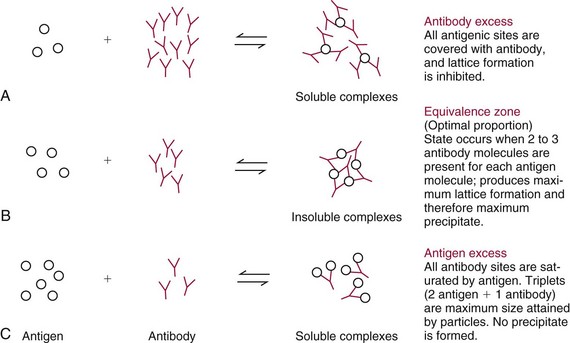
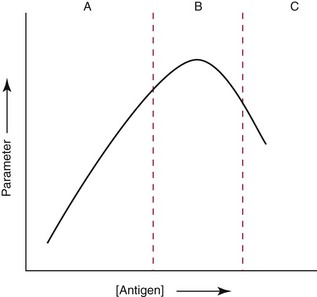
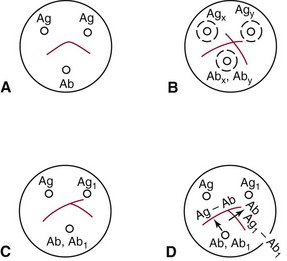
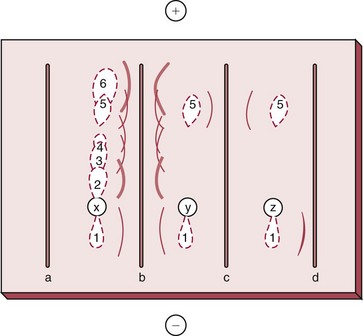
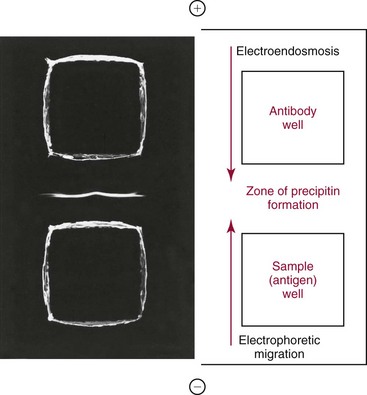
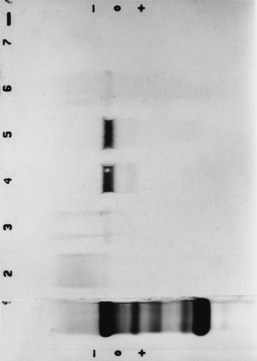
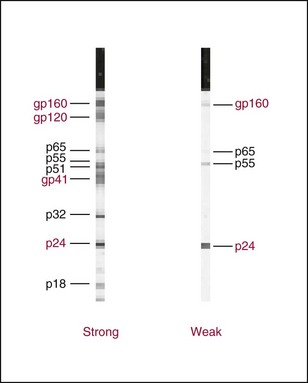
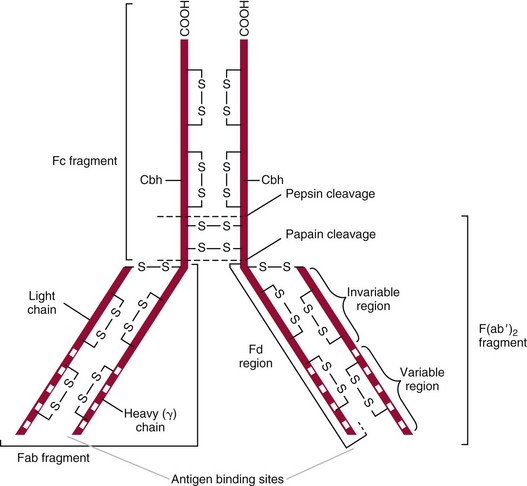
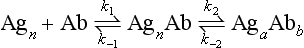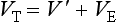Chapter 16 Larry J. Kricka, D. Phil., F.A.C.B., C. Chem., F.R.S.C., F.R.C.Path. and Jason Y. Park, M.D., Ph.D., F.A.C.P.* Antibodies are immunoglobulins that are capable of binding specifically to a wide array of natural and synthetic antigens, including proteins, carbohydrates, nucleic acids, lipids, and other molecules. Immunoglobulins consist of five general classes designated as immunoglobulin (Ig)G, IgA, IgM, IgD, and IgE. IgG is used most commonly in immunochemical reagents. A schematic diagram of the IgG molecule is shown in Figure 16-1. IgG is a glycoprotein [molecular weight (MW), 158,000 Da] composed of two heavy (γ) and two light (κ or λ) chains joined by disulfide bonds. Each chain (H or L) is the product of three (L) or four (H) distinct gene segments. These are the constant (C), joining (J), diversity (D), and variable (V) genes that undergo combinatorial joining during B-cell development. Several hundred germline V genes, 5 to 10 J genes, 15 D genes (H chain only), and a single C gene have been identified for each heavy or light chain class. During B-cell development, the V, D, and J (H chain) or V and J (L chain) undergo random rearrangement and splicing, and this recombined product then is spliced to the constant region gene. This combinatorial diversity, along with somatic mutations that occur at the splicing sites, generates a tremendous diversity of antibody specificities. When a B-cell clone expressing a particular antibody specificity on its surface is “selected” by an antigen, it expands and differentiates into a plasma cell that secretes the specific antibody. For more information on immunoglobulins, see Chapter 21. Polyclonal antiserum is raised in a normal animal host in response to immunogen administration. In contrast, monoclonal antibodies are produced in a very different manner and represent the product of a single clone or plasma cell line, rather than a heterogeneous mixture of antibodies produced by many plasma cell clones in response to immunization. Monoclonal antibodies are now widely used as reagents in immunoassay techniques.44,59 The usual method of production of monoclonal antibodies involves fusing antibody-producing plasma cells from the spleens or lymph nodes of mice that have been immunized several times over a 2- to 4-week period with a murine myeloma cell line from tissue culture in the presence of polyethylene glycol (PEG)(PEG promotes membrane and thus cell fusion through an unknown mechanism). The murine myeloma cell line is an immortalized plasma cell line. The murine myeloma cell lines most commonly used are deficient in the enzyme hypoxanthine guanine phosphoribosyl transferase (HGPRT) and therefore cannot synthesize purine bases from thymidine and hypoxanthine in the presence of aminopterin. Following fusion, the cells are placed into a selection medium containing hypoxanthine, aminopterin, and thymidine (HAT medium) to grow fused hybrid cell lines selectively. The fused hybrid cells can survive in a HAT medium, because these cells combine the immortality of the myeloma cell with the genetic material of the splenic plasma cell necessary for synthesis of thymidine in the presence of aminopterin. The unfused myeloma cells are killed by the HAT medium because they do not have the HGPRT gene, and the unfused spleen cells cannot be maintained in the culture. Colonies arising from the fused cells are then screened for antibody production, and cell lines secreting antibody of the desired specificity are cloned in subcultures. In this way, a single clonal line can be isolated that produces an antibody with a selected specificity and affinity