Renal System
The components of the renal system are the kidneys, ureters, bladder, and urethra. The kidneys, located retroperitoneally in the lumbar area, produce and excrete urine to maintain homeostasis. They regulate the volume, electrolyte concentration, and acid-base balance of body fluids; detoxify the blood and eliminate wastes; regulate blood pressure; and support red blood cell (RBC) production (erythropoiesis). The ureters are tubes that extend from the kidneys to the bladder; their only function is to transport urine to the bladder. The bladder is a muscular bag that serves as reservoir for urine until it leaves the body through the urethra.
Pathophysiologic changes
Wastes are eliminated from the body by urine formation. The process of urine formation occurs in the nephron, the functional unit of the kidney. Glomerular filtration, tubular reabsorption, and tubular secretion and excretion are all necessary for urine formation. Glomerular filtration is the process of filtering the blood as it flows through the kidneys. The glomerulus, a capillary network, filters plasma; the filtrate is either reabsorbed in the renal tubules or excreted as urine. Glomerular function depends on the permeability of the capillary walls, vascular pressure, and hydrostatic pressure. The normal glomerular filtration rate (GFR) is about 120 ml/minute. To prevent too much fluid from leaving the vascular system, tubular reabsorption opposes capillary filtration. Reabsorption takes place as capillary filtration progresses. When fluid filters through the capillaries, albumin, which doesn’t pass through capillary walls, remains behind. As the albumin concentration inside the capillaries increases, the capillaries begin to draw water back in by osmosis. This osmotic force controls the quantities of water and diffusible solutes that enter and leave the capillaries.
Factors that affect filtration or reabsorption affect total filtration effort. Capillary pressure and interstitial fluid colloid osmotic pressure affect filtration. Interstitial fluid pressure and plasma colloid osmotic pressure affect reabsorption.
Various factors can slow the GFR—altered renal perfusion; renal disease affecting the vessels, glomeruli, or tubules; or obstruction to urine flow. The results are retention of nitrogenous wastes (azotemia), such as blood urea nitrogen and creatinine, which can lead to acute renal failure.
CAPILLARY PRESSURE
The renal arteries branch into five segmental arteries, which supply different areas of the kidneys. The segmental arteries then branch into several divisions from which the afferent arterioles and vasa recta arise. Renal veins follow a similar branching pattern—characterized by stellate vessels and segmental branches—and empty into the inferior vena cava. The tubular system receives its blood supply from a peritubular capillary network. The ureteral veins follow the arteries and drain into the renal vein. The bladder receives blood through vesical arteries. Vesical veins unite to form the pudendal plexus, which empties into the iliac veins. A rich
lymphatic system drains the renal cortex, kidneys, ureters, and bladder.
lymphatic system drains the renal cortex, kidneys, ureters, and bladder.
Capillary pressure reflects mean arterial pressure (MAP). Increased MAP increases capillary pressure, which in turn increases the GFR. When MAP decreases, so do capillary pressure and GFR. Autoregulation of afferent and efferent arterioles minimizes and controls changes in capillary pressure, unless MAP exceeds 180 mm Hg or is less than 80 mm Hg.
The kidneys are innervated by sympathetic branches from the celiac plexus, upper lumbar splanchnic and thoracic nerves, and the intermesenteric and superior hypogastric plexuses, which surround the kidneys. Similar numbers of sympathetic and parasympathetic nerves from the renal plexus, superior hypogastric plexus, and intermesenteric plexus innervate the ureters. Nerves that arise from the inferior hypogastric plexus innervate the bladder. The parasympathetic nerve supply to the bladder controls urination.
Increased sympathetic activity and angiotensin II constrict afferent and efferent arterioles, decreasing the capillary pressure. Because these changes affect both the afferent and efferent arterioles, they have no net effect on GFR.
Inadequate renal perfusion accounts for 40% to 80% of cases of acute renal failure. Volume loss (as with GI hemorrhage, burns, diarrhea, and diuretic use), volume sequestration (as in pancreatitis, peritonitis, and rhabdomyolysis), or decreased effective circulating volume (as in cardiogenic shock and sepsis) may reduce circulating blood volume. Decreased cardiac output due to peripheral vasodilation (by sepsis or drugs) or profound renal vasoconstriction (as in severe heart failure, hepatorenal syndrome, or with such drugs as nonsteroidal anti-inflammatory drugs [NSAIDs]) also diminish renal perfusion.
Hypovolemia causes a decrease in MAP that triggers a series of neural and humoral responses: activation of the sympathetic nervous system and renin-angiotensin-aldosterone system, and release of arginine vasopressin.
Prostaglandin-mediated relaxation of afferent arterioles and angiotensin II-mediated constriction of efferent arterioles maintain GFR. GFR decreases steeply if MAP decreases to less than 80 mm Hg. Drugs that block prostaglandin production (such as NSAIDs) can cause severe vasoconstriction and acute renal failure during hypotension.
Prolonged renal hypoperfusion causes acute tubular necrosis. Processes involving large renal vessels, microvasculature, glomeruli, or tubular interstitium cause intrinsic renal disease. Emboli or thrombi, aortic dissection, or vasculitis can occlude renal arteries. Cholesterol-rich atheroemboli can occur spontaneously or follow aortic instrumentation. If they lodge in medium and small renal arteries, they trigger an eosinophil-rich inflammatory reaction.
 After age 40, a person’s renal function begins to diminish. If he lives to age 90, it may have decreased by as much as 50%. This change is reflected in a decreased GFR and is caused by age-related changes in the renal vasculature that disturb glomerular hemodynamics as well as by reduced cardiac output and atherosclerotic changes that reduce renal blood flow by more than 50%.
