Figure 37–1 Sites of action of antimicrobial drugs. Antimicrobial drugs include cell wall synthesis inhibitors, protein synthesis inhibitors, metabolic and nucleic acid inhibitors (e.g., inhibitors of folate synthesis, DNA gyrase, and RNA polymerase), and cell membrane inhibitors.
ANTIMICROBIAL ACTIVITY
The antimicrobial activity of a drug can be characterized in terms of its bactericidal or bacteriostatic effect, its spectrum of activity against important groups of pathogens, and its concentration- and time-dependent effects on sensitive organisms.
Bactericidal or Bacteriostatic Effect
A bactericidal drug kills sensitive organisms so that the number of viable organisms falls rapidly after exposure to the drug (Figure 37–2). In contrast, a bacteriostatic drug inhibits the growth of bacteria but does not kill them. For this reason, the number of bacteria remains relatively constant in the presence of a bacteriostatic drug, and immunologic mechanisms are required to eliminate organisms during treatment of an infection with this type of drug. (The same principle applies to a drug that kills or inhibits the growth of fungi and is referred to as a fungicidal drug or a fungistatic drug, respectively.)
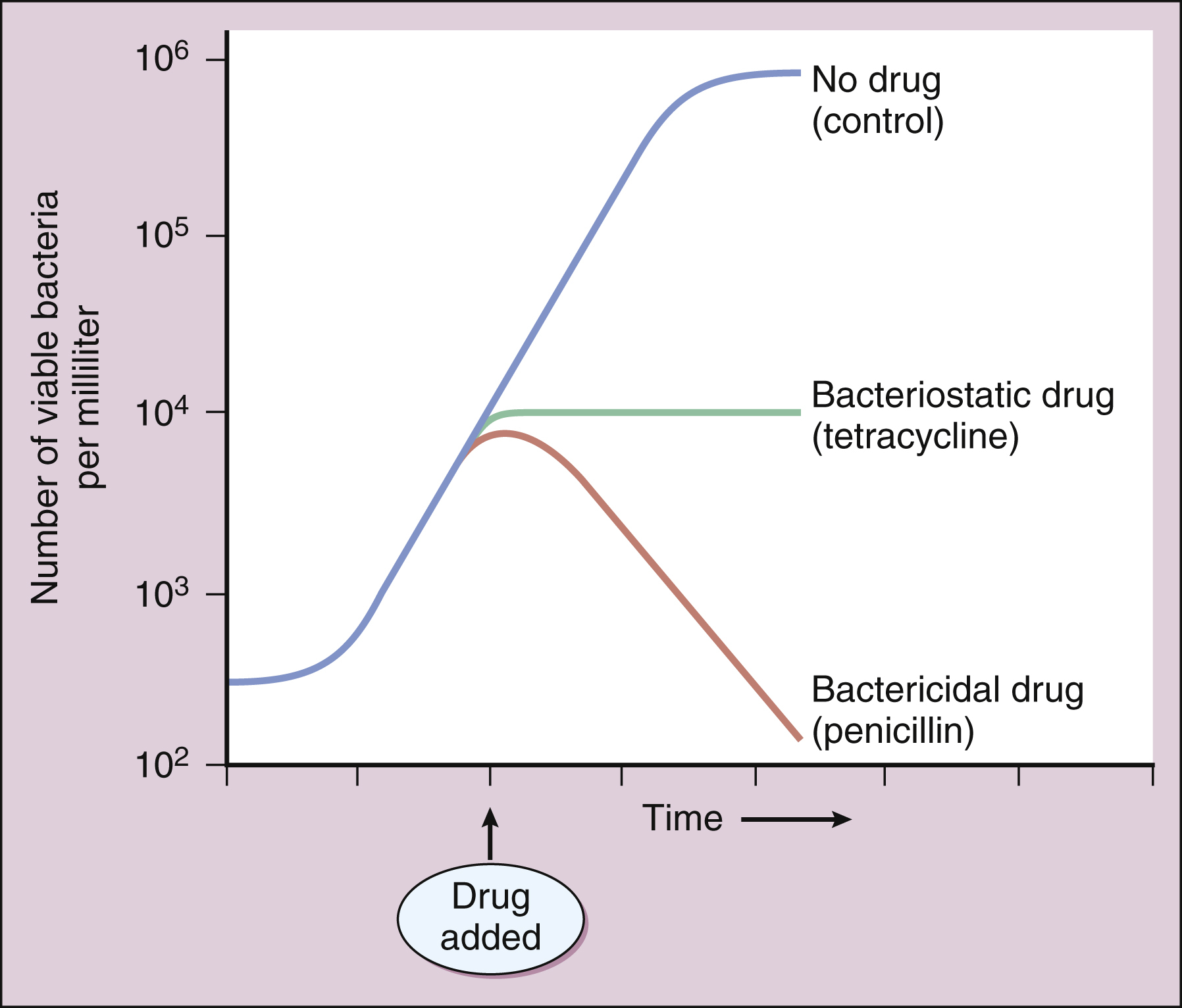
Figure 37–2 In vitro effects of bactericidal and bacteriostatic drugs. In the absence of an antimicrobial drug, bacteria exhibit logarithmic growth in a broth culture. The addition of a bacteriostatic drug (tetracycline) inhibits further growth but does not reduce the number of bacteria. The addition of a bactericidal drug (penicillin) reduces the number of viable bacteria.