for a single antigen. Phage display technology provides a new in vitro approach for producing antibodies (single chain Fv fragments, Fab fragments, and whole antibody molecules) that mimic the immune system but do not require B-cell immortalization.15,89 V genes coding for the heavy and light chain variable domains of immunoglobulin isolated from lymphocytes are amplified by the polymerase chain reaction and ligated into a filamentous bacteriophage vector to form combinatorial libraries of VH and VL genes. Individual bacteriophages display copies of a specific antibody on their surface, and the phage library can be screened for the antibody of a defined specificity using immobilized antigen (“panning”). Large libraries displaying antibodies formed from more than 1012 different VH and VL combinations can be constructed; this provides a rich source of antibodies with binding constants of 108 to 109 L/mol. Soluble antibody from selected bacteriophages can be secreted from infected bacteria (e.g., Escherichia coli) in yields greater than 500 mg/L. The strength of the binding of an antigen to an antibody depends on several forces acting cooperatively. These include van der Waals-London dipole-dipole interaction, hydrophobic interaction, and ionic coulombic bonding.42 Hydrophobic interactions result because the association of nonpolar groups is energetically favored in aqueous or other polar solutions. In proteins, hydrophobic interactions serve to bend and fold a molecule in a way that brings nonpolar R groups inside to the less polar interior; polar R groups are oriented outside toward the more polar aqueous environment. Thus hydrophobic bonding forms an interior, hydrophobic protein core, where most hydrophobic side chains can closely associate and weakly bind. Hydrophobic interaction enhances or stabilizes antigen-antibody binding but is not necessarily the major force in such binding.76 Given these forces, one would predict that changing pH, temperature, and ionic strength of the reaction medium should influence the binding of antigen and antibody. However, given a lower and upper limit of pH of 6 and 8 and an incubation temperature between 25 °C and 35 °C, these variables have only minimal effect on the rate of association and immune complex formation.58,78 In fact, extremes in pH (less than 4.0 and greater than 8.0) can cause inhibition of binding or dissociation of already formed antigen-antibody complexes. Changes in ionic strength will produce a significant effect on the rate of binding of antigen and antibody. This concept is studied further in the following sections. Cationic salts produce an inhibition of the binding of antibody with a cationic hapten.30 The order of inhibition by various cations is Cs+ > Rb+ > In general, the solubility of a protein in the presence of different linear polymers is inversely proportional to the molecular weight of the polymer (i.e., the higher the molecular weight of the polymer, the lower is the solubility of the protein). For example, in the presence of Dextran 500, the solubility of α-crystalline < fibrinogen < γ-globulin < albumin <<< tyrosine.52,53 Laurent thus proposed a steric exclusion mechanism to explain the effects of polymers on protein solubility.53 Assuming a fixed total volume (VT) of solvent being occupied by both polymer and protein and defining the volume occupied by polymer as VE (excluded volume, i.e., volume not accessible to proteins) and the volume occupied by protein as V′, then the relation Studies by Hellsing34,35 have provided support for the steric exclusion model and have demonstrated that (1) the composition of the immune complex formed is not affected by the presence of a polymer; (2) no complex is formed between the polymer and the antigen, antibody, or immune complex; (3) the polymer effect is dependent on the molecular weight of both antigen and polymer; and (4) the use of polymer in a reaction mixture can increase the precipitation of an immune complex with low-avidity antibody. Addition of polymer to a mixture of