After age 40, a person’s renal function begins to diminish. If he lives to age 90, it may have decreased by as much as 50%. This change is reflected in a decreased GFR and is caused by age-related changes in the renal vasculature that disturb glomerular hemodynamics as well as by reduced cardiac output and atherosclerotic changes that reduce renal blood flow by more than 50%.INTERSTITIAL FLUID COLLOID OSMOTIC PRESSURE
Few plasma proteins and RBCs are filtered out of the glomeruli, so interstitial fluid colloid osmotic pressure (the force of albumin in the interstitial fluid) remains low. Large quantities of plasma protein flow through glomerular capillaries. Size and surface charge keep albumin, globulin, and other large proteins from crossing the glomerular wall. Smaller proteins leave the glomerulus but are absorbed by the proximal tubule.
Injury to the glomeruli or peritubular capillaries can increase interstitial fluid colloid osmotic pressure, drawing fluid out of the glomerulus and the peritubular capillaries. Swelling and edema occur in Bowman’s space and the interstitial space surrounding the tubule. Increased interstitial fluid pressure opposes glomerular filtration, causes collapse of the surrounding nephrons and peritubular capillaries, and leads to hypoxia and renal cell injury or death. When cells die, intracellular enzymes that stimulate immune and inflammatory reactions are released. This further contributes to swelling and edema.
The resulting increase in interstitial fluid pressure can interfere with glomerular filtration and tubular reabsorption. Loss of glomerular filtration renders the kidney incapable of regulating blood volume and electrolyte composition. Diseases that damage the tubules alter their permeability, causing tubular proteinuria because small proteins can move from capillaries into tubules.
Normal glomerular cells, which are endothelial in nature, form a barrier that prevents cells and other particles from crossing the membrane. The basement membrane typically traps larger proteins. The channels of the basement membrane are coated with glycoproteins that are rich in glutamate, aspartate, and sialic acid.
This produces a negative charge barrier that impedes the passage of such anionic molecules as albumin. (See The glomerulus.)
This produces a negative charge barrier that impedes the passage of such anionic molecules as albumin. (See The glomerulus.)
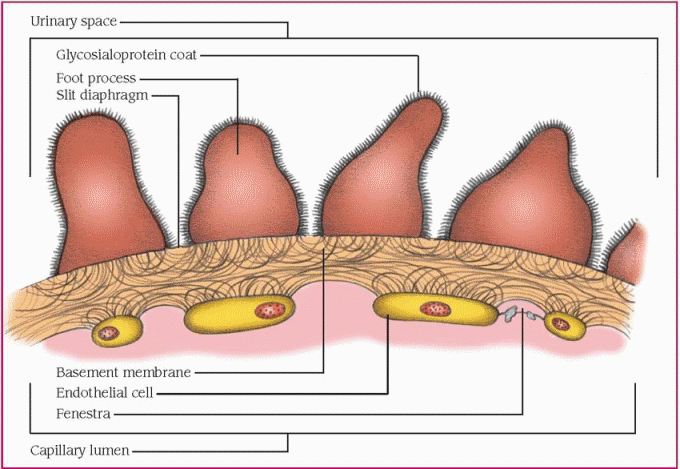
Glomerular disease disrupts the basement membrane, allowing large proteins to leak out. Damage to epithelial cells permits albumin leakage. Hypoalbuminemia, as in nephrotic syndrome, is the result of excessive loss of albumin in the urine, increased renal catabolism, and inadequate hepatic synthesis of albumin. Plasma oncotic pressure decreases and edema results as fluid moves from capillaries into the interstitium. Consequent activation of the renin-angiotensin system, arginine-vasopressin release, and sympathetic nervous system stimulation increases renal salt and water reabsorption, which further contributes to edema. The severity of edema is directly related to the degree of hypoalbuminemia, and is exacerbated by heart disease or peripheral vascular disease.
PLASMA COLLOID PRESSURE
Protein concentration of the plasma determines the plasma colloid pressure (the pulling force of albumin in the intravascular fluid), the major force influencing reabsorption of fluid into the capillaries. The plasma protein level can decrease as a result of liver disease, protein loss in the urine, and protein malnutrition.
As oncotic pressure decreases, less fluid moves back into the capillaries and fluid begins to accumulate in the tubular and peritubular areas. Swelling around the tubule causes collapse of the tubule and peritubular capillaries, hypoxia, and death of the nephrons.
Diminished plasma oncotic pressure and urine protein loss stimulate hepatic lipoprotein synthesis, and the resulting hyperlipidemia manifests as lipid bodies (fatty casts, oval fat bodies) in the urine. Metabolic disturbances result as other proteins are lost in the urine, including thyroxine-binding globulin, cholecalciferol-binding protein, transferrin, and metal-binding proteins. Urine losses of antithrombin III, decreased serum levels of proteins S and C, hyperfibrinogenemia, and enhanced platelet aggregation lead to a hypercoagulable state, as in nephrotic syndrome. Some patients also develop severe immunoglobulin G deficiency, which increases susceptibility to infection.
Congenital nephropathies and uropathies
The following congenital conditions can affect kidney function:
♦ Ectopic kidney—the kidney is located in the pelvic or thoracic area, causing reflux from the bladder into the ureters.
♦ Horseshoe kidney—the lower poles of the kidneys are fused by an isthmus.
♦ Obstructive uropathy—a pathologic condition of the kidney (abnormal vasculature, adhesions, kinks, or masses) that blocks the flow of urine, usually causing hydronephrosis.
♦ Renal dysplasia—the kidney is abnormally shaped, and the involved areas are nonfunctional.
♦ Renal hypoplasia—the kidney is small due to a reduction in the number of normally developed nephrons, and it may be unilateral or bilateral.
♦ Renal malrotation—the kidney is positioned abnormally.
