SIGNIFICANCE OF THE RENAL SINUS
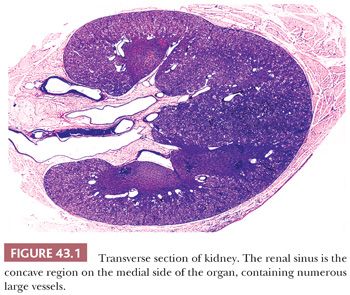
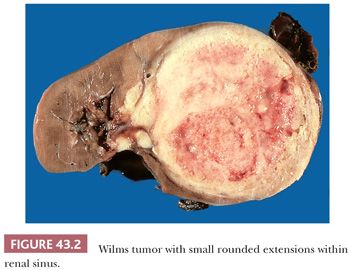
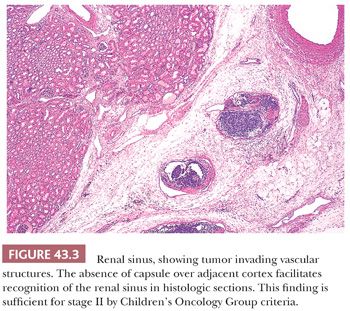
The renal sinus, illustrated in Figure 43.1, is the concave region on the medial aspect of the kidney, which contains the major renal vessels and portions of the pelvicalyceal system. Because it contains the major hematogenous and lymphatic drainage of the kidney, the renal sinus is the principal route by which tumor cells pass from the renal parenchyma to metastatic sites. Therefore, it is essential that this region be examined carefully in all neoplastic nephrectomy specimens. Tumor in sinus vessels can often be identified in the gross specimen (Fig. 43.2). In microscopic sections, the sinus is easily recognized when pelvicalyceal structures or medullary pyramids are present on the section. Less well known is the fact that renal cortex lining the sinus is unencapsulated, facilitating recognition of sections from the renal sinus that do not include pelvicalyceal structures (Fig. 43.3). Dense capsules surround the pelvis and calyces and extend along the adjacent medullary pyramids but usually disappear at the junction with cortex within the sinus (3,5). The renal sinus vessels extend into the kidney and merge with the larger intrarenal vessels. Precisely where this transition occurs is a somewhat subjective determination.
On earlier NWTS trials, the hilar plane was the major basis for distinguishing stage I from stage II in the renal sinus. This plane is an arbitrary one that connects the medial borders of the upper and lower poles and has no intrinsic anatomic significance. Not surprisingly, the hilar plane proved unsatisfactory as a staging criterion. It is usually distorted or obscured by tumor growth, is evaluable only during initial gross evaluation, and cannot be confirmed by secondary reviewers.
Beginning with NWTS Trial 5 (August 1995) and continuing today on COG protocols, the hilar plane was abandoned as a criterion for staging. Staging of tumor within the sinus is primarily determined by the presence or absence of tumor in venous or lymphatic vessels (5,6). Any tumor involving these vascular structures is upstaged, regardless of relations to the hilar plane. Figure 43.3 illustrates tumor in vessels of the renal sinus. Vascular involvement is considered unequivocal when tumor fills venous or lymphatic lumens, is attached to the intima, or invades the vessel wall. Less certain is the presence of tumor cells lying free in the lumen, raising the question of displacement artifact. The presence of prominent artifactual tumor spread elsewhere on the slide may favor artifact over true vascular involvement. Admixture of intraluminal tumor cells with blood favors true vascular invasion. Tumor in arterial lumens is assumed to represent artifactual displacement unless the arterial wall is invaded.
Soft tissue infiltration in the sinus, including the wall or lumen of the pelvicalyceal system, is acceptable for stage I, unless it potentially involves the medial sinus margin or invades vessels of the pelvic wall. Beware of bulky, rounded masses of apparent soft tissue invasion in the sinus because intravascular tumor can eventually efface vascular walls, mimicking soft tissue invasion.
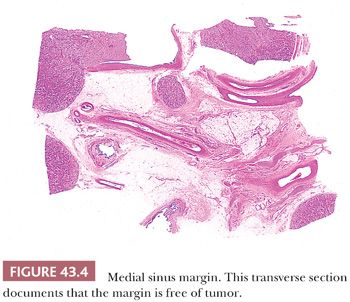
Whereas recognition of tumor in the renal sinus is usually not difficult, it is often challenging to evaluate the medial sinus margin. Involvement of resection margins is a criterion for stage III on the COG staging scheme. The medial margin is not demarcated by a fascia or other anatomic structure, and this region of the specimen consists of the ragged connective tissues surrounding the resected renal vessels. Sometimes it is possible to obtain an adequate transverse section through this region, especially after using the refrigerated fixation technique recommended earlier (Fig. 43.4).
A potential source of staging error is the renal vein margin. Veins usually retract when severed, and tumor protruding from the vein margin in the nephrectomy can produce a false impression of margin involvement. In this situation, the surgeon usually has a more accurate perception of whether tumor was entered during venous transection. The vein margin should be designated as positive when tumor invades the vein wall or small lymphatics in soft tissue surrounding the vein wall at that site or when the surgeon reports that tumor was entered during venous transection. Once again, effective communication between surgeon and pathologist can often prevent staging errors.
EVALUATION OF THE CAPSULE
The renal capsule, not the “tumor capsule,” is the defining criterion for invasion beyond the external surface of the kidney. The tumor capsule is often a composite of several layers that become fused as the tumor enlarges. A tumor growing within the kidney may sequentially acquire some or all of the following capsular investments: (a) an intrarenal pseudocapsule, composed of compressed or reactive connective tissue within the renal parenchyma; (b) the renal capsule; (c) a perirenal pseudocapsule, composed of compressed or reactive connective tissues in the perirenal adipose layers; and finally (d) the perirenal (Gerota) fascia. These layers tend to form a composite capsule that can defy precise analysis or lead to erroneous downstaging.
