Regulation of Renal Function and Vascular Volume
RENAL ANATOMY AND PHYSIOLOGY
The basic urine-forming unit of the kidney is the nephron, which consists of a filtering apparatus, the glomerulus, connected to a long tubular portion that reabsorbs and conditions the glomerular ultrafiltrate. Each human kidney is composed of ~1 million nephrons. Figure 25–1 illustrates subdivisions of the nephron.
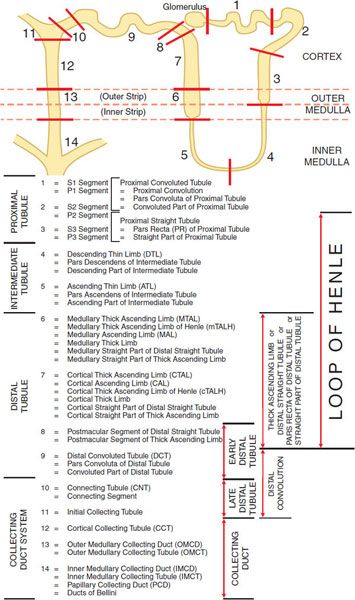
Figure 25–1 Anatomy and nomenclature of the nephron.
GLOMERULAR FILTRATION. In the glomerular capillaries, a portion of plasma water is forced through a filter that has 3 basic components: the fenestrated capillary endothelial cells, a basement membrane lying just beneath the endothelial cells, and the filtration slit diaphragms formed by epithelial cells that cover the basement membrane on its urinary space side. Solutes of small size flow with filtered water (solvent drag) into the urinary (Bowman’s) space, whereas formed elements and macromolecules are retained by the filtration barrier.
OVERVIEW OF NEPHRON FUNCTION. The kidney filters large quantities of plasma, reabsorbs substances that the body must conserve, and leaves behind or secretes substances that must be eliminated. The changing architecture and cellular differentiation along the length of a nephron is crucial to these functions (see Figure 25–1). The 2 kidneys in humans produce together ~120 mL of ultrafiltrate per minute, yet only 1 mL/min of urine is produced. Therefore, >99% of the glomerular ultrafiltrate is reabsorbed at a staggering energy cost. The kidneys consume 7% of total-body oxygen intake despite the fact that the kidneys make up only 0.5% of body weight.
The proximal tubule is contiguous with Bowman’s capsule and takes a tortuous path until finally forming a straight portion that dives into the renal medulla. Normally, ~65% of filtered Na+ is reabsorbed in the proximal tubule, and since this part of the tubule is highly permeable to water, reabsorption is essentially isotonic. Between the outer and inner strips of the outer medulla, the tubule abruptly changes morphology to become the descending thin limb (DTL), which penetrates the inner medulla, makes a hairpin turn, and then forms the ascending thin limb (ATL). At the juncture between the inner and outer medulla, the tubule once again changes morphology and becomes the thick ascending limb (TAL, with 3 segments noted in Figure 25–1). Together the proximal straight tubule, DTL, ATL, and TAL segments are known as the loop of Henle.
The DTL is highly permeable to water, yet its permeability to NaCl and urea is low. In contrast, the ATL is permeable to NaCl and urea but is impermeable to water. The TAL actively reabsorbs NaCl but is impermeable to water and urea. Approximately 25% of filtered Na+ is reabsorbed in the loop of Henle, mostly in the TAL, which has a large reabsorptive capacity. The TAL passes between the afferent and efferent arterioles and makes contact with the afferent arteriole by means of a cluster of specialized columnar epithelial cells known as the macula densa. The macula densa is strategically located to sense concentrations of NaCl leaving the loop of Henle. If the concentration of NaCl is too high, the macula densa sends a chemical signal (perhaps adenosine or ATP) to the afferent arteriole of the same nephron, causing it to constrict, thereby reducing the glomerular filtration rate (GFR). This homeostatic mechanism, known as tubuloglomerular feedback (TGF), protects the organism from salt and volume wasting. The macula densa also regulates renin release from the adjacent juxtaglomerular cells in the wall of the afferent arteriole.
Approximately 0.2 mm past the macula densa, the tubule changes morphology once again to become the distal convoluted tubule (DCT). Like the TAL, the DCT actively transports NaCl and is impermeable to water. Because these characteristics impart the ability to produce a dilute urine, the TAL and the DCT are collectively called the diluting segment of the nephron, and the tubular fluid in the DCT is hypotonic regardless of hydration status. However, unlike the TAL, the DCT does not contribute to the countercurrent-induced hypertonicity of the medullary interstitium (described below).
The collecting duct system (segments 10–14 in Figure 25–1) is an area of fine control of ultrafiltrate composition and volume. It is here that final adjustments in electrolyte composition are made, a process modulated by the adrenal steroid aldosterone. In addition, vasopressin (also called antidiuretic hormone [ADH]) modulates water permeability of this part of the nephron. The more distal portions of the collecting duct pass through the renal medulla, where the interstitial fluid is markedly hypertonic. In the absence of ADH, the collecting duct system is impermeable to water, and a dilute urine is excreted. In the presence of ADH, the collecting duct system is permeable to water, so water is reabsorbed. The movement of water out of the tubule is driven by the steep concentration gradient that exists between tubular fluid and medullary interstitium.