♦ Ureterocele—a prolapse of the end portion of the ureter into the bladder leads to an obstruction in urine flow.
STRUCTURAL VARIATIONS
Variations in normal anatomic structure of the urinary tract occur in 10% to 15% of the total population and range from minor and easily correctable to lethal. Ectopic kidneys, which result if the embryonic kidneys don’t ascend from the pelvis to the abdomen, function normally. If the embryonic kidneys fuse as they ascend, a single u-shaped kidney results, causing no symptoms in about one-third of affected people. The most common problems associated with horseshoe kidneys include hydronephrosis, infection, and calculus formation.
 Structural abnormalities of the renal system account for about 45% of renal failure in children.
Structural abnormalities of the renal system account for about 45% of renal failure in children.Urinary tract malformations are commonly associated with certain nonrenal anomalies. These characteristics include low-set and malformed ears, chromosomal disorders (especially trisomies 13 and 18), absent abdominal muscles, spinal cord and lower-extremity anomalies, imperforate anus or genital deviation, Wilms’ tumor, congenital ascites, cystic disease of the liver, and positive family history of renal disease (hereditary nephritis or cystic disease). (See Congenital nephropathies and uropathies.)
OBSTRUCTION
Obstruction along the urinary tract causes urine to accumulate behind the source of obstruction, leading to infection or damage. Obstructions may be congenital or acquired. Causes include tumors, calculi (stones), trauma, strictures (secondary to surgical intervention and scarring), edema, pregnancy, benign prostatic hyperplasia or carcinoma, inflammation of the GI tract, and loss of ureteral peristaltic activity or bladder muscle function.
Consequences of obstruction depend on the location and whether it’s unilateral or bilateral, partial or complete, and acute or chronic, as well as the cause. For example, obstruction of a ureter causes hydroureter, or an accumulation of urine within the ureter, which increases retrograde pressure to the renal pelvis and calyces. As urine accumulates in the renal collection system, hydronephrosis results. If the obstruction is complete and acute in nature, increasing pressure transmitted to the proximal tubule inhibits glomerular filtration. If the GFR declines to zero, the result is renal failure.
Chronic partial obstruction compresses structures as urine accumulates, and the result is papillary and medullary infarct. The kidneys initially increase in size, but progressive atrophy follows, with eventual loss of renal mass. The underlying tubular damage decreases the kidney’s ability to conserve sodium and water and excrete hydrogen ions and potassium; sodium and bicarbonate are wasted. Urine volume is excessive, even though the GFR has declined. The result is an increased risk of dehydration and metabolic acidosis.
Tubular obstruction, caused by renal calculi or scarring from repeated infection, can increase interstitial fluid pressure. As fluid accumulates in the nephron, it backs up into Bowman’s capsule and space. If the obstruction isn’t relieved, nephrons and capillaries collapse, and renal damage is irreversible. The papillae, which are the final site of urine concentration, are particularly affected.
Relief of the obstruction is usually followed by copious diuresis of sodium and water retained during the period of obstruction, and a return to a normal GFR. Excessive loss of sodium and water (more than 10 L/ day) is uncommon. If a GFR doesn’t recover quickly, diuresis may not be significant after the obstruction is relieved.
Unresolved obstruction can result in infection or even renal failure. Obstructions below the bladder cause urine to accumulate, forming a medium for bacterial growth.
 Urinary tract infections are most common in girls ages 7 to 11. This is a result of bacteria ascending the urethra.
Urinary tract infections are most common in girls ages 7 to 11. This is a result of bacteria ascending the urethra.Cystitis is an infection of the bladder that results in mucosal inflammation and congestion. The detrusor muscle becomes hyperactive, decreasing bladder capacity and leading to reflux into the ureters. This transient reflux can cause acute or chronic pyelonephritis if bacteria ascend to the kidney.
Bilateral obstruction of the ureters not relieved within 1 week of onset causes acute or chronic renal failure. Chronic renal failure progresses over weeks to months without symptoms until 90% of renal function is lost.
Disorders
Renal disorders include acute pyelonephritis, acute and chronic renal failure, acute tubular necrosis, congenital anomalies, glomerulonephritis, nephrotic syndrome, neurogenic bladder, polycystic kidney, renal calculi, renovascular hypotension, and vesicoureteral reflux.
ACUTE PYELONEPHRITIS
Acute pyelonephritis, also known as acute infective tubulointerstitial nephritis, is a sudden inflammation caused by bacteria that primarily affects the interstitial area and the renal pelvis or, less commonly, the renal tubules. It’s one of the most common renal diseases and may affect one or both kidneys. With treatment and continued follow-up care, the prognosis is good, and extensive permanent damage is rare.
Pyelonephritis is more common in females, probably because of a shorter urethra and the proximity of the urinary meatus to the vagina and the rectum—both conditions allow bacteria to reach the bladder more easily—and a lack of the antibacterial prostatic secretions produced in the male. Incidence increases with age and is higher in the following groups:
♦ sexually active women—intercourse increases the risk of bacterial contamination
♦ pregnant women—about 5% develop asymptomatic bacteriuria; if untreated, about 40% develop pyelonephritis
♦ people with diabetes—neurogenic bladder causes incomplete emptying and urinary stasis; glycosuria may support bacterial growth in the urine
♦ people with other renal diseases—compromised renal function aggravates susceptibility.
Causes
Acute pyelonephritis results from bacterial infection of the kidneys. Infecting bacteria usually are normal intestinal and fecal flora that grow readily in urine. The most common causative organism is Escherichia coli, but Klebsiella, Proteus, Pseudomonas, Staphylococcus aureus, and Enterococcus faecalis (formerly Streptococcus faecalis) may also cause this infection.