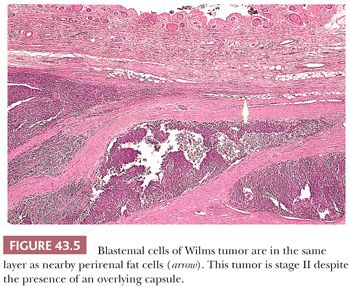
Stage I lesions may invade, but must not have penetrated, the renal capsule. Formation of a pseudocapsule in perirenal fat can obscure the fact that tumor cells have penetrated beyond the renal capsule. Sections taken from the edge, where protruding tumor abuts on the cortical surface, are often helpful in recognizing this situation because the position of the renal capsule can often be identified with precision. By tracking laterally from the outermost tumor cells, it is often possible to recognize that these cells are within the same tissue layer as perirenal fat (Fig. 43.5).
Some tumors are associated with a peritumoral granulation tissue response. This “inflammatory pseudocapsule” may cause adhesions to adjacent structures, mimicking tumor invasion. Inflammatory pseudocapsules can result from perirenal hemorrhage, tumor necrosis, pyelonephritis, or preoperative tumor rupture. Viable tumor cells are not commonly found within inflammatory pseudocapsules. An inflammatory pseudocapsule is not a basis for upstaging an otherwise stage I tumor, although its presence was associated with somewhat higher relapse rates for stage I WT in one study (7).
EVALUATION OF LYMPH NODES
Tumor involvement of abdominal lymph nodes is a basis for a stage III designation group. Nodal involvement by undifferentiated blastemal cells may be subtle because tumor cells may not appear much larger than lymphocytes. Even a single definite tumor cell in the marginal sinus of a regional node will mandate upstaging to stage III status. Sometimes immunostains for WT1 and leukocyte common antigen can be useful in determining whether a small cluster of cells in a nodal sinus represents tumor or lymphocytes (8). More often, the finer chromatin and nuclear molding exhibited by clusters of blastemal cells enables their recognition.
Several nodal changes may falsely suggest the presence of metastases. The most frequent of these “pseudometastatic” phenomena is the presence of aggregates of Tamm-Horsfall protein containing extravasated benign renal tubular epithelium. The epithelium of nephrons obstructed by tumor is often dislodged and transported to marginal sinuses of hilar nodes. Swollen endothelial cells in postcapillary venules and mesothelial inclusions can also be mistaken for metastatic tumor (8).
PEDIATRIC RENAL NEOPLASMS: DIFFERENTIAL DIAGNOSTIC CONSIDERATIONS
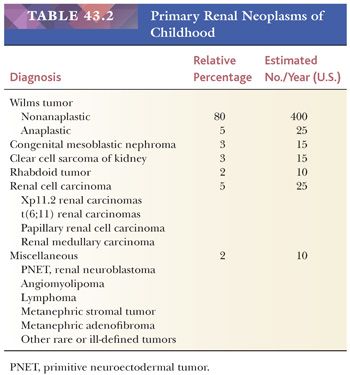
The major renal neoplasms of childhood are listed in Table 43.2, including their relative prevalence and the estimated annual incidence in the United States.
WILMS TUMOR (NEPHROBLASTOMA)
WT is the most likely consideration for any pediatric renal neoplasm, except during the first 3 months of life. WT and most other eponymous designations should be spelled without the possessive apostrophe (9).
CLINICAL FEATURES AND GENETICS
For general information concerning clinical and molecular aspects of WT, see Davidoff (1) and Dome et al. (2).
WT has a peak incidence between 2 and 5 years of age, with 90% being diagnosed by age 6 years (10,11). Rare cases are encountered in adults, but many tumors referred to as adult WT prove to be other neoplastic entities, including renal primitive neuroectodermal tumors, renal carcinoids, sarcomatoid renal cell carcinoma, papillary cell renal carcinoma, renal sarcomas (particularly primary renal synovial sarcoma), and metanephric adenomas. The concept that WT of adults has a worse prognosis than its pediatric counterpart is probably, in part, a result of diagnostic errors in cases reported in the literature (12).
Several dysmorphic syndromes are associated with a high risk of developing this neoplasm (1–3). No single cytogenetic or molecular abnormality has been consistently abnormal either in WT specimens or in patients predisposed to WT. The WT1 gene, located on chromosome 11p13, is consistently involved in the pathogenesis of the WAGR syndrome (WT associated with aniridia and genital anomalies) and the Denys-Drash syndrome (pseudohermaphroditism, severe glomerulopathy, and WT). Patients with WAGR syndrome usually have interstitial deletions of most or all of the 11p13 band, whereas patients with Denys- Drash syndrome are characterized by point mutations in the WT1 gene. However, the WT1 gene is mutated in only approximately 20% of sporadic WT specimens. A second WT locus maps to 11p15, where multiple imprinted candidate genes reside: these include IGF2, p57KIP2, and KVLQT1. This locus is implicated in the Beckwith-Wiedemann syndrome. WT occasionally shows a familial distribution, with a distribution usually suggesting autosomal dominant transmission, possibly by several distinct mutations (13).
Other somatic genetic alterations have been identified in WT. Inactivation of a tumor suppressor gene on the X chromosome, WTX, has been demonstrated in one-third of WTs (14). Moreover, WTs demonstrating loss of heterozygosity for chromosomes 1p and 16q have been shown to have a poor prognosis (15). Identification of the latter in current COG protocols triggers more intensive chemotherapy.
Many abnormal substances have been identified in the blood or urine of patients with WT, including acquired von Willebrand factor, active and inactive renin, erythropoietin, and neuron-specific enolase (16). These may contribute to unusual presenting symptoms in some patients, such as coagulopathy, hypertension, and polycythemia. None occurs with a frequency that would justify use as a screening test (16). Abnormal circulating mucin has been described with some WTs as well as several other malignant neoplasms. This appears as finely granular material on routinely stained blood smears and can cause anomalous results from automated hematology analyzers (16,17).
The prognosis for WT has dramatically improved from approximately 8% survival at the beginning of the century to more than 90% today. The most common sites of metastasis are regional lymph nodes, lungs, and liver (“the three L’s”). Spread to other organs is rare, even in advanced cases.
