrone sulfate circulate bound almost exclusively to albumin. As with testosterone, both free and albumin-bound fractions of E2 are thought to be biologically available,230 but measurement of this fraction has not been shown to be clinically important. The metabolism of E2 is chiefly an oxidative process dominated by three pathways, of which the fastest is oxidation of the β-hydroxy group at C-17 to a ketone (estradiol → estrone). This process is reversible; however, equilibrium favors the estrone species. Estrone is further oxidized along two pathways: the 2-hydroxylation pathway, leading to formation of catechol estrogens (2-hydroxyestrone, 2-hydroxyestradiol, and 2-hydroxyestriol and their corresponding methoxy derivatives), and the 16α-hydroxylation pathway, leading predominantly to formation of E350,333 (Figure 56-7). Figure 56-7 Main pathways of estradiol metabolism in humans. The circled area represents the site of chemical change. Normally, blood estrone concentrations parallel E2 concentrations throughout the menstrual cycle, but at one third to one half their magnitude. Estrone metabolism is affected by the pathophysiologic state. For example, obesity and hypothyroidism are associated with an increase in E3 formation, whereas low body weight and hyperthyroidism are associated with formation of catechol estrogens.50 Although assays for catechol estrogen measurement are available,55,196 they have no known current clinical value. In addition to the oxidative pathways already described, formation of estrogen conjugates has been reported as a major route of estrogen metabolism. The most abundant circulating estrogen conjugates are the sulfates, followed by the glucuronides, with estrone sulfate circulating at concentrations tenfold higher than unconjugated estrone.236 Initially, it was thought that sulfate conjugation would lead to an increase in polarity, making the compound more readily excretable; however, estrogen sulfates actually exhibit a longer half-life than do parent estrogens.236 These observations have led to the idea that estrone sulfate may serve as a precursor for the bioactive estrogens via desulfation and conversion to E2 by 17β-hydroxysteroid dehydrogenase. In contrast to estrogen sulfates, glucuronidation of estrogens generally is accepted to serve a classic excretory role. Estrogen glucuronides are detectable in both urine and bile. Progesterone, similar to the estrogens, is a female sex hormone. In conjunction with estrogens, it helps to regulate the accessory organs during the menstrual cycle.333,336 This hormone is especially important in preparing the uterus for implantation of the blastocyst and in maintaining pregnancy. In nonpregnant women, progesterone is secreted mainly by the corpus luteum. During pregnancy, the placenta becomes the major source of this hormone. Minor sources are the adrenal cortex in both sexes and the testes in men. The structural formula of progesterone, a C21 compound, is shown in Figure 56-8. Similar to the corticosteroids and testosterone, progesterone (pregn-4-ene-3,20-dione) contains a keto group (at C-3) and a double bond between C-4 and C-5 (Δ4); both structural characteristics are essential for progestational activity. The two-carbon sidechain (CH3CO) on C-17 does not seem to be very important for its physiologic action. Indeed, the synthetic compound 19-nortestosterone (see Figure 56-8) and its derivatives, which are widely used as oral contraceptives, are more potent progestational agents than progesterone itself. Biosynthesis of progesterone in ovarian tissues follows the same path from acetate to cholesterol through pregnenolone as it does in the adrenal cortex (see Figure 56-1).50,333,335 In luteal tissue, however, low-density lipoprotein cholesterol is thought to serve as the preferred precursor despite the potential of the corpus luteum to synthesize progesterone de novo from acetate.271 Initiation and control of luteal secretion of progesterone are regulated by LH and FSH.50,283,333 The important metabolic events leading to inactivation of progesterone are reduction and conjugation. The main metabolic pathway for the metabolism of progesterone is outlined in Figure 56-9. Metabolites of progesterone are classified into three groups based on the degree of reduction: 1. Pregnanediones. The C4-5 double bond is reduced, producing two compounds: pregnanedione (hydrogen atom at C-5 is in β-orientation) and allopregnanedione (hydrogen atom at C-5 is in α-orientation). 