Pathophysiology
Typically, the infection spreads from the bladder to the ureters, then to the kidneys, as in vesicoureteral reflux. Vesicoureteral reflux may result from congenital weakness at the junction of the ureter and the bladder. Bacteria refluxed to intrarenal tissues may create colonies of infection within 24 to 48 hours. Infection may also result from instrumentation (such as catheterization, cystoscopy, or urologic surgery), from a hematogenic infection (as in septicemia or endocarditis), or possibly from lymphatic infection.
Pyelonephritis may also result from an inability to empty the bladder (for example, in patients with neurogenic bladder), urinary stasis, or urinary obstruction due to tumors, strictures, or benign prostatic hyperplasia.
Signs and symptoms
Typical clinical features include:
♦ urinary urgency and frequency, burning during urination, dysuria, nocturia, and hematuria (usually microscopic but may be gross) caused by urinary tract irritation
♦ cloudy urine that has an ammonia-like or fishy odor resulting from bacteria in the urine and subsequent leukocyte response
♦ a temperature of 102° F (38.9° C) or higher, shaking chills, nausea and vomiting, flank pain, anorexia, and general fatigue caused by the infection.
These signs and symptoms characteristically develop rapidly over a few hours or a few days. Although they may disappear within days, even without treatment, residual bacterial infection is likely and may cause the signs and symptoms to recur later.
 Elderly patients may exhibit GI or pulmonary symptoms rather than the usual febrile responses to pyelonephritis. In children younger than age 2, fever, vomiting, nonspecific abdominal complaints, or failure to thrive may be the only signs of acute pyelonephritis.
Elderly patients may exhibit GI or pulmonary symptoms rather than the usual febrile responses to pyelonephritis. In children younger than age 2, fever, vomiting, nonspecific abdominal complaints, or failure to thrive may be the only signs of acute pyelonephritis.Complications
♦ Septic shock
♦ Chronic pyelonephritis
♦ Chronic renal insufficiency
Chronic pyelonephritis
Chronic pyelonephritis is a persistent kidney inflammation that can scar the kidneys and may lead to chronic renal failure. Its cause may be bacterial, metastatic, or urogenous. This disease is most common in patients who are predisposed to recurrent acute pyelonephritis, such as those with urinary obstructions or vesicoureteral reflux.
Patients with chronic pyelonephritis may have a childhood history of unexplained fevers or bed-wetting. Clinical effects may include flank pain, anemia, low urine specific gravity, proteinuria, leukocytes in urine and, especially in late stages, hypertension. Uremia rarely develops from chronic pyelonephritis unless structural abnormalities exist in the excretory system. Bacteriuria may be intermittent. When no bacteria are found in the urine, diagnosis depends on excretory urography (renal pelvis may appear small and flattened) and renal biopsy.
Effective treatment of chronic pyelonephritis requires control of hypertension, elimination of the existing obstruction (when possible), and long-term antimicrobial therapy.
Diagnosis
Diagnosis requires urinalysis and culture. Typical findings include:
♦ pyuria (pus in urine)—Urine sediment reveals the presence of leukocytes singly, in clumps, and in casts and, possibly, a few red blood cells.
♦ significant bacteriuria—Urine culture reveals more than 100,000 organisms per microliter of urine.
♦ low specific gravity and osmolality—These findings result from a temporarily decreased ability to concentrate urine.
♦ slightly alkaline urine pH—These findings result from a temporarily decreased ability to concentrate urine.
♦ proteinuria, glycosuria, and ketonuria—These conditions are less common.
Computed tomography (CT) scan also helps in the evaluation of acute pyelonephritis. CT scan of the kidneys, ureters, and bladder may reveal calculi, tumors, or cysts in the kidneys and the urinary tract. Excretory urography may show asymmetrical kidneys.
Treatment
Treatment centers on antibiotic therapy appropriate to the specific infecting organism after identification by urine culture and sensitivity studies.
When the infecting organism can’t be identified, therapy usually consists of a broad-spectrum antibiotic, such as ampicillin or cephalexin. If the patient is pregnant or elderly, antibiotics must be prescribed cautiously. A urinary analgesic, such as phenazopyridine, is also appropriate.
Symptoms may disappear after several days of antibiotic therapy. Although urine usually becomes sterile within 48 to 72 hours, the course of such therapy is 10 to 14 days. Follow-up treatment includes reculturing urine 1 week after drug therapy stops, then periodically for the next year to detect residual or recurring infection. Most patients with uncomplicated infections respond well to therapy and don’t suffer reinfection.
If infection is caused by an obstruction or a vesicoureteral reflux, an antibiotic may be less effective; surgery may then be necessary to relieve the obstruction or correct the anomaly. Patients at high risk for recurring urinary tract and kidney infections, such as those with prolonged use of an indwelling catheter or maintenance antibiotic therapy, require long-term follow-up. Recurrent episodes of acute pyelonephritis can eventually result in chronic pyelonephritis. (See Chronic pyelonephritis.)
Special considerations
Patient care is supportive during antibiotic treatment of underlying infection.
♦ Administer an antipyretic for fever.
♦ Force fluids to achieve urine output of more than 2,000 ml/day. This helps to empty the bladder of contaminated urine and prevents calculi formation. Don’t encourage intake of more than 3 qt (3 L) because this may decrease the effectiveness of the antibiotic.
♦ Provide a diet to prevent calculus formation (low sodium, low meat [all types], and increased potassium).
♦ Teach proper technique for collecting a cleancatch urine specimen. Be sure to refrigerate or culture a urine specimen within 30 minutes of collection to prevent overgrowth of bacteria.
♦ Stress the need to complete prescribed antibiotic therapy even after symptoms subside. Encourage long-term follow-up care for high-risk patients. (See Preventing acute pyelonephritis.)