GROSS FEATURES
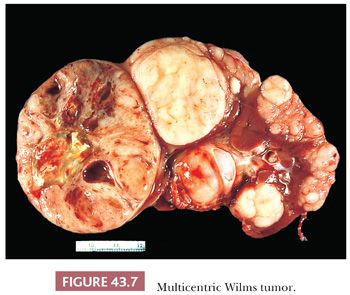
WT typically presents as a unicentric, spherical mass that is sharply demarcated from renal parenchyma (Figs. 43.2 and 43.6). Approximately 10% are multicentric (Fig. 43.7). Multicentricity is associated with an increased likelihood of tumor formation in the remaining kidney. Bilateral renal tumors are initially present in 5% to 6% of cases.
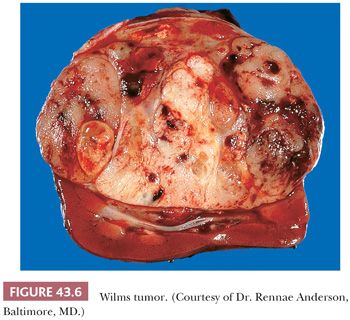
The sectioned surface of WT is usually pale gray and uniform, but focal hemorrhage, necrosis, and cyst formation are commonly encountered. The texture is usually soft and friable, contributing to displacement artifacts during specimen manipulation, but tumors with abundant stroma may have a dense, myomatous consistency. Calcification is relatively uncommon. Prominent septa often impart a nodular appearance.
WT can arise anywhere in the cortex or medulla, usually compressing and distorting renal parenchyma around its margin. Rarely, exophytic tumors connected to the renal surface by a narrow pedicle may mimic extrarenal WT. Polypoid masses in the pelvicalyceal lumen may occur either as extensions from the intrarenal mass or as separate tumors arising within the pelvic wall. The renal vein and its branches are often filled by tumor thrombus that can extend via the inferior vena cava into the right atrium.
MICROSCOPIC FEATURES
WTs are rivaled only by teratomas in their diversity of cell and tissue types and stages of differentiation. Space permits illustration and description of only a few of the histologic appearances of WT, and other sources should be consulted for additional illustrations and descriptive details (3,4). Most specimens exhibit, to a variable degree, the so-called triphasic appearance, including cells of blastemal, stromal, and epithelial lineage (Fig. 43.8). However, monophasic and biphasic lesions are relatively common, consisting of any one or two of these cell lineages.
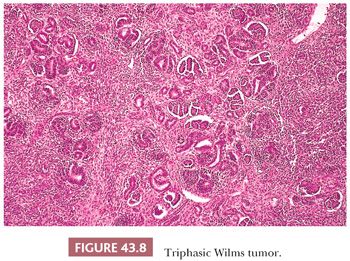
Each of the three major cell types can present a spectrum of patterns and degrees of differentiation, accounting for the remarkable diversity of appearances characterizing various WTs. Generally, however, there is relatively little variation in histologic appearance from region to region within the same tumor.
Blastemal Patterns
The blastemal cells of WT are small, closely packed cells with a high nuclear-to-cytoplasmic ratio, revealing little or no evidence of differentiation toward epithelial or stromal cell types at the light microscopic level. Their nuclei are usually round or oval, with moderately coarse chromatin, and mold to each other in the same fashion as do the nuclei of small cell carcinoma of the lung. Nucleoli are relatively inconspicuous. Blastemal cells form several distinctive aggregation patterns, which can be divided into two broad categories based on their structure and degree of invasiveness.
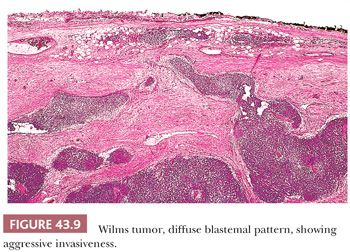
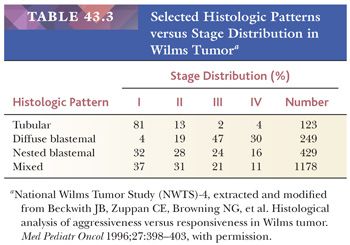
Diffuse Blastemal Pattern. Monomorphous, relatively noncohesive sheets of blastemal cells with aggressively invasive margins characterize this pattern (Fig. 43.9). It is the most consistently aggressive pattern of WT (18). As shown in Table 43.3, this pattern rarely is seen with stage I disease, and a majority of cases present as stage III or IV tumors. Fortunately, most tumors with the diffuse blastemal pattern prove responsive to modern therapeutic protocols (18), so this pattern remains in the “favorable histology” category. In metastatic sites, the diffuse blastemal pattern can be readily confused with other small blue cell tumors of childhood, and ultrastructural or other special studies may be required to establish the correct diagnosis.
Nested Blastemal Patterns. These patterns are characterized by sharply outlined nests of blastemal cells in a myxoid mesenchymal background. They usually lack the invasive behavior seen with the diffuse blastemal pattern and have sharply defined margins at the advancing edge of the tumor (Fig. 43.10) Several organized patterns may be seen, of which the following three are the most common:
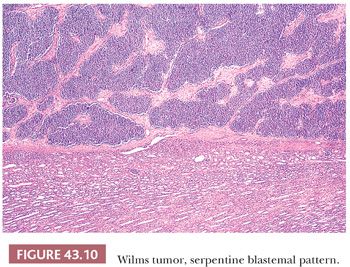
Serpentine blastema. This pattern features serpiginous, anastomosing cords of blastemal cells in a loose, stellate, or spindled stroma (Fig. 43.10). It is among the most distinctive patterns in WT and can help to distinguish WT from other blue cell neoplasms of childhood.
Nodular blastema. This resembles the serpentine pattern but has rounded blastemal nests instead of cell cords.
Basaloid blastema. The basaloid form resembles the serpentine or nodular patterns, except that the blastemal cell clusters are outlined by a palisaded layer of cuboidal or columnar cells, reminiscent of the architecture of cutaneous basal cell carcinoma.
Epithelial Patterns
Epithelial differentiation in WT produces a variety of cell types and degrees of differentiation, as listed in Table 43.4. Most of these recapitulate events in normal nephrogenesis (homologous differentiation). Others are foreign to the normal kidney at any stage of development (heterologous differentiation).