A bactericidal drug is usually preferable to a bacteriostatic drug for the treatment of most bacterial infections. This is because bactericidal drugs usually produce a more rapid microbiologic response and more clinical improvement and are less likely to elicit microbial resistance. Bactericidal drugs have actions that induce lethal changes in microbial metabolism or block activities that are essential for microbial viability. For example, drugs that inhibit the synthesis of the bacterial cell wall (e.g., penicillins) prevent the formation of a structure that is required for the survival of bacteria. At the same time, penicillins activate autolytic enzymes that destroy bacteria. In contrast to bactericidal drugs, bacteriostatic drugs usually inhibit a metabolic reaction that is needed for bacterial growth but is not necessary for cell viability. For example, sulfonamides block the synthesis of folic acid, which is a cofactor for enzymes that synthesize DNA components and amino acids.
Drugs that reversibly inhibit bacterial protein synthesis (e.g., tetracyclines) are also bacteriostatic, whereas drugs that irreversibly inhibit protein synthesis (e.g., streptomycin) are usually bactericidal.
Some drugs can be either bactericidal or bacteriostatic, depending on their concentration and the bacterial species against which they are used.
Antimicrobial Spectrum
The spectrum of antimicrobial activity of a drug is the primary determinant of its clinical use. Antimicrobial agents that are active against a single species or a limited group of pathogens (e.g., gram-positive bacteria) are called narrow-spectrum drugs, whereas agents that are active against a wide range of pathogens are called broad-spectrum drugs. Agents that have an intermediate range of activity are sometimes called extended-spectrum drugs.
If the specific pathogen that is responsible for an infection is known, a narrow-spectrum drug is the logical choice to treat the infection. This is because a broad-spectrum drug is more likely to cause superinfection by eradicating organisms that make up the normal flora of the gut or respiratory tract. In cases in which an infection is serious and the responsible pathogen is not yet known, however, a broad-spectrum bactericidal drug (e.g., imipenem) is often used for initial treatment.
Concentration- and Time-Dependent Effects
Antimicrobial drugs exhibit various concentration- and time-dependent effects that influence their clinical efficacy, dosage, and frequency of administration. Examples of these effects are the minimal inhibitory concentration (MIC), the concentration-dependent killing rate (CDKR), and the postantibiotic effect (PAE).
The MIC is the lowest concentration of a drug that inhibits bacterial growth. Based on the MIC, a particular strain of bacteria can be classified as susceptible or resistant to a particular drug (see below).
An example of a CDKR is shown in (Figure 37–3A). Some aminoglycosides (e.g., tobramycin) and some fluoroquinolones (e.g., ciprofloxacin) exhibit a CDKR against a large group of gram-negative bacteria, including Pseudomonas aeruginosa and members of the family Enterobacteriaceae. In contrast, penicillins and other β-lactam antibiotics usually do not exhibit a CDKR.