2. Pregnanolones. The keto group at C-3 is reduced, producing hydroxyl groups in α- or β-orientation. However, most urinary pregnanolones exist in the α-configuration. 3. Pregnanediols. The keto group at C-20 is also reduced. As in the previous case, metabolites containing the 20-hydroxyl group in α-orientation are quantitatively more important. In fact, urinary measurement of pregnanediol (5β-pregnane-3α,20α-diol) can be used as an index of endogenous production of progesterone, because this metabolite is quantitatively very significant, and its concentration correlates with most clinical conditions. In the genotypic female, lack of testosterone and AMH causes regression of the wolffian ducts and maintenance of the Müllerian ducts, thus forming the female reproductive tract. Gonadotropin activity in utero is suppressed because of high concentrations of circulating estrogens derived from the mother.50,333 When the placenta separates, concentrations of fetal sex steroids drop abruptly. Serum E2 in neonates is decreased to basal concentrations within 5 to 7 days after birth and persists at this concentration until puberty. The negative feedback action of steroids is now removed, and gonadotropins are released. Postnatal peaks of LH and FSH are measurable for a few months after birth, peaking at 2 to 5 months and then dropping to basal concentrations. During childhood, circulating concentrations of sex steroids and gonadotropins are low and are similar for both sexes. However, in patients with hypogonadism (Turner syndrome), LH and FSH concentrations are higher than in healthy children.50,333 The transition from sexual immaturity appears to begin with diminished sensitivity of the pituitary gland or hypothalamus, or both, to the negative feedback effect of sex steroids. The mechanism for this change is unclear. As puberty approaches, nocturnal secretion of gonadotropins occurs. Concentrations for LH, FSH, and gonadal steroids rise gradually over several years before stabilizing at adult concentrations when full sexual maturity is reached. In girls, puberty is considered precocious if onset of pubertal development (secondary sex characteristics) occurs before the age of 8 years (see later section on precocious puberty), and delayed if no development has occurred by the age of 13 years, or if menarche has not occurred by age 16.5 years.67,205 It was reported in 2003 that the median age of menarche in the United States is 12.43 years, which is 0.34 year earlier than that reported in 1973.57,189 This study also found that the median age at menarche of non-Hispanic black (12.06 years) girls is significantly earlier than that of non-Hispanic white (12.55 years) and Mexican-American (12.25 years) girls.57 Adrenarche precedes puberty by a few years. In girls, the rise in adrenal androgen concentrations (DHEA, DHEA-S, and androstenedione) begins at age 6 to 7 years.231 This rise in adrenal androgen concentrations lasts until late puberty. A cortical androgen-stimulating hormone may contribute to the rise in adrenal androgens at puberty in both sexes.231 In girls, puberty is associated with elevations in estrogen secretion by the ovary in response to gonadotropin concentrations that increase in response to GnRH. Estrogen secretion by the ovary increases, causing enlargement of the uterus and breasts. In the breast, estrogen enhances growth of ducts; progesterone augments this effect. As the breast develops, estrogen increases adipose tissue around the lactiferous duct system, contributing to the further enlargement of breast tissue.50,333 These physiologic and physical processes associated with puberty in girls culminate in menarche—the beginning of menstrual function and the first menstrual period.
Reproductive Endocrinology and Related Disorders
Male Reproductive Biology
Hypothalamic-Pituitary-Gonadal Axis
Androgens
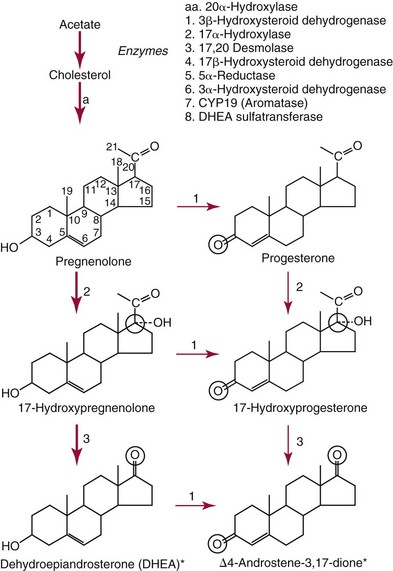
Biosynthesis of Testosterone
Androgen Transport in Blood
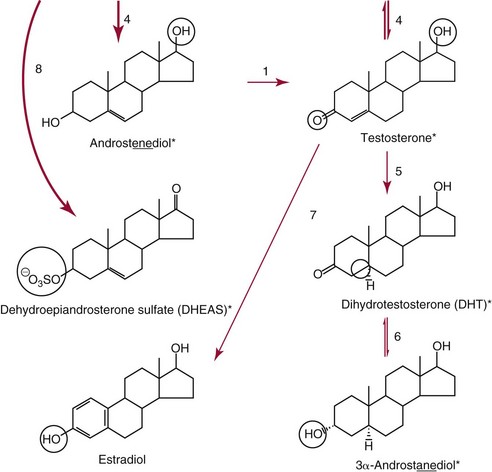
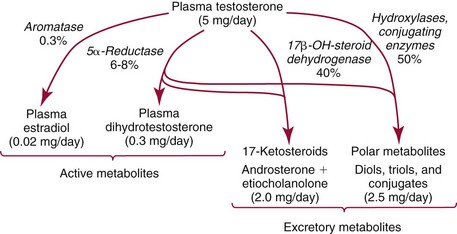
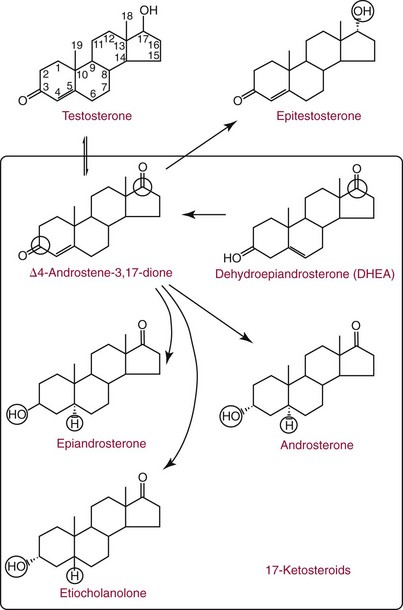
Metabolism of Testosterone
Testosterone Concentrations
Male Reproductive Abnormalities
Hypogonadotropic Hypogonadism
Hypergonadotropic Hypogonadism
Defects in Androgen Action
Erectile Dysfunction
Gynecomastia
Female Reproductive Biology
Estrogens
Chemistry
Biosynthesis
Biosynthesis During Pregnancy
Transport in Blood
Metabolism
Progesterone
Chemistry
Biosynthesis
Metabolism
Female Reproductive Development
Fetal
Postnatal
Puberty
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Reproductive Endocrinology and Related Disorders