The hypertonicity of the medullary interstitium plays a vital role in the ability of mammals and birds to concentrate urine, which is accomplished by a combination of the unique topography of the loop of Henle and the specialized permeability features of the loop’s subsegments. The “passive countercurrent multiplier hypothesis” proposes that active transport in the TAL concentrates NaCl in the interstitium of the outer medulla. Because this segment of the nephron is impermeable to water, active transport in the ascending limb dilutes the tubular fluid. As the dilute fluid passes into the collecting-duct system, water is extracted if, and only if, ADH is present. Since the cortical and outer medullary collecting ducts have a low permeability to urea, urea is concentrated in the tubular fluid. The inner medullary collecting duct, however, is permeable to urea, so urea diffuses into the inner medulla, where it is trapped by countercurrent exchange in the vasa recta. Because the DTL is impermeable to salt and urea, the high urea concentration in the inner medulla extracts water from the DTL and concentrates NaCl in the tubular fluid of the DTL. As the tubular fluid enters the ATL, NaCl diffuses out of the salt-permeable ATL, thus contributing to the hypertonicity of the medullary interstitium.
GENERAL MECHANISM OF RENAL EPITHELIAL TRANSPORT. There are multiple mechanisms by which solutes may cross cell membranes (see Figure 5–4). The kinds of transport achieved in a nephron segment depend mainly on which transporters are present and whether they are embedded in the luminal or basolateral membrane. Figure 25–2 presents a general model of renal tubular transport that be summarized as follows:
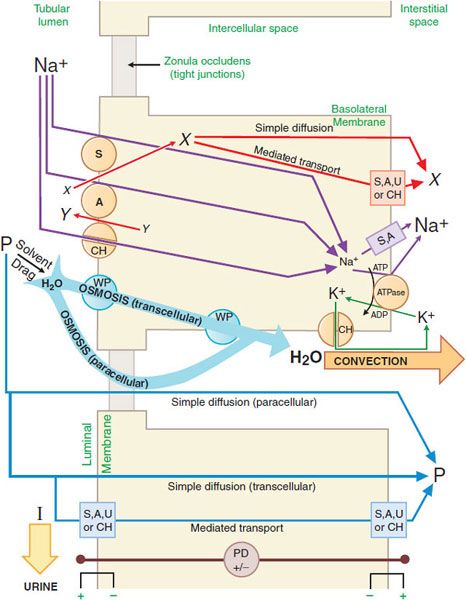
Figure 25–2 Generic mechanism of renal epithelial cell transport (see text for details). S, symporter; A, antiporter; CH, ion channel; WP, water pore; U, uniporter; ATPase, Na+, K+-ATPase (sodium pump); X and Y, transported solutes; P, membrane-permeable (reabsorbable) solutes; I, membrane-impermeable (nonreabsorbable) solutes; PD, potential difference across indicated membrane or cell.
1. Na+, K+-ATPase (sodium pump) in the basolateral membrane transports Na+ into the intercellular and interstitial spaces and K+ into the cell, establishing an electrochemical gradient for Na+ across the cell membrane directed inward.
2. Na+ can diffuse down this Na+ gradient across the luminal membrane via Na+ channels and via membrane symporters that use the energy stored in the Na+ gradient to transport solutes out of the tubular lumen and into the cell (e.g., Na+-glucose, 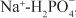 , and Na+-amino acid) and antiporters (e.g., Na+-H+) that move solutes into the lumen as Na+ moves out of the tubular lumen and into the cell.
, and Na+-amino acid) and antiporters (e.g., Na+-H+) that move solutes into the lumen as Na+ moves out of the tubular lumen and into the cell.
3. Na+ exits the basolateral membrane into intercellular and interstitial spaces via the Na+ pump.
4. The action of Na+-linked symporters in the luminal membrane causes the concentration of substrates for these symporters to rise in the epithelial cell. These substrate/solute gradients then permit simple diffusion or mediated transport (e.g., symporters, antiporters, uniporters, and channels) of solutes into the intercellular and interstitial spaces.
5. Accumulation of Na+ and other solutes in the intercellular space creates a small osmotic pressure differential across the epithelial cell. In water-permeable epithelium, water moves into the intercellular spaces driven by the osmotic pressure differential. Water moves through aqueous pores in both the luminal and the basolateral cell membranes, as well as through tight junctions (paracellular pathway). Bulk water flow carries some solutes into the intercellular space by solvent drag.
6. Movement of water into the intercellular space concentrates other solutes in the tubular fluid, resulting in an electrochemical gradient for these substances across the epithelium. Membrane-permeable solutes then move down their electrochemical gradients into the intercellular space by both the trans-cellular (e.g., simple diffusion, symporters, antiporters, uniporters, and channels) and paracellular pathways. Membrane-impermeable solutes remain in the tubular lumen and are excreted in the urine with an obligatory amount of water.
7. As water and solutes accumulate in the intercellular space, hydrostatic pressure increases, thus providing a driving force for bulk water flow. Bulk water flow carries solute out of the intercellular space into the interstitial space and, finally, into the peritubular capillaries.
ORGANIC ACID AND ORGANIC BASE SECRETION
The kidney is a major organ involved in the elimination of organic chemicals from the body. Organic molecules may enter the renal tubules by glomerular filtration or may be actively secreted directly into tubules. The proximal tubule has a highly efficient transport system for organic acids and an equally efficient but separate transport system for organic bases. Current models for these secretory systems are illustrated in Figure 25–3. Both systems are powered by the sodium pump in the basolateral membrane, involve secondary and tertiary active transport, and use a facilitated-diffusion step. There are at least 9 different organic acid and 5 different organic base transporters (see Chapter 5). A family of organic anion transporters (OATs) links countertransport of organic anions with dicarboxylates (Figure 25–3A).