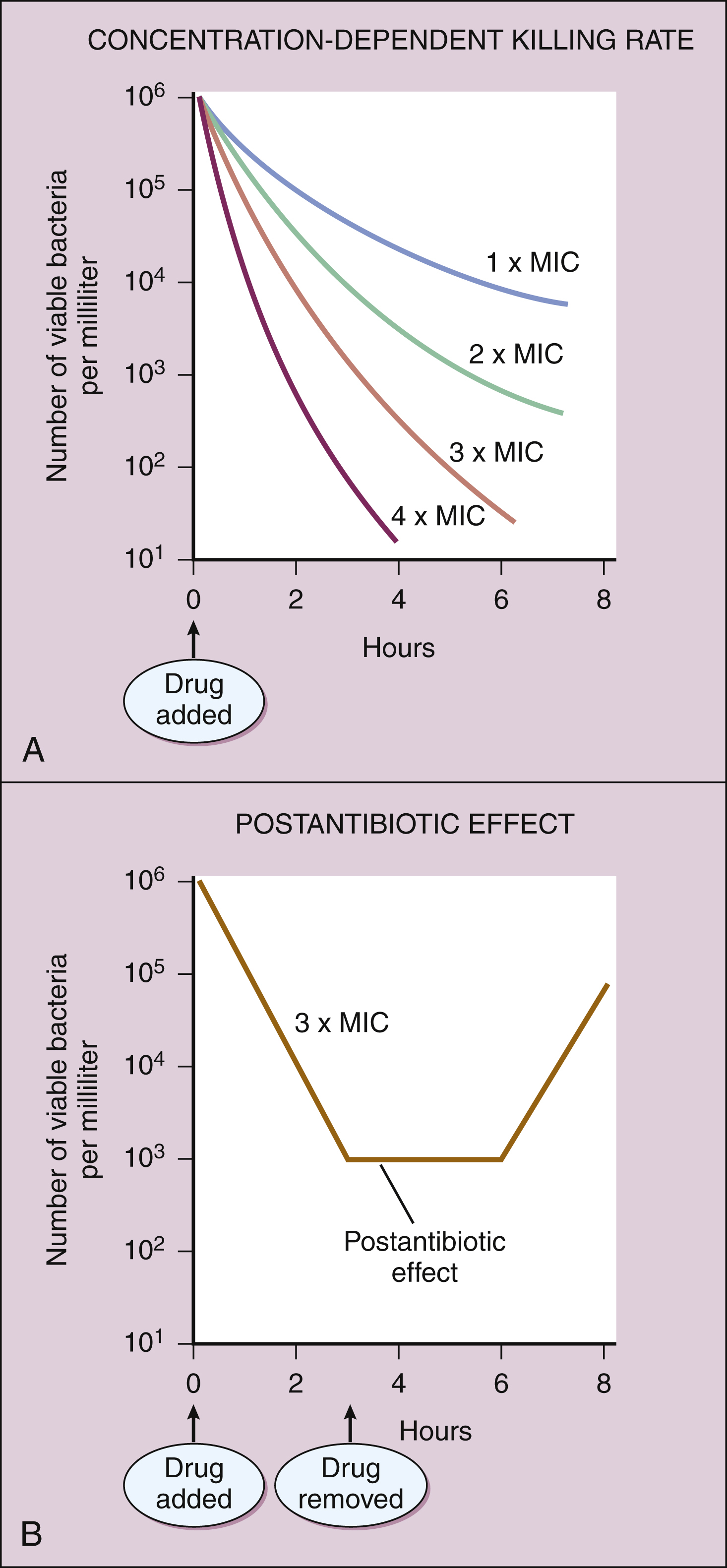
Figure 37–3 Concentration- and time-dependent effects of antimicrobial drugs. (A) When an aminoglycoside (e.g., tobramycin) is added to a culture of a gram-negative bacterium (e.g., Escherichia coli), it will exhibit a concentration-dependent killing rate. In this example, 106 bacteria were incubated with different concentrations of tobramycin, ranging from one to four times the minimal inhibitory concentration (MIC). (B) When tobramycin is removed from the culture, bacterial growth continues to be inhibited for several hours.
After an antibacterial drug is removed from a bacterial culture, evidence of a persistent effect on bacterial growth may exist. This effect (see Fig. 37–3B) is called the PAE. Most bactericidal antibiotics exhibit a PAE against susceptible pathogens. For example, penicillins show a PAE against gram-positive cocci, and aminoglycosides show a PAE against gram-negative bacilli. Because aminoglycosides exhibit both a CDKR and a PAE, treatment regimens have been developed in which the entire daily dose of an aminoglycoside is given at one time. Theoretically, the high rate of bacterial killing produced by these regimens would more rapidly eliminate bacteria, and the PAE would prevent any remaining bacteria from replicating for several hours after the drug has been eliminated from the body.