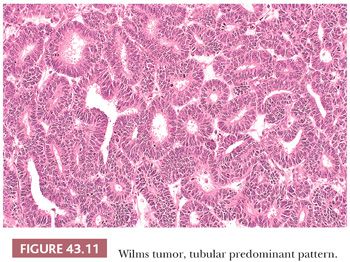
Tubular differentiation is the most frequent epithelial pattern. This ranges from vague hints at tubular formation in blastemal foci to highly differentiated tubules resembling nephronic elements in mature kidneys. True tubular lumens are present in most specimens (Fig. 43.11), and their presence strongly favors WT over renal primitive neuroectodermal tumor (PNET), clear cell sarcoma of the kidney, or neuroblastoma. As shown in Table 43.3, tumors in which tubular differentiation is predominant tend to be less aggressive, and most present at stage I (18). Although not usually invasive, tubular-predominant WT often shows rapid growth, and it is not uncommon to find huge tumors that remain encapsulated within the kidney.
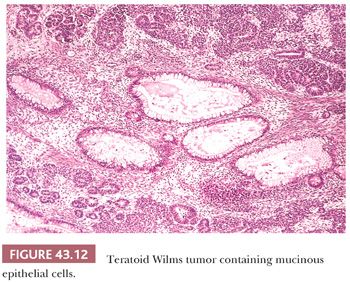
Glomerular differentiation, which is present in many WTs, ranges from simple papillary formations barely suggesting glomerulogenesis to mature tumor glomeruli closely resembling those of normal kidneys. Large, complex papillary structures sometimes lead to diagnostic confusion with papillary renal carcinomas. Heterologous epithelial cell types include mucinous differentiation (Fig. 43.12) and squamous cells.
Stromal Patterns
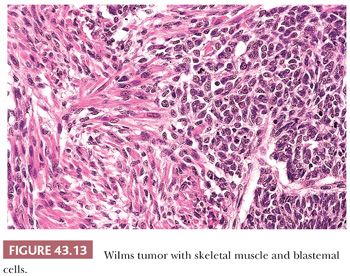
Table 43.5 lists the principal stromal cell types seen in WTs. Immature myxoid and spindled mesenchymal cells are present in most specimens. Skeletal muscle is the most common heterologous cell type and usually attains a high degree of differentiation (Fig. 43.13). The presence of skeletal muscle in WT must not be confused with renal rhabdoid tumor, which is discussed later in this chapter. Although the term fetal rhabdomyomatous nephroblastoma is sometimes used to describe WTs with abundant skeletal muscle (19,20), we are not enthusiastic about the use of subdesignations for WT patterns. The structural diversity of WT could ultimately engender an endless number of separate designations for various morphologic patterns. Only subtypes with distinctive clinical or biologic implications deserve separate designations.
EFFECTS OF THERAPY ON WILMS TUMOR HISTOLOGY
Modern chemotherapy has been extremely effective in the management of most WT cases, and chemotherapy before nephrectomy is now used for many cases of bilateral, metastatic, or unresectable lesions. Chemotherapy usually results in massive necrosis of immature and actively proliferating cell types in WT, whereas slowly replicating and differentiated cell types may be unaffected. For example, tumors composed mostly of mature skeletal muscle show little clinical regression with chemotherapy, despite ablation of all malignant cells (20,21). The presence of mature or slowly replicating cells with few mitotic figures does not necessarily imply treatment failure, and treated WT cases can be staged according to the distribution of viable, mitotically active foci of persistent tumor (20,21). The presence of actively proliferating tumor (particularly blastema) after chemotherapy indicates resistance and is associated with a diminished prognosis. Cells with anaplastic nuclear changes are generally unaffected by chemotherapy and have the same significance as in untreated WT (21). Using these principles, posttherapy WT specimens are classified into one of four categories in COG protocols: complete necrosis (<1% viable tumor), blastemal predominant (>1/3 viable and 2/3 of viable WT is blastemal), intermediate (neither of the previous criteria applies), or anaplastic. This categorization of treated tumors may alter therapy for patients with bilateral WT (6).
ULTRASTRUCTURAL AND IMMUNOHISTOCHEMICAL STUDIES
Ultrastructural study is rarely necessary to establish a diagnosis of WT but can occasionally be very helpful in distinguishing WT from other undifferentiated neoplasms. The ultrastructural features of WT and other childhood renal tumors are detailed elsewhere (22,23).
Immunohistochemistry has been used productively in studies of WT biology and differentiation but, to date, has limited usefulness in diagnosis. The diversity of cell lines and degrees of differentiation imparts a correspondingly varied profile of immunohistochemical results. Blastemal cells may yield either positive or negative results for vimentin and cytokeratin, whereas various differentiating cell lines will give results according to their patterns of differentiation. Positive results for desmin with negative stains for other muscle markers have been suggested as a characteristic feature of WT blastemal cells and might prove helpful in distinguishing this entity from other undifferentiated tumors of childhood (24), although this feature is shared with desmoplastic small round cell tumor. Immunoreactivity for WT1 protein is typically limited to the blastema and epithelial components of WT, with the stroma being negative (25). Hence, absence of labeling for WT1, particularly in a stromarich tumor, does not exclude the diagnosis of WT. Although WT1 immunoreactivity may help distinguish WT from PNET, one should be aware that desmoplastic small round cell tumor (26) and other adult neoplasms, such as malignant mesothelioma, typically label for WT1. Hence, although useful, WT1 immunoreactivity cannot in and of itself establish or disprove a diagnosis of WT.
ANAPLASTIC NUCLEAR CHANGES: THE MARKER OF UNFAVORABLE HISTOLOGY IN WILMS TUMOR
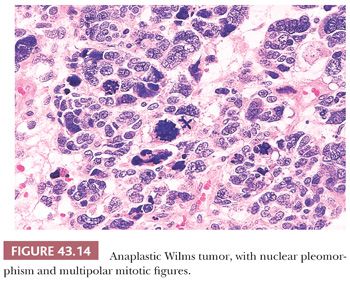
Approximately 5% of WTs contain cells with huge hyperchromatic nuclei, associated with multipolar mitotic figures (2,27–29). Anaplastic nuclear change is the only criterion of “unfavorable histology” in WTs, and all WTs lacking this feature are designated as having “favorable histology.” Anaplasia is almost never seen in WT diagnosed during the first year and is rare in the second year of life. Its relative frequency increases after that age, and it is found in approximately 10% of WTs diagnosed after age 5 years.