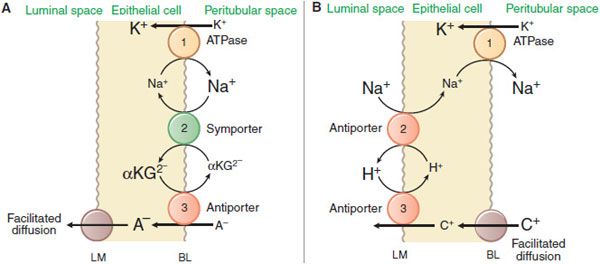
Figure 25–3 Mechanisms of organic acid (A) and organic base (B) secretion in the proximal tubule. The numbers 1, 2, and 3 refer to primary, secondary, and tertiary active transport. A–, organic acid [anion]; C+, organic base [cation]; αKG2-, α-ketoglutarate but also other dicarboxylates. BL and LM indicate basolateral and luminal membranes, respectively.
RENAL HANDLING OF SPECIFIC ANIONS AND CATIONS
Reabsorption of Cl– generally follows reabsorption of Na+. In segments of the tubule with low-resistance tight junctions (i.e., “leaky” epithelium), such as the proximal tubule and TAL, Cl– movement can occur paracellularly. Cl– crosses the luminal membrane by antiport with formate and oxalate (proximal tubule), symport with Na+/K+ (TAL), symport with Na+ (DCT), and antiport with 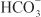 (collecting-duct system). Cl– crosses the basolateral membrane by symport with K+ (proximal tubule and TAL), antiport with Na+/
(collecting-duct system). Cl– crosses the basolateral membrane by symport with K+ (proximal tubule and TAL), antiport with Na+/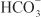 (proximal tubule), and Cl– channels (TAL, DCT, collecting-duct system).
(proximal tubule), and Cl– channels (TAL, DCT, collecting-duct system).
Eighty to ninety percent of filtered K+ is reabsorbed in the proximal tubule (diffusion and solvent drag) and TAL (diffusion), largely through the paracellular pathway. The DCT and collecting-duct system secrete variable amounts of K+ by a channel-mediated pathway. Modulation of the rate of K+ secretion in the collecting-duct system, particularly by aldosterone, allows urinary K+ excretion to be matched with dietary intake. The transepithelial potential difference (VT), lumen-positive in the TAL and lumen-negative in the collecting-duct system, drives K+ reabsorption and secretion, respectively.
Most of the filtered Ca2+ (~70%) is reabsorbed by the proximal tubule by passive diffusion through a paracellular route. Another 25% of filtered Ca2+ is reabsorbed by the TAL in part by a paracellular route driven by the lumen-positive VT and in part by active transcellular Ca2+ reabsorption modulated by parathyroid hormone (PTH; see Chapter 44). Most of the remaining Ca2+ is reabsorbed in DCT by a transcellular pathway. The transcellular pathway in the TAL and DCT involves passive Ca2+ influx across the luminal membrane through Ca2+ channels (TRPV5), followed by Ca2+ extrusion across the basolateral membrane by a Ca2+-ATPase. Also, in DCT and CNT, Ca2+ crosses the basolateral membrane by Na+-Ca2+ exchanger (antiport). Inorganic phosphate (Pi) is largely reabsorbed (80% of filtered load) by the proximal tubule. The Na+-Pi symporter uses the free energy of the Na+ electrochemical gradient to transport Pi into the cell. The Na+-Pi symporter is inhibited by PTH.
The renal tubules reabsorb 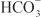 and secrete protons (tubular acidification), thereby participating in acid–base balance. These processes are described in the section on carbonic anhydrase inhibitors.
and secrete protons (tubular acidification), thereby participating in acid–base balance. These processes are described in the section on carbonic anhydrase inhibitors.
PRINCIPLES OF DIURETIC ACTION
Diuretics are drugs that increase the rate of urine flow; clinically useful diuretics also increase the rate of Na+ excretion (natriuresis) and of an accompanying anion, usually Cl–. Most clinical applications of diuretics are directed toward reducing extracellular fluid volume by decreasing total-body NaCl content.
Although continued diuretic administration causes a sustained net deficit in total-body Na+, the time course of natriuresis is finite because renal compensatory mechanisms bring Na+ excretion in line with Na+ intake, a phenomenon known as diuretic braking. These compensatory mechanisms include activation of the sympathetic nervous system, activation of the renin–angiotensin–aldosterone axis, decreased arterial blood pressure (which reduces pressure natriuresis), renal epithelial cell hypertrophy, increased renal epithelial transporter expression, and perhaps alterations in natriuretic hormones such as atrial natriuretic peptide. The net effects on extracellular volume and body weight are shown in Figure 25–4.
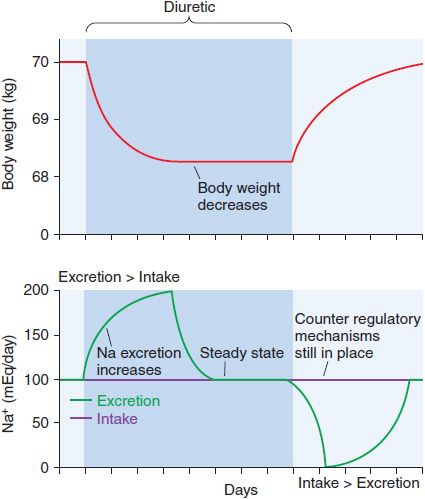
Figure 25–4 Changes in extracellular fluid volume and weight with diuretic therapy. The period of diuretic administration is shown in the shaded box along with its effects on body weight in the upper part of the figure and Na+ excretion in the lower half of the figure. Initially, when Na+ excretion exceeds intake, body weight and extracellular fluid volume (ECFV) decrease. Subsequently, a new steady state is achieved where Na+ intake and excretion are equal but at a lower ECFV and body weight. This results from activation of the renin-angiotensin-aldosterone system (RAAS) and sympathetic nervous system (SNS), “the braking phenomenon.” When the diuretic is discontinued, body weight and ECFV rise during a period where Na+ intake exceeds excretion. A new steady state is then reached as stimulation of the RAAS and SNS wane.