ACUTE RENAL FAILURE
Acute renal failure, the sudden interruption of renal function, can be caused by obstruction,
poor circulation, or underlying kidney disease. Whether prerenal, intrarenal, or postrenal, it usually passes through three distinct phases: oliguric, diuretic, and recovery. About 2% to 5% of hospitalized patients develop acute renal failure. The condition is usually reversible with treatment, but if not treated, it may progress to end-stage renal disease, prerenal azotemia, and death.
poor circulation, or underlying kidney disease. Whether prerenal, intrarenal, or postrenal, it usually passes through three distinct phases: oliguric, diuretic, and recovery. About 2% to 5% of hospitalized patients develop acute renal failure. The condition is usually reversible with treatment, but if not treated, it may progress to end-stage renal disease, prerenal azotemia, and death.
Causes
Acute renal failure may be prerenal, intrarenal, or postrenal.
Prerenal failure
♦ Antihypertensive medications (angiotensinconverting enzyme inhibitors and angiotensin receptor blockers)
♦ Arrhythmias that cause reduced cardiac output
♦ Arterial embolism
♦ Arterial or venous thrombosis
♦ Ascites
♦ Burns
♦ Cardiac tamponade
♦ Cardiogenic shock
♦ Dehydration
♦ Disseminated intravascular coagulation
♦ Diuretic overuse
♦ Eclampsia
♦ Heart failure
♦ Hemorrhage
♦ Hypercalcemia
♦ Hypoalbuminemia
♦ Hypotension
♦ Hypovolemic shock
♦ Malignant hypertension
♦ Myocardial infarction
♦ Nephrotoxic antibiotics (amphotericin)
♦ Nonsteroidal anti-inflammatory agents
♦ Pulmonary embolism
♦ Radiocontrast agents
♦ Sepsis
♦ Trauma
♦ Tumor
♦ Vasculitis
♦ Vasopressor agents (epinephrine)
Intrarenal failure
♦ Acute glomerulonephritis
♦ Acute interstitial nephritis
♦ Acute pyelonephritis
♦ Acute tubular necrosis
♦ Bilateral renal vein thrombosis
♦ Crush injuries
♦ Disseminated intravascular coagulation
♦ Malignant nephrosclerosis
♦ Myopathy
♦ Nephrotoxins
Follow these measures to help prevent acute pyelonephritis:
♦ Observe strict sterile technique during catheter insertion and care.
♦ If the patient is female, tell her she can prevent bacterial contamination by wiping the perineum from front to back after defecation.
♦ If the patient has a history of urinary tract infections, suggest routine checkups.
♦ Teach the patient to recognize the signs and symptoms of infection, such as cloudy urine, burning on urination, urgency, and frequency, especially when accompanied by flank pain and a low-grade fever.
♦ Obstetric complications
♦ Papillary necrosis
♦ Polyarteritis nodosa
♦ Poorly treated prerenal failure
♦ Rapidly progressive glomerulonephritis
♦ Renal myeloma
♦ Scleroderma
♦ Sickle cell disease
♦ Systemic lupus erythematosus
♦ Transfusion reaction
♦ Vasculitis
Postrenal failure
♦ Benign prostatic hyperplasia
♦ Bladder obstruction
♦ Ureteral obstruction
♦ Urethral obstruction
Pathophysiology
The pathophysiology of prerenal, intrarenal, and postrenal failure differs.
Prerenal failure
Prerenal failure ensues when a condition that diminishes blood flow to the kidneys leads to hypoperfusion. Examples include hypovolemia, hypotension, vasoconstriction, or inadequate cardiac output. Azotemia (excess nitrogenous waste products in the blood) develops in 40% to 80% of acute renal failure cases.
When renal blood flow is interrupted, so is oxygen delivery. The ensuing hypoxemia and ischemia can rapidly and irreversibly damage the kidney. The tubules are most susceptible to hypoxemia’s effects.
The effects of acute renal failure can be seen across most body systems. For this reason, a multidisciplinary approach to care is needed.
Renal system
♦ Oliguria occurs as a result of a decreased glomerular filtration rate (GFR).
♦ Hyperkalemia occurs as a result of a decreased GFR and metabolic acidosis.
♦ Hyperphosphatemia and hypocalcemia occur because the kidney can’t excrete phosphorus.
♦ Hypotension and dehydration may occur during the diuretic phase and lead to further ischemia of the kidneys.
Cardiovascular system
♦ Hypertension and edema occur with fluid accumulation and hypervolemia.
♦ Fluid overload may cause pulmonary and peripheral edema, possibly leading to heart failure because of how these conditions increase the heart’s workload.
♦ Acute pulmonary edema and hypertensive crisis may result from nephron loss and decreased kidney size, causing decreased blood flow to the kidneys.
♦ Arrhythmias and cardiac arrest may result from hyperkalemia.
Endocrine and metabolic systems
♦ A hypermetabolic state caused by energy demands causes tissue catabolism and alterations in the blood glucose level.
♦ Metabolic acidosis occurs because the kidney can’t excrete hydrogen ions and reabsorb sodium and bicarbonate.
GI system
♦ Nausea, vomiting, and anorexia occur with uremia.
♦ GI bleeding may occur with coagulation abnormalities and uremic gastric irritation.
Immune and hematologic systems
♦ Anemia occurs because of decreased erythropoiesis, glomerular filtration of erythrocytes, or bleeding associated with platelet dysfunction.
♦ Infection and sepsis may occur because of decreased white blood cell-mediated immunity.
♦ A hypercoagulable state results from anticoagulant abnormalities, which leads to bleeding and clotting difficulties.
Integumentary system
♦ Accumulation of uremic toxins leads to dryness, pruritus, pallor, purpura, and (rarely) the deposition of the uremic toxins on the skin (uremic frost).