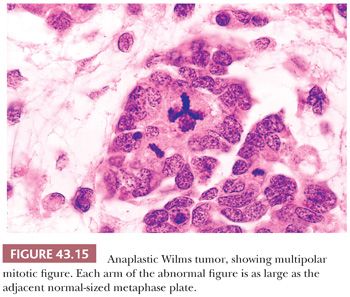
Anaplastic nuclear change reflects extreme polyploidy and is usually apparent under low magnification (10× objective) (Fig. 43.14). The features of anaplasia include (a) markedly enlarged tumor cell nuclei with increased chromatin content and (b) multipolar or obviously polyploid mitotic figures. Multipolar mitotic figures are the most unequivocal criterion for polyploidy. We have considered as anaplastic the few WT specimens in which polyploid mitotic figures were found in the absence of severe nuclear gigantism. It is important to distinguish multipolar mitotic figures from minor mitotic anomalies resulting from uneven separation of metaphase plates. This error will be avoided if we require that each limb of a mitotic figure be as large as or larger than diploid mitotic plates, as shown in Figure 43.15.
Focal and Diffuse Anaplasia
Initially, the presence of anaplastic nuclear changes in any region of a WT was considered prognostically unfavorable. It subsequently was thought that anaplasia was an indicator of increased resistance to adjuvant chemotherapy and not necessarily a marker of increased tumor aggressiveness, so stage I anaplastic WT and WT with limited intrarenal foci of anaplasia (focal anaplasia [FA]) would be predicted to have an excellent prognosis. This concept implies that the prognosis for a patient with anaplastic WT is determined by the completeness of surgical removal of anaplastic cells (18). However, more recent data on patients with stage I anaplastic WT have challenged this concept (30). Regardless, the definition of FA has therapeutic implications on current protocols and includes only those WTs meeting all of the following criteria: (a) anaplasia confined to one or more discrete sites within the primary tumor and not present in extrarenal sites and (b) tumor cells outside anaplastic foci show no “nuclear unrest” (nuclear or mitotic abnormalities that approach, but do not quite attain, that degree of severity required for a designation of anaplasia).
In some specimens, the criteria of anaplasia are unequivocally met on only one or two slides, but other sections reveal widespread nuclear unrest. These appearances suggest that the cells are in transition toward anaplastic morphology. For this reason, nuclear unrest that is widespread or present in extrarenal sites excludes a case from the FA category.
Any WT not meeting this revised definition of FA is designated as diffuse anaplasia (DA). Any one of the following situations will justify assignment to the DA category:
1. Nonlocalized anaplastic change
2. Anaplastic change or nuclear unrest in invasive sites, or any extrarenal deposits
3. Localized anaplastic change in a tumor that also has severe nuclear unrest elsewhere
4. Anaplasia in a random biopsy specimen
5. Anaplasia involving the edge of one or more sections, and the site(s) from which the sections were taken cannot be determined
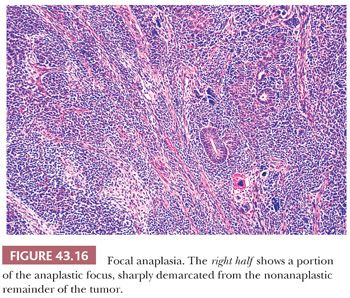
Anaplastic foci are clearly demarcated from adjacent nonanaplastic tumor (Fig. 43.16). Whereas most cases in the FA category have a single focus, tumors may have multiple small foci of FA, when each anaplastic focus is clearly demarcated and entirely surrounded by nonanaplastic tumor (28). Anaplastic foci are usually a few millimeters in diameter but can be larger if their localized nature is clearly documented. When anaplastic changes extend to the edge of several sections, it is important to determine whether the sections showing anaplasia were all from the same focus. This determination depends on careful documentation of the site from which each tumor section was obtained and is one reason this point was emphasized in the first part of this chapter.
The following caveats concerning anaplastic WT deserve emphasis:
1. DA is usually apparent in the majority of tumor sections. Anaplastic cells were found in more than half the tumor sections in 77% of specimens with DA and in more than onefourth of slides in 98% of cases (28).
2. Once anaplasia has been identified in a case of WT, the threshold for designating its presence elsewhere can be lowered. A single gigantic nucleus or polyploid mitosis is sufficient evidence that anaplasia also is present in another site (27,28).
3. Enlarged nuclei in differentiated skeletal muscle cells of a WT do not signify anaplasia unless accompanied by multipolar mitotic figures. These may represent a degenerative phenomenon similar to that frequently identified in skeletal muscle cells of the extremities in reaction to injury. However, severe nuclear enlargement in embryonal muscle fibers has the same significance as in other cell types of WT.
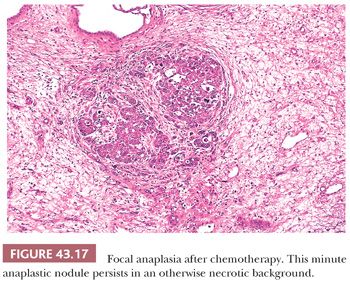
4. The cytologic criteria for anaplasia are the same in specimens removed after chemotherapy. The recognition of FA is usually easier in specimens from treated patients. These foci, being generally resistant, continue to enlarge during the period of therapy. Even minute anaplastic foci stand out in sharp contrast in the necrotic or atrophic background (Fig. 43.17).
Molecular Correlates of Anaplasia
Anaplasia is tightly correlated with the presence of p53 gene mutations. Whereas favorable histology WTs virtually never harbor p53 mutations, the majority of anaplastic WTs do (31). Furthermore, microdissection experiments have demonstrated that p53 mutations are usually restricted to the anaplastic portions of WT with FA (32). Because an apoptotic pathway induced by chemotherapy depends on functional p53, the association of p53 mutations with the resistance to chemotherapy that is the hallmark of anaplastic WT makes sense. At a practical level, p53 protein overexpression by immunohistochemistry correlates well but not perfectly with morphologically defined anaplasia (33).