Diuretics may modify renal handling of other cations (e.g., K+, H+, Ca2+, and Mg2+), anions (e.g., Cl–, 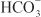 , and
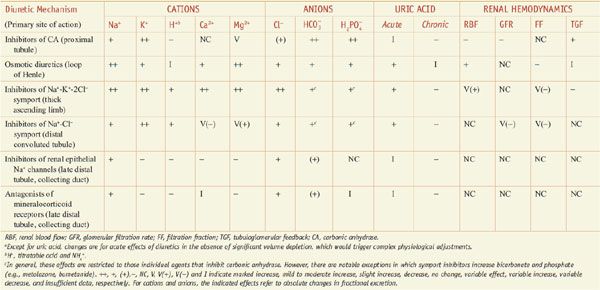
, and 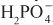 ), and uric acid. In addition, diuretics may alter renal hemodynamics indirectly. Table 25–1 gives a comparison of the general effects of the major diuretic classes.
), and uric acid. In addition, diuretics may alter renal hemodynamics indirectly. Table 25–1 gives a comparison of the general effects of the major diuretic classes.
Table 25–1
Excretory and Renal Hemodynamic Effects of Diureticsa
INHIBITORS OF CARBONIC ANHYDRASE
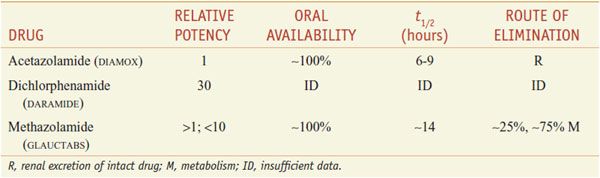
There are 3 orally administered carbonic anhydrase inhibitors—acetazolamide, dichlorphenamide (not marketed in the U.S.), and methazolamide (Table 25–2).
Table 25–2
Inhibitors of Carbonic Anhydrase
MECHANISM AND SITE OF ACTION. Proximal tubular epithelial cells are richly endowed with the zinc metalloenzyme carbonic anhydrase, which is found in the luminal and basolateral membranes (type IV carbonic anhydrase), as well as in the cytoplasm (type II carbonic anhydrase) (Figure 25–5). Carbonic anhydrase plays a role in NaHCO3 reabsorption and acid secretion.
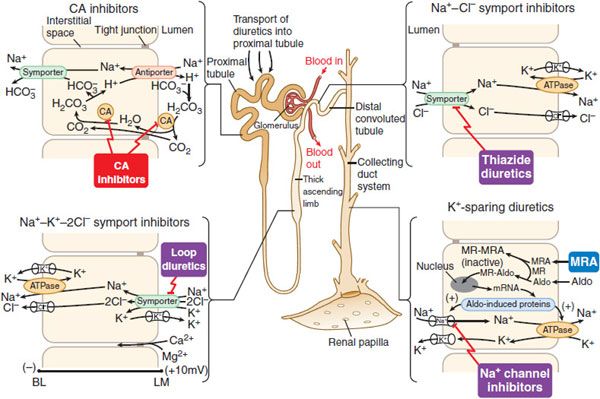
Figure 25–5 Sites and mechanisms of action of diuretics. Three important features are noteworthy:
In the proximal tubule, the free energy in the Na+ gradient established by the basolateral Na+ pump is used by a Na+-H+ antiporter (a.k.a. Na+-H+ exchanger [NHE]) in the luminal membrane to transport H+ into the tubular lumen in exchange for Na+. In the lumen, H+ reacts with filtered 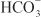 to form H2CO3, which decomposes rapidly to CO2 and water in the presence of carbonic anhydrase in the brush border. Carbonic anhydrase reversibly accelerates this reaction several thousand fold. CO2 is lipophilic and rapidly diffuses across the luminal membrane into the epithelial cell, where it reacts with water to form H2CO3, a reaction catalyzed by cytoplasmic carbonic anhydrase. Continued operation of the Na+-H+ antiporter maintains a low proton concentration in the cell, so H2CO3 ionizes spontaneously to form H+ and
to form H2CO3, which decomposes rapidly to CO2 and water in the presence of carbonic anhydrase in the brush border. Carbonic anhydrase reversibly accelerates this reaction several thousand fold. CO2 is lipophilic and rapidly diffuses across the luminal membrane into the epithelial cell, where it reacts with water to form H2CO3, a reaction catalyzed by cytoplasmic carbonic anhydrase. Continued operation of the Na+-H+ antiporter maintains a low proton concentration in the cell, so H2CO3 ionizes spontaneously to form H+ and 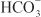 , creating an electrochemical gradient for
, creating an electrochemical gradient for 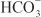 across the basolateral membrane. The electrochemical gradient for
across the basolateral membrane. The electrochemical gradient for 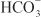 is used by a Na+–
is used by a Na+–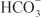 symporter (a.k.a. the Na+–
symporter (a.k.a. the Na+–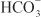 co-transporter [NBC]) in the basolateral membrane to transport NaHCO3 into the interstitial space. The net effect of this process is transport of NaHCO3 from the tubular lumen to the interstitial space, followed by movement of water (isotonic reabsorption). Removal of water concentrates Cl– in the tubular lumen, and consequently, Cl– diffuses down its concentration gradient into the interstitium by the paracellular pathway.
co-transporter [NBC]) in the basolateral membrane to transport NaHCO3 into the interstitial space. The net effect of this process is transport of NaHCO3 from the tubular lumen to the interstitial space, followed by movement of water (isotonic reabsorption). Removal of water concentrates Cl– in the tubular lumen, and consequently, Cl– diffuses down its concentration gradient into the interstitium by the paracellular pathway.