Musculoskeletal system
♦ Muscle weakness may result from hyperkalemia.
♦ Pathologic bone fractures may be caused by prolonged hypocalcemia.
Neurologic system
♦ Altered mental status and peripheral neuropathies are related to the effects of uremic toxins on the highly sensitive nerve cells.
♦ Headache, drowsiness, irritability, and seizures result from central nervous system involvement and, without treatment, may progress to coma.
Respiratory system
♦ Tachypnea and labored breathing result from anemia, causing tissue hypoxia. Respiratory rate and effort also increase to compensate for metabolic acidosis.
Collaborative management
A renal specialist or nephrologist can help evaluate, treat, and manage the patient’s kidney function. Respiratory and cardiology specialists may be consulted depending on the patient’s history and complications he may develop. Nutritional therapy may be involved to help institute necessary restrictions or supplementations. Physical and occupational therapy may be necessary to help with energy conservation and rehabilitation, depending on the patient’s condition and length of stay. If a prolonged hospital stay is expected and the patient requires long-term or home care, social services may be consulted early on in the patient’s care. The patient may also benefit from psychological or spiritual counseling if he’s acutely ill or if he’ll require continued care on discharge.
Azotemia is a consequence of renal hypoperfusion. The impaired blood flow results in a decreased glomerular filtration rate (GFR) and increased tubular reabsorption of sodium and water. A decrease in the GFR causes electrolyte imbalance and metabolic acidosis. Usually, restoring renal blood flow and glomerular filtration reverses azotemia.
Intrarenal failure
Intrarenal failure, also called intrinsic or parenchymal renal failure, results from damage to the filtering structures of the kidneys. Causes of intrarenal failure are classified as nephrotoxic, inflammatory, or ischemic. When the damage is caused by nephrotoxicity or inflammation, the delicate layer under the epithelium (the basement membrane) becomes irreparably damaged, typically leading to chronic renal failure. Severe or prolonged lack of blood flow caused by ischemia may lead to renal damage (ischemic parenchymal injury) and excess nitrogen in the blood (intrinsic renal azotemia).
Acute tubular necrosis, the precursor to intrarenal failure, can result from ischemic damage to renal parenchyma during unrecognized or poorly treated prerenal failure; or from obstetric complications, such as eclampsia, postpartum renal failure, septic abortion, or uterine hemorrhage.
The fluid loss causes hypotension, which leads to ischemia. The ischemic tissue
generates toxic oxygen-free radicals, which cause swelling, injury, and necrosis.
generates toxic oxygen-free radicals, which cause swelling, injury, and necrosis.
Another cause of acute failure is the use of nephrotoxins, including analgesics, anesthetics, heavy metals, radiographic contrast media, organic solvents, and antimicrobials, particularly aminoglycoside antibiotics. These drugs accumulate in the renal cortex, causing renal failure that manifests well after treatment or other toxin exposure. The necrosis caused by nephrotoxins tends to be uniform and limited to the proximal tubules, whereas ischemia necrosis tends to be patchy and distributed along various parts of the nephron.
Postrenal failure
Bilateral obstruction of urine outflow leads to postrenal failure. The cause may be in the bladder, ureters, or urethra.
Bladder obstruction can result from:
♦ anticholinergic use
♦ autonomic nerve dysfunction
♦ infection
♦ tumors.
Ureteral obstructions, which restrict urine flow from kidneys to bladder, can result from:
♦ blood clots
♦ calculi
♦ edema or inflammation
♦ necrotic renal papillae
♦ retroperitoneal fibrosis or hemorrhage
♦ surgery (accidental ligation and strictures)
♦ tumor or uric acid crystals.
Urethral obstruction can result from prostatic hyperplasia, tumor, or strictures.
The three types of acute renal failure (prerenal, intrarenal, or postrenal) usually pass through three distinct phases: oliguric, diuretic, and recovery. (See How acute renal failure affects the body.)
Oliguric phase
Oliguria may result from one or several factors. Necrosis of the tubules can cause sloughing of cells, cast formations, and ischemic edema. The resulting tubular obstruction causes a retrograde increase in pressure and a decrease in GFR. Renal failure can occur within 24 hours from this effect. Glomerular filtration may remain normal in some cases of renal failure, but tubular reabsorption of filtrate may be accelerated. In this instance, ischemia may increase tubular permeability and cause backleak. Another concept is that intrarenal release of angiotensin II or redistribution of blood flow from the cortex to the medulla may constrict the afferent arterioles, increasing glomerular permeability and decreasing the GFR.
Urine output may remain at less than 30 ml/hour or 400 ml/day for a few days to weeks. Before damage occurs, the kidneys respond to decreased blood flow by conserving sodium and water.
Damage impairs the kidney’s ability to conserve sodium. Fluid (water) volume excess, azotemia (elevated serum levels of urea, creatinine, and uric acid), and electrolyte imbalance occur. Ischemic or toxic injury leads to the release of mediators and intrarenal vasoconstriction. Medullary hypoxia results in the swelling of tubular and endothelial cells, adherence of neutrophils to capillaries and venules, and inappropriate platelet activation. Increasing
ischemia and vasoconstriction further limit perfusion.
ischemia and vasoconstriction further limit perfusion.
Injured cells lose polarity, and the ensuing disruption of tight junctions between the cells promotes backleak of filtrate. Ischemia impairs the function of energy-dependent membrane pumps, and calcium accumulates in the cells. This excess calcium further stimulates vasoconstriction and activates proteases and other enzymes. Untreated prerenal oliguria may lead to acute tubular necrosis.