antigen and antibody causes a notable increase in the rate of immune complex growth, especially during the early phase of the reaction. Numerous polymer species have been tested (Box 16-1) for applications in immunochemical methods. The most desirable characteristics of the polymer are high molecular weight, a high degree of linearity (minimal branching), and high aqueous solubility. Most investigators have found PEG 6000 in concentrations of 3 to 5 g/dL to be most useful. If the number of antibody-combining sites is notably greater than the antigen-epitope sites ([Ab] >>> [Ag]), then antigen-binding sites are quickly saturated by antibodies before cross-linking can occur, along with the formation of small insoluble antigen-antibody complexes (Figure 16-2, A). When an antibody is in moderate excess (i.e., [Ab] > [Ag]), the probability of cross-linking of Ag by Ab is more likely, and hence large insoluble complex formation is favored (Figure 16-2, B). When [Ag] is in great excess, large complexes would be less probable (Figure 16-2, C). This model describes the results observed when antigens and antibodies are mixed in various concentration ratios. The curve shown in Figure 16-3 is a schematic diagram of the classical precipitin curve. Although the concentration of total antibody is constant, the concentration of free antibody [Ab]f (i.e., not bound to antigen) and free antigen [Ag]f varies throughout the range for any given Ag/Ab ratio. A low Ag/Ab ratio exists in A of Figure 16-3 (zone of antibody excess). Under these conditions, [Ab]f exists in solution, but [Ag]f does not. As total antigen increases, the size of the immune complexes increases up to equivalence (Figure 16-3, B), where little or no [Ab]f or [Ag]f exists. This is the zone of maximum immune complex size. This equivalence zone does not represent a ratio of exact molar equivalence of reactants but is the optimal combining ratio for cross-linking in the particular system under evaluation. As Ag/Ab increases (Figure 16-3, C), the immune complex size will decrease and [Ag]f will increase (zone of antigen excess). No [Ab]f should exist in this area of the curve. However, for a given Ag/Ab ratio, the population of immune complexes formed at equilibrium will be heterogeneous with respect to size and composition. The simplest method uses an agar dish or slide with holes cut as shown in Figure 16-4. When the same antigen is in adjacent wells, the lines of precipitation fuse and are continuous—this is a reaction of identity (Figure 16-4, A). When the precipitin bands cross each other, this is a reaction of nonidentity (Figure 16-4, B); if the two antigens are related but are not identical, a reaction of partial identity is observed (Figure 16-4, C). Here the cardinal point is that the precipitate serves as a barrier that does not block unrelated diffusing reactants. As shown in Figure 16-4, D, when two related antigens, Ag and Ag1, are in separate wells, and the respective antibodies, Ab and Ab1, are in the third well, an AgAb precipitate forms on one side and blocks further diffusion of Ab from the antibody well. However, on the other side, the Ag1Ab1 precipitate does not stop Ab from migrating further and forming an AgAb spur. If several antigens of interest exist in a solution (e.g., spinal fluid or serum), the various protein species can be separated and identified by IEP. This technique has been used extensively for the study of antigen mixtures and for evaluation of the specificity of antiserum.43 The procedure is performed using an agarose gel medium poured onto a thin plastic sheet. The sample to be analyzed is placed in a reservoir in the gel, and an electrical field is applied across the gel surface. During electrophoresis, the proteins in the serum are separated according to their electrophoretic mobilities (Figure 16-5). Following electrophoresis, an antiserum against the protein of interest is placed in a trough parallel and adjacent to the electrophoresed sample. Simultaneous diffusion of the antigen from the separated