Carbonic anhydrase inhibitors potently inhibit both the membrane-bound and cytoplasmic forms of carbonic anhydrase, resulting in nearly complete abolition of NaHCO3 reabsorption in the proximal tubule. Because of the large excess of carbonic anhydrase in proximal tubules, a high percentage of enzyme activity must be inhibited before an effect on electrolyte excretion is observed. Although the proximal tubule is the major site of action of carbonic anhydrase inhibitors, carbonic anhydrase also is involved in secretion of titratable acid in the collecting duct system, which is a secondary site of action for this class of drugs.
EFFECTS ON URINARY EXCRETION. Inhibition of carbonic anhydrase is associated with a rapid rise in urinary 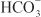 excretion to ~35% of filtered load. This, along with inhibition of titratable acid and NH4+ secretion in the collecting-duct system, results in an increase in urinary pH to ~8 and development of a metabolic acidosis. However, even with a high degree of inhibition of carbonic anhydrase, 65% of
excretion to ~35% of filtered load. This, along with inhibition of titratable acid and NH4+ secretion in the collecting-duct system, results in an increase in urinary pH to ~8 and development of a metabolic acidosis. However, even with a high degree of inhibition of carbonic anhydrase, 65% of 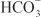 is rescued from excretion. The loop of Henle has a large reabsorptive capacity and captures most of the Cl– and a portion of the Na+. Thus, only a small increase in Cl– excretion occurs,
is rescued from excretion. The loop of Henle has a large reabsorptive capacity and captures most of the Cl– and a portion of the Na+. Thus, only a small increase in Cl– excretion occurs, 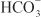 being the major anion excreted along with the cations Na+ and K+. The fractional excretion of Na+ may be as much as 5%, and the fractional excretion of K+ can be as much as 70%. The increased excretion of K+ is in part secondary to increased delivery of Na+ to the distal nephron, as described in the section on inhibitors of Na+ channels. The effects of carbonic anhydrase inhibitors on renal excretion are self-limiting, probably because the resulting metabolic acidosis decreases the filtered load of
being the major anion excreted along with the cations Na+ and K+. The fractional excretion of Na+ may be as much as 5%, and the fractional excretion of K+ can be as much as 70%. The increased excretion of K+ is in part secondary to increased delivery of Na+ to the distal nephron, as described in the section on inhibitors of Na+ channels. The effects of carbonic anhydrase inhibitors on renal excretion are self-limiting, probably because the resulting metabolic acidosis decreases the filtered load of 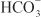 to the point that the uncatalyzed reaction between CO2 and water is sufficient to achieve
to the point that the uncatalyzed reaction between CO2 and water is sufficient to achieve 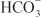 reabsorption.
reabsorption.
EFFECTS ON RENAL HEMODYNAMICS. By inhibiting proximal reabsorption, carbonic anhydrase inhibitors increase delivery of solutes to the macula densa. This triggers TGF, which increases afferent arteriolar resistance and reduces renal blood flow (RBF) and GFR.
OTHER ACTIONS. These agents have extrarenal sites of action. Carbonic anhydrase in the ciliary processes of the eye mediates formation of 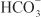 in aqueous humor. Inhibition of carbonic anhydrase decreases the rate of formation of aqueous humor and consequently reduces intraocular pressure. Acetazolamide frequently causes paresthesias and somnolence, suggesting an action of carbonic anhydrase inhibitors in the CNS. The efficacy of acetazolamide in epilepsy is due in part to the production of metabolic acidosis; however, direct actions of acetazolamide in the CNS also contribute to its anticonvulsant action. Owing to interference with carbonic anhydrase activity in erythrocytes, carbonic anhydrase inhibitors increase CO2 levels in peripheral tissues and decrease CO2 levels in expired gas. Acetazolamide causes vasodilation by opening vascular Ca2+-activated K+ channels; however, the clinical significance of this effect is unclear.
in aqueous humor. Inhibition of carbonic anhydrase decreases the rate of formation of aqueous humor and consequently reduces intraocular pressure. Acetazolamide frequently causes paresthesias and somnolence, suggesting an action of carbonic anhydrase inhibitors in the CNS. The efficacy of acetazolamide in epilepsy is due in part to the production of metabolic acidosis; however, direct actions of acetazolamide in the CNS also contribute to its anticonvulsant action. Owing to interference with carbonic anhydrase activity in erythrocytes, carbonic anhydrase inhibitors increase CO2 levels in peripheral tissues and decrease CO2 levels in expired gas. Acetazolamide causes vasodilation by opening vascular Ca2+-activated K+ channels; however, the clinical significance of this effect is unclear.
ABSORPTION AND ELIMINATION. See Table 25–2 for pharmacokinetic data.