CYSTIC VARIANTS OF WILMS TUMOR
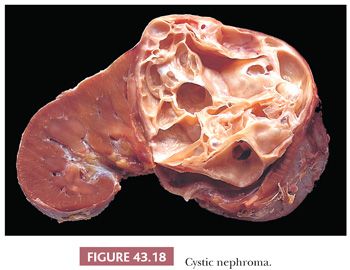
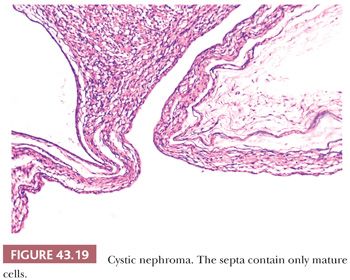
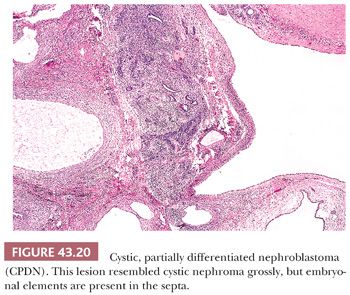
Scattered cysts are commonly encountered in conventional WT, but rarely, the tumor is composed entirely of cystic spaces and delicate septa, without an expansile solid component. These lesions are encapsulated and clearly demarcated from adjacent renal parenchyma (Fig. 43.18). For tumors that are entirely composed of mature cells, as shown in Figure 43.19, the term cystic nephroma (CN) is recommended (34). This term conveys the concept of a benign cystic neoplasm more clearly than the alternative designation “multilocular cyst.” If the septa contain embryonal cell types, the designation “cystic, partially differentiated nephroblastoma” (CPDN) is appropriate (Fig. 43.20). CPDN has traditionally been considered a transitional stage in the development of pediatric CN, and these lesions may represent a “hyperfavorable end” of the Wilms tumor spectrum. The epithelial cells lining the cysts of CN and CPDN range from flattened to columnar and often are of the “hobnail” type, with prominent acidophilic cytoplasm that is more abundant at the base than the apex. The cysts often have distinct walls composed of maturing spindle cells.
The distinction of CN from CPDN has little or no clinical importance when the lesion has been cleanly and completely resected because both lesions are curable by resection alone. If resection is incomplete or associated with tumor spill, a slight potential for recurrence may exist for each (34,35). Lesions designated CN also occur in adults, but the latter occur predominantly in adult females and may be related to mixed epithelial and stromal tumor (36).
The differential diagnosis of CN and CPDN includes localized (segmental) variants of polycystic renal disease (37), which can produce grossly cystic masses localized to a portion of one kidney. These differ from CN and CPDN in that cysts are mingled with renal cortical and medullary elements, and the abnormal region is not demarcated by a distinct pseudocapsule. CN and CPDN are sharply encapsulated neoplasms, with no normal renal elements inside the capsule. Localized cystic renal dysplasia can be distinguished by the presence of typical features or renal dysplasia, including primitive ducts, maldeveloped nephrons, and the frequent presence of cartilage. Localized lymphatic lesions of the kidney, including cystic lymphangiectasis and lymphangioma, can mimic the gross appearances of these tumors, as can an occasional multicystic clear cell sarcoma of kidney, synovial sarcoma, or congenital mesoblastic nephroma. Multilocular cystic renal cell carcinoma, characterized by cells with clear cytoplasm lining the cysts (38), is rarely encountered in pediatric patients outside of the setting of von HippelLindau syndrome. This neoplasm has at this writing never been associated with metastasis and thus is regarded by most as a neoplasm of low malignant potential (39). Cystic WT is distinguished by the presence of solid, expansile regions that replace or distort the cystic spaces rather than being passively molded by the cysts.
NEPHROGENIC RESTS AND NEPHROBLASTOMATOSIS
In more than 30% of kidneys resected for WT, the renal parenchyma contains one or more regions of persistent embryonal tissue, representing precursor lesions of WT. These lesions have been given many names in the past; Beckwith has suggested the generic term nephrogenic rests (NRs). This term applies to all putative precursor lesions of WT or their nonneoplastic derivatives (40,41). The presence of multiple or diffusely distributed NRs is termed nephroblastomatosis. This term is most commonly applied when the rests are in an active state of cellular proliferation and are large enough to be grossly visible or apparent on imaging studies (42).
NRs have a dynamic life history that can produce a variety of appearances. This feature led to complex schemes of classification, with varying terminology. Beckwith’s classification is based on the assumption that the structure of NRs reflects both the dynamic state and the history of an individual rest. This scheme was presented in more detail elsewhere (3,40,41), and only a brief summary can be provided here.
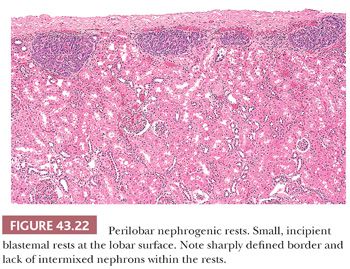
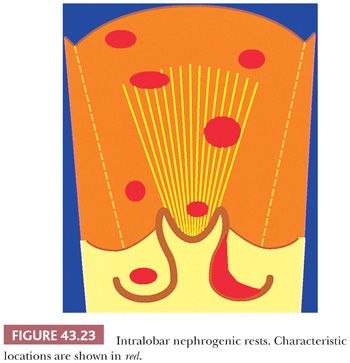
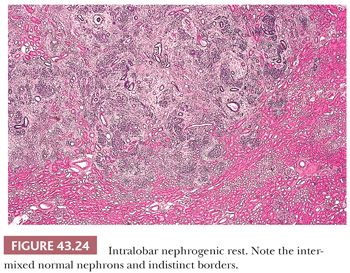
Two fundamental categories of NRs are recognized based on their topographic relation to the renal lobe. These are designated perilobar nephrogenic rests (PLNRs) (Figs. 43.21 and 43.22) and intralobar nephrogenic rests (ILNRs) (Figs. 43.23 and 43.24). ILNRs may occur anywhere in the renal lobe, including the peripheral cortex. They often also occur within the renal sinus, including in the walls of the pelvicalyceal system. In these latter sites, the rests are outside of the confines of the renal lobe, rendering the term intralobar somewhat misleading. ILNRs have a more varied structure than do PLNRs and are characterized by their interstitial location between nephrons, whereas PLNRs usually are discrete structures that are clearly demarcated from adjacent nephrons. The major features distinguishing ILNRs from PLNRs are summarized in Table 43.6.