sample and the antibody from the trough leads to the formation of precipitin arcs, whose shape and position are characteristic of individual separated proteins within the specimen. By comparison with a known control separated on the same plate, individual proteins can be tentatively identified. This technique, also known as two-dimensional IEP, is a variation of IEP wherein electrophoresis is also used in the second dimension to drive the antigen into a gel containing antibodies specific for the antigens of interest.50 This technique is more sensitive and produces higher resolution than IEP. For more information on CRIE, see the previous edition of this textbook. With CIE, two parallel lines of wells are punched in the agar. One row is filled with antigen solution, and the opposing row is filled with antibody solution (Figure 16-6). If the solutions were allowed to passively diffuse, a precipitin line would form between the opposing wells in 18 to 24 hours. With CIE, this process is accelerated by applying a voltage across the gel so that the antigen and the antibody move toward each other. Qualitative information (i.e., identification of antigen) is provided within 1 to 2 hours. Historically, this method has found application in the detection of bacterial antigens in blood, urine, and cerebrospinal fluid. This technique has gained widespread acceptance as an immunochemical method for identifying proteins.3 As in IEP and CRIE, a first-dimension electrophoresis is performed in agarose gel to separate proteins in the sample. Subsequently, antiserum spread directly on the gel causes the protein(s) of interest to precipitate. The immune precipitate is trapped within the gel matrix, and all other nonprecipitated proteins are removed by washing the gel. The gel may then be stained for identification of the proteins. IF is technically more efficient than IEP or CRIE, and it produces patterns that are interpreted more easily. The usefulness of IF, which is now widely used for the evaluation of myeloma proteins, is illustrated in Figure 16-7. The techniques discussed previously use direct evaluation of the immunoprecipitation of the protein(s) in a gel. However, certain media, such as polyacrylamide, do not lend themselves to direct immunoprecipitation, nor is there always sufficient antigen concentration to produce an immunoprecipitate that will be retained in the gel during subsequent processing. Under these circumstances, a technique termed Western blotting can be used (see Chapter 12).11,26 This technique involves an electrophoresis step followed by transfer of separated proteins onto an overlying strip of nitrocellulose or a nylon membrane by a process called electro-blotting. Once the proteins are in the membrane, they can be detected using antibodies labeled with probes, such as radioactive isotopes or enzymes. When such probes are used, the limits of detection can be 10 to 100 times lower than when direct immunoprecipitation and staining of proteins are conducted. This technique is analogous to Southern blotting (electrophoresed DNA blotted onto a membrane), and Northern blotting (electrophoresed RNA blotted onto a membrane). An example of this technique applied to the detection of human immunodeficiency virus type 1 (HIV-1) antibodies is shown in Figure 16-8. HIV-1 antigens are separated according to molecular weight by gel electrophoresis and then are transferred to a nitrocellulose membrane by electro-blotting. A serum sample is then incubated with a strip of the membrane. HIV-1 antibodies in the sample bind to the viral antigens transferred to the strip as discrete bands. After washing, the strip is incubated with an alkaline phosphatase antihuman immunoglobulin conjugate. After a further washing step, the nitrocellulose strip is incubated with an alkaline phosphatase substrate solution (5-bromo-4-chloro-3-indolyl phosphate and nitroblue tetrazolium) to reveal where anti–HIV-1 antibody is bound to specific viral antigens fixed in the membrane. Protein transfer and immobilization following