TOXICITY, ADVERSE EFFECTS, CONTRAINDICATIONS, DRUG INTERACTIONS. Serious toxic reactions to carbonic anhydrase inhibitors are infrequent; however, these drugs are sulfonamide derivatives and, like other sulfonamides, may cause bone marrow depression, skin toxicity, sulfonamide-like renal lesions, and allergic reactions. With large doses, many patients exhibit drowsiness and paresthesias. Most adverse effects, contraindications, and drug interactions are secondary to urinary alkalinization or metabolic acidosis, including: (1) diversion of ammonia of renal origin from urine into the systemic circulation, a process that may induce or worsen hepatic encephalopathy (the drugs are contraindicated in patients with hepatic cirrhosis); (2) calculus formation and ureteral colic owing to precipitation of calcium phosphate salts in an alkaline urine; (3) worsening of metabolic or respiratory acidosis (the drugs are contraindicated in patients with hyperchloremic acidosis or severe chronic obstructive pulmonary disease); (4) reduction of the urinary excretion rate of weak organic bases.
THERAPEUTIC USES. The efficacy of carbonic anhydrase inhibitors as single agents is low. The combination of acetazolamide with diuretics that block Na+ reabsorption at more distal sites in the nephron causes a marked natriuretic response in patients with low basal fractional excretion of Na+ (<0.2%) who are resistant to diuretic monotherapy. Even so, the long-term usefulness of carbonic anhydrase inhibitors often is compromised by the development of metabolic acidosis. The major indication for carbonic anhydrase inhibitors is open-angle glaucoma. Two products developed specifically for this use are dorzolamide (TRUSOPT, others) and brinzolamide (AZOPT), which are available only as ophthalmic drops. Carbonic anhydrase inhibitors also may be employed for secondary glaucoma and preoperatively in acute angle-closure glaucoma to lower intraocular pressure before surgery (see Chapter 64). Acetazolamide also is used for the treatment of epilepsy (see Chapter 21). Acetazolamide can provide symptomatic relief in patients with high-altitude illness or mountain sickness. Acetazolamide also is useful in patients with familial periodic paralysis. The mechanism for the beneficial effects of acetazolamide in altitude sickness and familial periodic paralysis may be related to the induction of a metabolic acidosis. Finally, carbonic anhydrase inhibitors can be useful for correcting a metabolic alkalosis, especially one caused by diuretic-induced increases in H+ excretion.
OSMOTIC DIURETICS
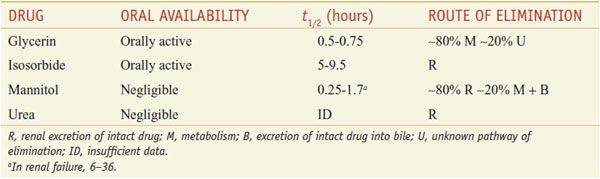
Osmotic diuretics are freely filtered at the glomerulus, undergo limited reabsorption by the renal tubule, and are relatively inert pharmacologically. Osmotic diuretics are administered in doses large enough to increase significantly the osmolality of plasma and tubular fluid. Table 25–3 lists 4 osmotic diuretics—glycerin (OSMOGLYN), isosorbide, mannitol (OSMITROL, others), and urea (currently not available in the U.S.).
Table 25–3
Osmotic Diuretics
MECHANISM AND SITE OF ACTION. Osmotic diuretics act both in proximal tubule and loop of Henle, with the latter being the primary site of action. By extracting water from intracellular compartments, osmotic diuretics expand extracellular fluid volume, decrease blood viscosity, and inhibit renin release. These effects increase RBF, and the increase in renal medullary blood flow removes NaCl and urea from the renal medulla, thus reducing medullary tonicity. A reduction in medullary tonicity causes a decrease in the extraction of water from the DTL, which in turn limits the concentration of NaCl in the tubular fluid entering the ATL. This latter effect diminishes the passive reabsorption of NaCl in the ATL. In addition osmotic diuretics inhibit Mg2+ reabsorption in the TAL.
EFFECTS ON URINARY EXCRETION. Osmotic diuretics increase urinary excretion of nearly all electrolytes, including Na+, K+, Ca2+, Mg2+, Cl–, 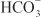 , and phosphate.
, and phosphate.
EFFECTS ON RENAL HEMODYNAMICS. Osmotic diuretics increase RBF by a variety of mechanisms, but total GFR is little changed.
ABSORPTION AND ELIMINATION. Pharmacokinetic data on the osmotic diuretics are gathered in Table 25–3. Glycerin and isosorbide can be given orally, whereas mannitol and urea must be administered intravenously.
TOXICITY, ADVERSE EFFECTS, CONTRAINDICATIONS, AND DRUG INTERACTIONS. Osmotic diuretics are distributed in the extracellular fluid and contribute to the extracellular osmolality. Thus, water is extracted from intracellular compartments, and the extracellular fluid volume becomes expanded. In patients with heart failure or pulmonary congestion, this may cause frank pulmonary edema. Extraction of water also causes hyponatremia, which may explain the common adverse effects, including headache, nausea, and vomiting. Conversely, loss of water in excess of electrolytes can cause hypernatremia and dehydration. Osmotic diuretics are contraindicated in patients who are anuric owing to severe renal disease. Urea may cause thrombosis or pain if extravasation occurs, and it should not be administered to patients with impaired liver function because of the risk of elevation of blood ammonia levels. Both mannitol and urea are contraindicated in patients with active cranial bleeding. Glycerin is metabolized and can cause hyperglycemia.
THERAPEUTIC USES. One use for mannitol is in the treatment of dialysis disequilibrium syndrome. Overly removing solutes from the extracellular fluid by hemodialysis results in a reduction in the osmolality of extracellular fluid. Consequently, water moves from the extracellular compartment into the intracellular compartment, causing hypotension and CNS symptoms (headache, nausea, muscle cramps, restlessness, CNS depression, and convulsions). Osmotic diuretics increase the osmolality of the extracellular fluid compartment and thereby shift water back into the extracellular compartment. By increasing the osmotic pressure of plasma, osmotic diuretics extract water from the eye and brain. All osmotic diuretics are used to control intraocular pressure during acute attacks of glaucoma and for short-term reductions in intraocular pressure both preoperatively and postoperatively in patients who require ocular surgery. Also, mannitol and urea are used to reduce cerebral edema and brain mass before and after neurosurgery.