Diuretic phase
As the kidneys become unable to conserve sodium and water, the diuretic phase, marked by increased urine secretion of more than 400 ml/24 hours, ensues. The GFR may be normal or increased, but tubular support mechanisms are abnormal. Excretion of dilute urine causes dehydration and electrolyte imbalances. A high blood urea nitrogen (BUN) level produces osmotic diuresis and consequent deficits of potassium, sodium, and water. The diuretic phase may last days or weeks.
Recovery phase
If the cause of the diuresis is corrected, azotemia gradually disappears and recovery occurs. The recovery phase is a gradual return to normal or near-normal renal function over 3 to 12 months.
 Even with treatment, the elderly patient is particularly susceptible to volume overload, precipitating acute pulmonary edema, hypertensive crisis, hyperkalemia, and infection.
Even with treatment, the elderly patient is particularly susceptible to volume overload, precipitating acute pulmonary edema, hypertensive crisis, hyperkalemia, and infection.Signs and symptoms
Acute renal failure is a critical illness. Its early signs and symptoms are oliguria, azotemia and, rarely, anuria. Electrolyte imbalance, metabolic acidosis, and other severe effects follow, as the patient becomes increasingly uremic and renal dysfunction disrupts other body systems.
♦ GI—anorexia, nausea, vomiting, diarrhea or constipation, stomatitis, bleeding, hematemesis, dry mucous membranes, uremic breath
♦ Central nervous system—headache, drowsiness, irritability, confusion, peripheral neuropathy, seizures, coma
♦ Integumentary—dryness, pruritus, pallor, purpura and, rarely, uremic frost
♦ Cardiovascular—early in the disease, hypotension; later, hypertension, arrhythmias, fluid overload, heart failure, systemic edema
♦ Respiratory—pulmonary edema, Kussmaul’s respirations
♦ Hematologic—anemia, altered clotting mechanisms.
Complications
Renal failure affects many body processes. Complications may include:
♦ fever and chills (common), which indicate infection
♦ metabolic acidosis, due to decreased excretion of hydrogen ions
♦ anemia due to erythropoietinemia, glomerular filtration of erythrocytes, or bleeding associated with platelet dysfunction; tissue hypoxia, stimulating increased ventilation and work of breathing
♦ sepsis because of decreased white blood cell-mediated immunity
♦ heart failure due to fluid overload and anemia, which cause additional workload to the heart
♦ hypercoagulable state due to abnormalities in quantities or function of anticoagulant proteins, coagulation factor, platelet, or endothelial mediators, resulting in bleeding or clotting difficulties
♦ altered mental status and peripheral sensation due to effects on the highly sensitive cells of nerves secondary to retained toxins, hypoxia, electrolyte imbalance, and acidosis.
Diagnosis
♦ Blood studies showing elevated BUN, serum creatinine, and potassium levels; decreased hematocrit and bicarbonate and hemoglobin levels; and low blood pH
♦ Urine studies showing casts, cellular debris, and decreased specific gravity; in glomerular diseases, proteinuria and urine osmolality close to serum osmolality; a urine sodium level less than 20 mEq/L if oliguria results from decreased perfusion, and more than 40 mEq/L if the cause is intrarenal
♦ Creatinine clearance test measuring the GFR and reflecting the number of remaining functioning nephrons
♦ Electrocardiogram (ECG) showing tall, peaked T waves; widening QRS complex; and disappearing P waves if hyperkalemia is present
♦ Ultrasonography, plain films of the abdomen, kidney-ureter-bladder radiography, excretory urography, renal scan, retrograde pyelography, computed tomographic scans, and nephrotomography
Treatment
♦ High-calorie diet low in sodium and potassium and with restricted protein to meet metabolic needs (protein intake must be adequate enough to prevent malnourishment)
♦ Careful monitoring of electrolyte levels; I.V. therapy to maintain and correct fluid and electrolyte balance
♦ Fluid restriction to minimize edema (although fluid restoration and maintenance may be needed during the diuretic phase)
♦ Renal-dose dopamine (1 to 5 mcg/kg/minute) given I.V. to enhance renal perfusion
♦ Diuretic therapy to treat oliguric phase
♦ Sodium polystyrene sulfonate (Kayexalate) by mouth or enema to reverse hyperkalemia with mild hyperkalemic symptoms (malaise, loss of appetite, muscle weakness)
♦ Hypertonic glucose with insulin I.V., for more severe hyperkalemic symptoms (numbness and tingling and ECG changes)
♦ Sodium bicarbonate I.V. to treat metabolic acidosis
♦ Hemodialysis, continuous renal replacement therapy, or peritoneal dialysis to correct electrolyte and fluid imbalances
♦ Discontinuation of nephrotoxic agents (antibiotics, radioactive agents, and other such drugs)
♦ Discontinuation or adjustment of all medications cleared by renal excretion
Special considerations
Patient care includes careful monitoring and dietary education.
♦ Measure and record intake and output, including body fluids, such as wound drainage, nasogastric output, and diarrhea. Weigh the patient daily.
♦ Assess the hemoglobin level and hematocrit, and administer packed red blood cells, as ordered.
♦ Monitor vital signs. Watch for and report signs and symptoms of pericarditis (pleuritic chest pain, tachycardia, pericardial friction rub), inadequate renal perfusion (hypotension), and acidosis.
♦ Maintain proper electrolyte balance. Strictly monitor the potassium level.
 Watch the patient for symptoms of hyperkalemia (malaise, anorexia, paresthesia, or muscle weakness) and ECG changes (tall, peaked T waves; widening QRS complex; and disappearing P waves), and report them immediately. Avoid administering medications that contain potassium.