separation by electrophoresis, isoelectric focusing, sodium dodecyl sulfate polyacrylamide electrophoresis (see Chapter 12 for further discussion of these techniques), or other methods provide a powerful tool for the analytical study of proteins present in low concentrations in cell culture or body fluids. When applied to antigen assays, concentrations of antigen as low as 500 ng/mL or 2.5 ng/band in the gel can be detected by this method. The detection limit of the technique can be lowered to approximately 100 pg by using chemiluminescence labels on the antibody.10,47 With this technique, a concentration gradient is established for a single reactant, usually the antigen. The antibody is uniformly dispersed in the gel matrix. Antigen is allowed to passively diffuse from a well into the gel, and immune precipitation occurs until antibody excess exists. The antigen-antibody interaction is manifested by a defined ring of precipitation around the antigen well, and ring diameter will increase with increased antigen concentration. Calibrators are run at the same time as the sample, and a calibration curve of ring diameter versus concentration is plotted. Under equilibrium conditions, a linear relationship exists between antigen concentration and the square of the precipitin ring diameter.57 In addition, the precision of the measurement of the ring diameter is better when equilibrium is established. However, quantitative data can also be derived by reading the ring diameter before equilibrium is established.23 This approach, although less precise, is often more practical for a clinical laboratory if time is at a premium. Antigen concentrations are calculated in both the preequilibrium and equilibrium methods23,57 by plotting the square of the precipitin ring diameter against calibrating antigen concentrations. RID can be made more sensitive by using PEG to enhance precipitin line formation or by using 125I- or enzyme-labeled reagents.33,61,69 In electroimmunoassay, as in RID, a single concentration gradient is established for the antigen, but in this case, an applied voltage is used to drive the antigen from the application well into a homogeneous suspension of antibody in the gel (Figure 16-9).51 Unlike RID, this produces a unidirectional migration of antigen and results in a lower limit of detection for electroimmunoassay methods. The height of the resulting rocket-shaped precipitin line is proportional to the antigen concentration. Quantitation is affected by using calibrators on the same plate and estimating the concentrations of unknowns from the heights of the “rockets” obtained. The calibration curve is linear only over a narrow concentration range, so samples may have to be diluted or concentrated as necessary. Electroimmunoassay methods produce the best results, with antigens having a strong anodic mobility and intermediate to low molecular weight. Proteins such as transferrin, C3, or IgG, with low anodal mobility or virtually no net charge at pH 8.6 (the most common pH used for the method), can be modified by carbamylation or can be run at a lower pH to make their measurements by electroimmunoassay feasible. Other modifications, such as use of an intermediate gel that causes precipitation of C3, allow measurement of C3d in human serum and illustrate the exceptional versatility of this method.9
Principles of Immunochemical Techniques
Basic Concepts
Antibodies
Immunogens
Antigen-Antibody Binding
Hydrophobic Interaction
Coulombic Bonds
Factors Influencing Binding
Ion Species and Ionic Strength Effects