INHIBITORS OF NA+-K+-2CL– SYMPORT (LOOP DIURETICS, HIGH-CEILING DIURETICS)
These diuretics inhibit activity of the Na+-K+-2Cl– symporter in the TAL of the loop of Henle, hence the moniker loop diuretics. Although the proximal tubule reabsorbs ~65% of filtered Na+, diuretics acting only in the proximal tubule have limited efficacy because the TAL has the capacity to reabsorb most of the rejectate from the proximal tubule. In contrast, inhibitors of Na+-K+-2Cl– symport in the TAL, sometimes called high-ceiling diuretics, are highly efficacious because (1) ~25% of the filtered Na+ load normally is reabsorbed by the TAL, and (2) nephron segments past the TAL do not possess the resorptive capacity to rescue the flood of rejectate exiting the TAL.
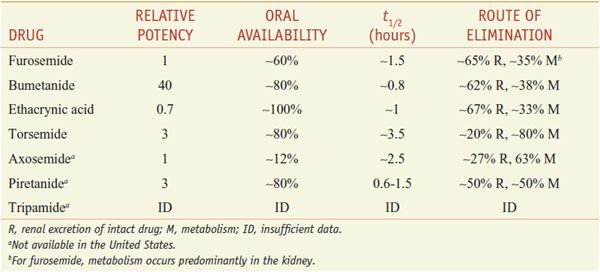
Of the inhibitors of Na+-K+-2Cl– symport (Table 25–4), only furosemide (LASIX), bumetanide (BUMEX), ethacrynic acid (EDECRIN), and torsemide (DEMADEX) are available currently in the U.S. Furosemide and bumetanide contain a sulfonamide moiety. Ethacrynic acid is a phenoxyacetic acid derivative; torsemide is a sulfonylurea. All loop diuretics except torsemide are available as oral and injectable formulations.
Table 25–4
Inhibitors of Na+–K+–2Cl– Symport (Loop Diuretics, High-Ceiling Diuretics)
MECHANISM AND SITE OF ACTION. These agents act primarily in the TAL, where the flux of Na+, K+, and Cl– from the lumen into epithelial cells is mediated by a Na+-K+-2Cl– symporter (Figure 25–5). Inhibitors of Na+-K+-2Cl– symport block its function, bringing salt transport in this segment of the nephron to a virtual standstill. Evidence suggests that these drugs attach to the Cl– binding site located in the symporter’s transmembrane domain. Inhibitors of Na+K+-2Cl– symport also inhibit Ca2+ and Mg2+ reabsorption in the TAL by abolishing the transepithelial potential difference that is the dominant driving force for reabsorption of these cations. Na+-K+-2Cl– symporters are found in many secretory and absorbing epithelia. The Na+-K+-2Cl– symporters are of 2 varieties. The “absorptive” symporter (called ENCC2, NKCC2, or BSCl) is expressed only in the kidney, is localized to the apical membrane and subapical intracellular vesicles of the TAL, and is regulated by cyclic AMP/PKA. The “secretory” symporter (called ENCC3, NKCCl, or BSC2) is a “housekeeping” protein that is expressed widely and, in epithelial cells, is localized to the basolateral membrane. The affinity of loop diuretics for the secretory symporter is somewhat less than for the absorptive symporter (e.g., 4-fold difference for bumetanide).
EFFECTS ON URINARY EXCRETION. Loop diuretics increase urinary Na+ and Cl– excretion profoundly (i.e., up to 25% of the filtered Na+ load) and markedly increase Ca2+ and Mg2+ excretion. Furosemide has weak carbonic anhydrase–inhibiting activity and thus increases urinary excretion of 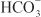 and phosphate. All inhibitors of Na+-K+-2Cl– symport increase urinary K+ and titratable acid excretion. This effect is due in part to increased Na+ delivery to the distal tubule (the mechanism by which increased distal Na+ delivery enhances K+ and H+ excretion is discussed in the section on Na+ channel inhibitors). Other mechanisms contributing to enhanced K+ and H+ excretion include flow-dependent enhancement of ion secretion by the collecting duct, nonosmotic vasopressin release, and activation of the renin–angiotensin–aldosterone axis.
and phosphate. All inhibitors of Na+-K+-2Cl– symport increase urinary K+ and titratable acid excretion. This effect is due in part to increased Na+ delivery to the distal tubule (the mechanism by which increased distal Na+ delivery enhances K+ and H+ excretion is discussed in the section on Na+ channel inhibitors). Other mechanisms contributing to enhanced K+ and H+ excretion include flow-dependent enhancement of ion secretion by the collecting duct, nonosmotic vasopressin release, and activation of the renin–angiotensin–aldosterone axis.
Acutely, loop diuretics increase uric acid excretion; their chronic administration results in reduced uric acid excretion. Chronic effects of loop diuretics on uric acid excretion may be due to enhanced proximal tubule transport or secondary to volume depletion or to competition between diuretic and uric acid for the organic acid secretory mechanism in proximal tubule. Asymptomatic hyperuricemia is a common consequence of loop diuretics, but painful episodes of gout are rarely reported. By blocking active NaCl reabsorption in the TAL, inhibitors of Na+-K+-2Cl– symport interfere with a critical step in the mechanism that produces a hypertonic medullary interstitium. Therefore, loop diuretics block the kidney’s ability to concentrate urine. Also, because the TAL is part of the diluting segment, inhibitors of Na+-K+-2Cl– symport markedly impair the kidney’s ability to excrete a dilute urine during water diuresis.