Watch the patient for symptoms of hyperkalemia (malaise, anorexia, paresthesia, or muscle weakness) and ECG changes (tall, peaked T waves; widening QRS complex; and disappearing P waves), and report them immediately. Avoid administering medications that contain potassium.♦ Assess the patient frequently, especially during emergency treatment to lower the potassium level. If the patient receives hypertonic glucose and insulin infusions, monitor potassium and glucose levels. If you give sodium polystyrene sulfonate rectally, make sure the patient doesn’t retain it and become constipated, to prevent bowel perforation.
♦ Maintain nutritional status. Provide a highcalorie, restricted-protein, low-sodium, and low-potassium diet, with vitamin supplements. Give the anorexic patient small, frequent meals.
♦ Use sterile technique because the patient with acute renal failure is highly susceptible to infection. Don’t allow personnel with upper respiratory tract infections to care for the patient.
♦ Prevent complications of immobility by encouraging frequent coughing and deep breathing and by performing passive range-of-motion exercises. Help the patient walk as soon as possible. Add lubricating lotion to the patient’s bath water to combat skin dryness.
♦ Provide good mouth care frequently because mucous membranes are dry. If stomatitis occurs, an antibiotic solution may be ordered. Have the patient swish the solution around in his mouth before swallowing.
♦ Monitor for GI bleeding by guaiac-testing stools for blood. Administer medications carefully, especially antacids and stool softeners. Use aluminum hydroxide-based antacids; magnesium-based antacids can cause the serum magnesium level to rise to a critical level.
♦ Use appropriate safety measures, such as side rails and restraints (only when absolutely necessary), because the patient with central nervous system involvement may be dizzy or confused.
♦ Provide emotional support to the patient and his family. Reassure them by clearly explaining procedures.
♦ During peritoneal dialysis, position the patient carefully. Elevate the head of the bed to reduce pressure on the diaphragm and aid respiration. Be alert for signs of infection (cloudy drainage, elevated temperature) and, rarely, bleeding. If pain occurs, reduce the amount of dialysate. If the patient has diabetes, monitor his blood glucose level periodically and administer insulin, as ordered. Watch for complications, such as peritonitis, atelectasis, hypokalemia, pneumonia, and shock.
♦ If the patient requires hemodialysis, or continuous renal replacement therapy (CRRT), check the blood access site (arteriovenous fistula, subclavian or femoral catheter) every 2 hours for patency and signs of clotting (palpate for a thrill; auscultate for a bruit). Don’t use the arm with the shunt or fistula for taking blood pressures or drawing blood. Weigh the patient before beginning dialysis. During dialysis and CRRT, monitor vital signs, clotting times, blood flow, the function of the vascular access site, and arterial and venous pressures. Watch for complications, such as septicemia, embolism, hepatitis, and rapid fluid and electrolyte loss. After dialysis, monitor vital signs and the vascular access site; weigh the patient;
watch for signs of fluid and electrolyte imbalances.
watch for signs of fluid and electrolyte imbalances.
♦ Use standard precautions when handling blood and body fluids.
ACUTE TUBULAR NECROSIS
Acute tubular necrosis (ATN), also known as acute tubulointerstitial nephritis, is the most common cause of acute renal failure in critically ill patients. ATN injures the nephron’s tubular segment, causing renal failure and uremic syndrome. Mortality ranges from 40% to 70%, depending on complications from underlying diseases. Nonoliguric forms of ATN have a better prognosis.
Causes
♦ Diseased tubular epithelium that allows leakage of glomerular filtrate across the membranes and reabsorption of filtrate into the blood
♦ Obstruction of urine flow by the collection of damaged cells, casts, red blood cells (RBCs), and other cellular debris within the tubular walls
♦ Ischemic injury to glomerular epithelial cells, resulting in cellular collapse and decreased glomerular capillary permeability
♦ Ischemic injury to vascular endothelium, eventually resulting in cellular swelling and tubular obstruction
Pathophysiology
ATN results from ischemic or nephrotoxic injury, most commonly in debilitated patients, such as the critically ill or those who have undergone extensive surgery. In ischemic injury, disruption of blood flow to the kidneys may result from circulatory collapse, severe hypotension, trauma, hemorrhage, dehydration, cardiogenic or septic shock, surgery, anesthetics, or reactions to transfusions. Nephrotoxic injury may follow ingestion of certain chemical agents, such as contrasts administered during radiologic procedures or administration of antibiotics (aminoglycosides), or result from a hypersensitive reaction of the kidneys. Because nephrotoxic ATN doesn’t damage the basement membrane of the nephron, it’s potentially reversible. However, ischemic ATN can damage the epithelial and basement membranes and can cause lesions in the renal interstitium. (See Understanding acute tubular necrosis.)
Signs and symptoms
ATN is usually difficult to recognize in its early stages because effects of the critically ill patient’s primary disease may mask the symptoms of ATN. However, signs and symptoms may include:
♦ decreased urine output, generally the first recognizable effect because of reduced renal blood flow and glomerular filtration
♦ hyperkalemia because of disturbed renal tubular absorption
♦ uremic syndrome with oliguria (or, rarely, anuria) and confusion, which may progress to uremic coma caused by renal failure
♦ dry mucous membranes and skin due to the accumulation of uremic toxins
♦ central nervous system signs and symptoms, such as lethargy, twitching, or seizures resulting from effects of uremic toxins on highly sensitive nerve cells
♦ bibasilar crackles, tachycardia, and other signs of fluid overload.
Complications
♦ Heart failure
♦ Uremic pericarditis
♦ Pulmonary edema
♦ Uremic lung
♦ Metabolic acidosis
♦ Anemia
♦ Anorexia, intractable vomiting
♦ Poor wound healing due to debilitation
 Fever and chills may signal the onset of an infection, which is the leading cause of death in ATN.
Fever and chills may signal the onset of an infection, which is the leading cause of death in ATN.Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