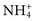 > K+ > Na+ > Li+. This order corresponds to the decreasing ionic radius and the increasing radius of hydration. Similar results were found for anionic haptens and anionic salts. For example, the order of inhibition of binding for anionic salts is CNS− > NO−3 > I− > Br− > Cl− > F−, which again is in the order of decreasing ionic radius and increasing radius of hydration. If the competition theory as suggested by these experiments is correct, one would expect the degree of inhibition to be a concentration-dependent phenomenon, and indeed the rate of formation of immune complexes is slower in normal saline (NaCl, 0.15 mol/L) than the same reaction carried out in deionized water. Given the previous observation, F− should be the anion of choice for immunochemical reaction buffers. In fact, F− does provide a modest improvement over Cl−, but the advantage is so small that laboratories rarely substitute toxic fluoride ion for innocuous chloride ion in buffer solutions. A small but measurable difference in the initial rate of combination of antigen and antibody can be seen for phosphate as compared with tris buffer. In general, the most notable differences in reaction rates for the various anionic species evaluated are seen when t is less than 5 minutes. When reactions are evaluated at longer times (i.e., t greater than 5 minutes), the difference in the rate of Ag-Ab complex formation is relatively small for different anionic species.58
> K+ > Na+ > Li+. This order corresponds to the decreasing ionic radius and the increasing radius of hydration. Similar results were found for anionic haptens and anionic salts. For example, the order of inhibition of binding for anionic salts is CNS− > NO−3 > I− > Br− > Cl− > F−, which again is in the order of decreasing ionic radius and increasing radius of hydration. If the competition theory as suggested by these experiments is correct, one would expect the degree of inhibition to be a concentration-dependent phenomenon, and indeed the rate of formation of immune complexes is slower in normal saline (NaCl, 0.15 mol/L) than the same reaction carried out in deionized water. Given the previous observation, F− should be the anion of choice for immunochemical reaction buffers. In fact, F− does provide a modest improvement over Cl−, but the advantage is so small that laboratories rarely substitute toxic fluoride ion for innocuous chloride ion in buffer solutions. A small but measurable difference in the initial rate of combination of antigen and antibody can be seen for phosphate as compared with tris buffer. In general, the most notable differences in reaction rates for the various anionic species evaluated are seen when t is less than 5 minutes. When reactions are evaluated at longer times (i.e., t greater than 5 minutes), the difference in the rate of Ag-Ab complex formation is relatively small for different anionic species.58
Polymer Effect54
Types of Reactions
The Precipitin Reaction
Qualitative Methods
Passive Gel Diffusion
Immunoelectrophoresis (IEP)
Crossed Immunoelectrophoresis (CRIE)
Counterimmunoelectrophoresis (CIE)
Immunofixation (IF)
Western Blotting
Quantitative Methods
Radial Immunodiffusion and Electroimmunoassay
Radial Immunodiffusion Immunoassay
Electroimmunoassay
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Principles of Immunochemical Techniques
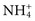 . The attraction between the charged groups is greatest in a medium with a low dielectric constant caused by reduced interaction of the solvent or other solute (salts) with the macromolecular ions. In a medium of high dielectric constant (aqueous solutions containing added salt), a diffuse double layer of charged particles will tend to shield the attraction of the charged species in the reactive sites of the antigen and antibody. This inhibition under certain circumstances can considerably reduce the binding constant for many antigen-antibody systems.
. The attraction between the charged groups is greatest in a medium with a low dielectric constant caused by reduced interaction of the solvent or other solute (salts) with the macromolecular ions. In a medium of high dielectric constant (aqueous solutions containing added salt), a diffuse double layer of charged particles will tend to shield the attraction of the charged species in the reactive sites of the antigen and antibody. This inhibition under certain circumstances can considerably reduce the binding constant for many antigen-antibody systems.