Rests of either major category may be further classified based on their developmental fates. An individual NR may undergo any of the following fates, several of which often occur sequentially:
1. It may remain unchanged in size or composition, even for many years, as a tiny, microscopic blastemal focus similar to that in Figure 43.22 (dormant NR).
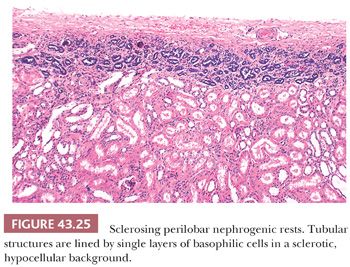
2. The most common fate of NRs is maturation, sclerosis, and eventual disappearance (sclerosing NRs and obsolescent NRs). A typical sclerosing PLNR is shown in Figure 43.25.
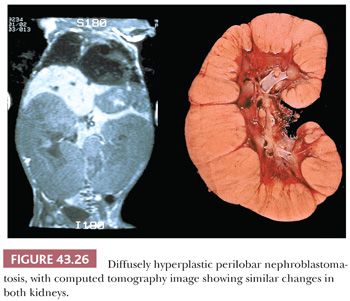
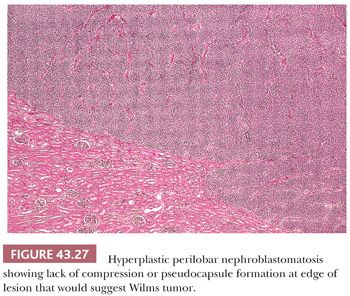
3. Hyperplastic NRs represent coordinated proliferation of all susceptible cells of the rest, as distinguished from a clonal neoplastic process originating in a single cell of the rest. Hyperplasia may produce large masses that can exhibit active growth and numerous mitotic figures, and a section from the interior of a hyperplastic NR may be indistinguishable from WT. Diffuse hyperplastic growth, involving all or most cells of a rest, tends to preserve the original shape of the rest. When PLNRs form a continuous layer of embryonal cells at the lobar surface, hyperplastic proliferation will produce a thick “rind” of abnormal tissue at the renal surface, as seen in Figure 43.26. Ovoid and lenticular masses result from hyperplasia of NRs that originally had these shapes. An irregular-shaped, multinodular appearance will result if only some of the cells are capable of proliferation (41). An important feature distinguishing actively hyperplastic NRs from WT is the usual absence of a pseudocapsule at the interface between hyperplastic NRs and renal parenchyma (Fig. 43.27).
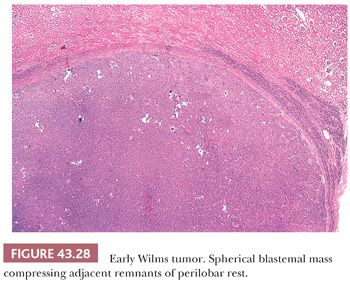
Neoplastic induction is assumed to represent a clonal event originating in single cells of a rest, resulting in WT or benign adenomas. Rapidly growing tumors originating at a single point (or cell) will tend to grow equally in all directions, forming spherical, expansile nodules with compressed rest remnants often present at the periphery (Fig. 43.28). Very rarely, anaplasia has been reported in an NR (43).
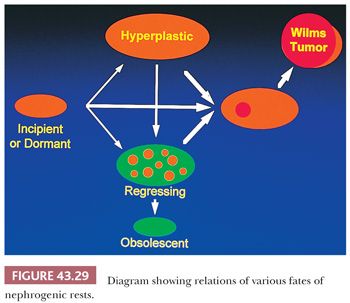
Relations between the various developmental fates of NRs are shown diagrammatically in Figure 43.29. An individual rest commonly progresses through several of these stages sequentially. For example, an incipient or dormant rest may undergo hyperplastic proliferation, followed by a phase of growth arrest, maturation, and regression. This will result in a large but inactive-appearing lesion. Ultimately, one or more cells within the regressing rest may be induced to form WT. The arrows in Figure 43.29 show the possible pathways a rest might follow in its journey toward obsolescence or neoplasia.
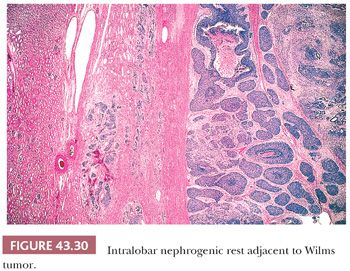
ILNRs are most often found at the tumor-kidney interface, where they can be misinterpreted as infiltrating tumor cells or effaced by tumor compression. A helpful feature distinguishing ILNRs at the edge of a WT is the poorly defined, irregular outer border of the ILNR, which contrasts with the sharp, pushing interface between the WT and the rest within which it arose (Fig. 43.30).
The presence of NRs in a kidney removed for WT is correlated with an increased risk for subsequent tumor formation in the remaining kidney (40). The type of rest and age of the patient modify this risk (44). When a carefully sampled kidney is free of rests, the risk of this unfortunate event is extremely low. The possibility of subsequent WT developing in the remaining kidney should be considered in planning the followup of patients whose nephrectomy specimen reveals the presence of NRs in addition to WT. Of note, a higher percentage of WTs arising from nephroblastomatosis are anaplastic (42).
DIFFERENTIAL DIAGNOSIS OF WILMS TUMOR
Triphasic Wilms Tumor
This pattern rarely presents a problem in diagnosis, except when small biopsy specimens are obtained from large retroperitoneal masses of uncertain origin. In this setting, other mixed neoplasms might deserve consideration, including teratoma, hepatoblastoma, pancreatoblastoma, teratoma, mesothelioma, synovial sarcoma, and intra-abdominal desmoplastic small round cell tumor (45,46). In the absence of nephrogenic differentiation or the distinctive blastemal aggregation patterns cited earlier, ancillary studies such as immunohistochemistry or electron microscopy may be required to distinguish some of these lesions from WT. Molecular detection of the distinctive EWSWT1 gene fusion of desmoplastic small round cell tumor (47) or the SYTSSX gene fusion of synovial sarcoma (48) can establish these diagnoses. Epithelioid morphology in clear cell sarcoma of the kidney presents another consideration in the differential diagnosis, as discussed later in this chapter.