EFFECTS ON RENAL HEMODYNAMICS. If volume depletion is prevented by replacing fluid losses, inhibitors of Na+-K+-2Cl– symport generally increase total RBF and redistribute RBF to the midcortex. The mechanism of the increase in RBF is not known, but may involve prostaglandins: nonsteroidal anti-inflammatory drugs (NSAIDs) attenuate the diuretic response to loop diuretics in part by preventing prostaglandin-mediated increases in RBF. Loop diuretics block TGF by inhibiting salt transport into the macula densa so that the macula densa no longer can detect NaCl concentrations in the tubular fluid. Therefore, unlike carbonic anhydrase inhibitors, loop diuretics do not decrease GFR by activating TGF. Loop diuretics are powerful stimulators of renin release. This effect is due to interference with NaCl transport by the macula densa and, if volume depletion occurs, to reflex activation of the sympathetic nervous system and stimulation of the intrarenal baroreceptor mechanism.
OTHER ACTIONS. Loop diuretics, particularly furosemide, acutely increase systemic venous capacitance and thereby decrease left ventricular filling pressure. This effect, which may be mediated by prostaglandins and requires intact kidneys, benefits patients with pulmonary edema even before diuresis ensues. High doses of inhibitors of Na+-K+-2Cl– symport can inhibit electrolyte transport in many tissues, but this effect is clinically important only in the inner ear.
ABSORPTION AND ELIMINATION. Table 25–4 presents some pharmacokinetic properties of the agents. Because these drugs are bound extensively to plasma proteins, delivery of these drugs to the tubules by filtration is limited. However, they are secreted efficiently by the organic acid transport system in the proximal tubule, and thereby gain access to the Na+-K+-2Cl– symporter in the luminal membrane of the TAL. Approximately 65% of furosemide is excreted unchanged in urine, and the remainder is conjugated to glucuronic acid in the kidney. Thus, in patients with renal disease, the elimination t1/2 of furosemide is prolonged. Bumetanide and torsemide have significant hepatic metabolism, so liver disease can prolong the elimination t1/2 of these loop diuretics. Oral bioavailability of furosemide varies (10-100%). In contrast, oral availabilities of bumetanide and torsemide are reliably high. Heart failure patients have fewer hospitalizations and better quality of life with torsemide than with furosemide, perhaps because of its more reliable absorption.
As a class, loop diuretics have short elimination half lives; prolonged-release preparations are not available. Consequently, often the dosing interval is too short to maintain adequate levels of loop diuretics in the tubular lumen. Note that torsemide has a longer t1/2 than other agents available in the U.S. As the concentration of loop diuretic in the tubular lumen declines, nephrons begin to avidly reabsorb Na+, which often nullifies the overall effect of the loop diuretic on total-body Na+. This phenomenon of “postdiuretic Na+ retention” can be overcome by restricting dietary Na+ intake or by more frequent administration of the loop diuretic.
TOXICITY, ADVERSE EFFECTS, CONTRAINDICATIONS, DRUG INTERACTIONS. Most adverse effects are due to abnormalities of fluid and electrolyte balance. Overzealous use of loop diuretics can cause serious depletion of total-body Na+. This may manifest as hyponatremia and/or extracellular fluid volume depletion associated with hypotension, reduced GFR, circulatory collapse, thromboembolic episodes, and in patients with liver disease, hepatic encephalopathy. Increased Na+ delivery to the distal tubule, particularly when combined with activation of the renin–angiotensin system, leads to increased urinary K+ and H+ excretion, causing a hypochloremic alkalosis. If dietary K+ intake is not sufficient, hypokalemia may develop, and this may induce cardiac arrhythmias, particularly in patients taking cardiac glycosides. Increased Mg2+ and Ca2+ excretion may result in hypomagnesemia (a risk factor for cardiac arrhythmias) and hypocalcemia (rarely leading to tetany). Loop diuretics should be avoided in postmenopausal osteopenic women, in whom increased Ca2+ excretion may have deleterious effects on bone metabolism.
Loop diuretics can cause ototoxicity that manifests as tinnitus, hearing impairment, deafness, vertigo, and a sense of fullness in the ears. Hearing impairment and deafness are usually, but not always, reversible. Ototoxicity occurs most frequently with rapid intravenous administration and least frequently with oral administration. Ethacrynic acid appears to induce ototoxicity more often than do other loop diuretics and should be reserved for use only in patients who cannot tolerate other loop diuretics. Loop diuretics also can cause hyperuricemia (occasionally leading to gout) and hyperglycemia (infrequently precipitating diabetes mellitus) and can increase plasma levels of LDL cholesterol and triglycerides while decreasing plasma levels of HDL cholesterol. Other adverse effects include skin rashes, photosensitivity, paresthesias, bone marrow depression, and GI disturbances. Contraindications to the use of loop diuretics include severe Na+ and volume depletion, hypersensitivity to sulfonamides (for sulfonamide-based loop diuretics), and anuria unresponsive to a trial dose of loop diuretic.
Drug interactions may occur when loop diuretics are coadministered with:
• Aminoglycosides, carboplatin, paclitaxel, and others (synergism of ototoxicity)
• Anticoagulants (increased anticoagulant activity)
• Digitalis glycosides (increased digitalis-induced arrhythmias)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