WT with extensive heterologous differentiation (“teratoid WT”) is easily confused with immature teratoma. Renal teratomas are extremely rare, and some of the reported cases represent teratoid WT. Teratomas are characterized by heterologous organoid differentiation (recognizable differentiation into organs or body parts other than the one in which the tumor arises, such as gut wall or trachea). WT, like other mixed tumors, may contain one or more cell types found in another organ, but these have histioid differentiation, characterized by random juxtaposition of various cell types, without the consistent structural organization characteristic of developing or mature organs.
Blastemal Wilms Tumor versus Other Small Blue Cell Tumors of Childhood
This problem is most likely to arise when dealing with biopsy specimens from metastatic sites or from large abdominal tumors of uncertain origin. The distinct aggregation patterns of blastemal WT, the presence of nuclear molding, or the focal presence of tubular differentiation will often reveal the diagnosis. Early tubular differentiation in WT lacks lumens and therefore can sometimes be confused with rosettes of neuroblastoma or PNET. However, early tubular structures usually have cells neatly aligned to form a single layer around the future lumen, whereas neuroblastic rosettes are aggregated more randomly around the central zone and are less consistently aligned. The presence of lumens even focally confirms tubular differentiation, whereas neurofibrils are diagnostic of a neuroblastic pseudorosette. Neuroblastic rosettes do rarely occur in WT but usually only in teratoid WTs, which are not likely to be confused with neuroblastomas or PNETs. Immunostains, electron microscopy, molecular diagnostic techniques, cytogenetics, circulating tumor markers, and other special studies may sometimes be required to confirm the nature of a small blue cell tumor.
Epithelial-Predominant Wilms Tumor versus Papillary Renal Cell Carcinoma
This dilemma is most often encountered in tumors from adolescents or adults and is sometimes difficult to resolve with certainty. The papillary adenomas may resemble epithelial-predominant NRs, and epithelialpredominant WTs can have a predominantly papillary architecture. Sometimes, papillary renal cell carcinomas have a predominant tubular or solid component (49). Molecular or cytogenetic studies may be helpful because papillary renal cell carcinoma characteristically contains increased copies of chromosomes 7 and 17 and, in tumors from male patients, deletion of Y (50,51). Unequivocal glomerular differentiation, characteristic blastemal aggregation patterns, or the presence of heterologous cell types can confirm the diagnosis of WT. Positive staining for cytokeratin 7 is a useful marker for papillary renal cell carcinoma (52), but we have encountered focal positive staining for this epitope in many WTs. Nuclear labeling for WT1 protein may distinguish WT, NRs, and metanephric adenoma from papillary renal cell carcinoma (53) because, of these four lesions, only the latter does not label.
Epithelial Wilms Tumor versus Conventional (Clear Cell) Renal Carcinoma
Composite tumors with regions of both WT and clear cell renal carcinoma have been reported (54). Our experience with these composite neoplasms suggests that the nuclear grade of the carcinomatous component has the same significance as in conventional clear cell renal carcinomas. Those WTs containing grade I or II clear cell renal carcinomas will generally behave as WTs, whereas higher grade carcinomatous elements are more likely to have carcinomatous metastases and resistant tumor biology. A limited biopsy from one of these composite tumors may reveal only one component, leading to an apparent diagnostic discrepancy when subsequent specimens reveal the other component.
Stromal-Predominant Wilms Tumor versus Congenital Mesoblastic Nephroma and Sarcomatous Renal Tumors
This topic is discussed in the sections on these entities.
CONGENITAL MESOBLASTIC NEPHROMA
Congenital mesoblastic nephroma (CMN) is a distinctive infantile renal neoplasm composed of monomorphous spindle cells (55,56). Although it comprises only 2% to 3% of pediatric renal neoplasms, CMN is the most common renal neoplasm in the first 3 months of life. Sixty-two percent are diagnosed in the first 3 months of life, and the median age is 2 months. Ninety percent are diagnosed in the first year. Cases reported as CMNs of adults and adolescents are, in our opinion, unconvincing examples of this neoplasm, and this diagnosis should be considered suspect in any patient beyond the second year of life. CMN is generally associated with good outcome, and most cases are treated by nephrectomy alone. However, recurrences and/or metastases do occur in approximately 5% of cases. Cellular histology, stage, and vascular invasion predict relapse (57).
In recent years, a substantial proportion of the CMNs have been detected during fetal sonography. Hydramnios is common during gestation, and nonimmunologic fetal hydrops has been reported. Hyperreninism, hypercalcemia, and coagulopathy or shock from tumor rupture during delivery also have been reported in some cases.
GROSS APPEARANCE
CMN arises unicentrically and usually appears to have arisen deep within the parenchyma near the renal sinus. The renal sinus and adjacent structures on the medial side of the kidney are major sites of extrarenal spread of CMN. Surgeons and pathologists must pay particular attention to the medial margin of the resection specimen in dealing with potential CMN specimens. This is no simple matter. The medial specimen margin is notoriously difficult to evaluate, and it is rarely possible to be certain that it is free of involvement by CMN.
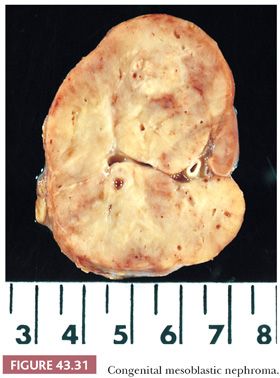
The appearance of the sectioned surface is variable. Some specimens (particularly classic CMN) have a tough, whorled appearance resembling leiomyoma, but cellular CMNs are more frequently soft, friable tumors (Fig. 43.31). Hemorrhage, necrosis, and cyst formation are present to some degree in a majority of specimens. These findings are too often present in cases with good outcome to serve as useful markers of adverse prognosis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