MICROBIAL SENSITIVITY AND RESISTANCE
Laboratory Tests for Microbial Sensitivity
Microbial sensitivity to drugs can be determined by various means, including the broth dilution test, the disk diffusion method (Kirby-Bauer test), and the Etest method. These laboratory procedures are described in Box 37–1.
BOX 37–1 LABORATORY DETERMINATION OF MICROBIAL SENSITIVITY TO ANTIBIOTICS
Microbial sensitivity to drugs can be determined by various means, including the broth dilution test, the disk diffusion method, and the Etest method.
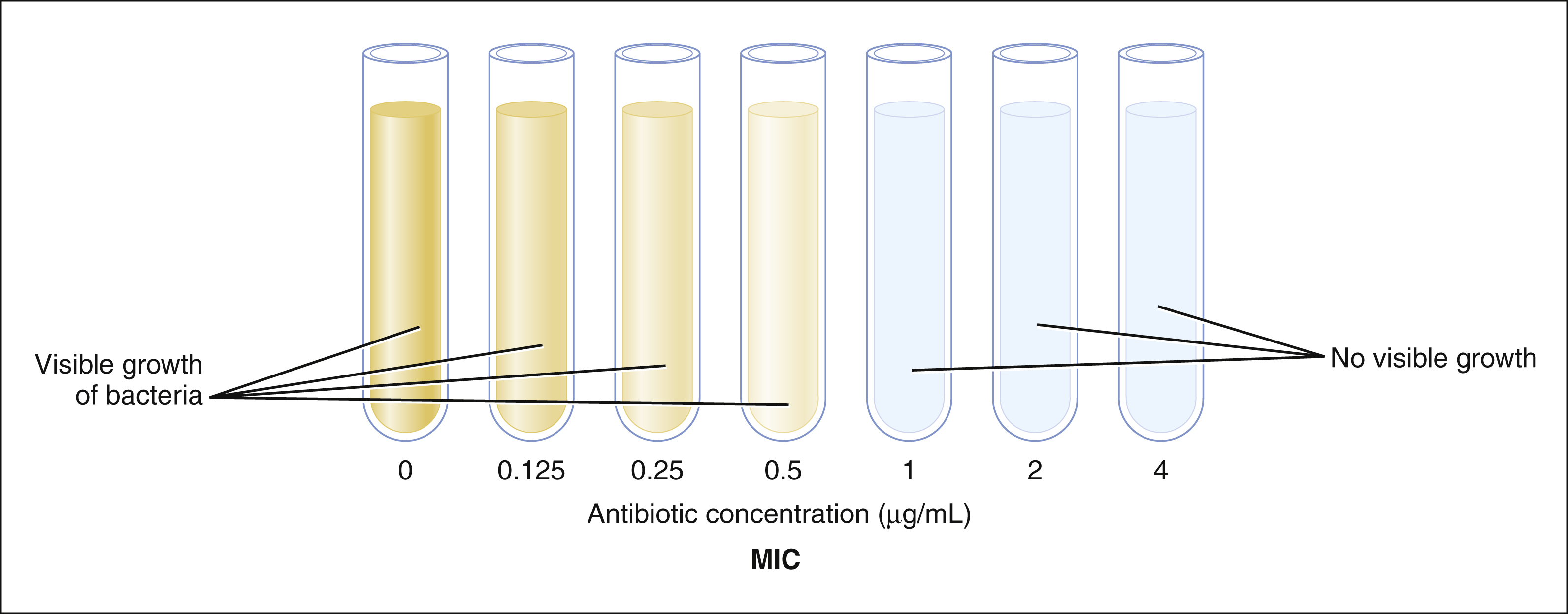
Broth Dilution Test
Tubes that contain a nutrient broth are inoculated with equal numbers of bacteria and serially diluted concentrations of an antibiotic. After incubation, the minimal inhibitory concentration (MIC) is identified as the lowest antibiotic concentration that prevents visible growth of bacteria. On the basis of the MIC, the organism is classified as having susceptibility, intermediate sensitivity, or resistance to the drug tested. In the following example, the MIC is 1 μg/mL, and the organism is susceptible to the drug.
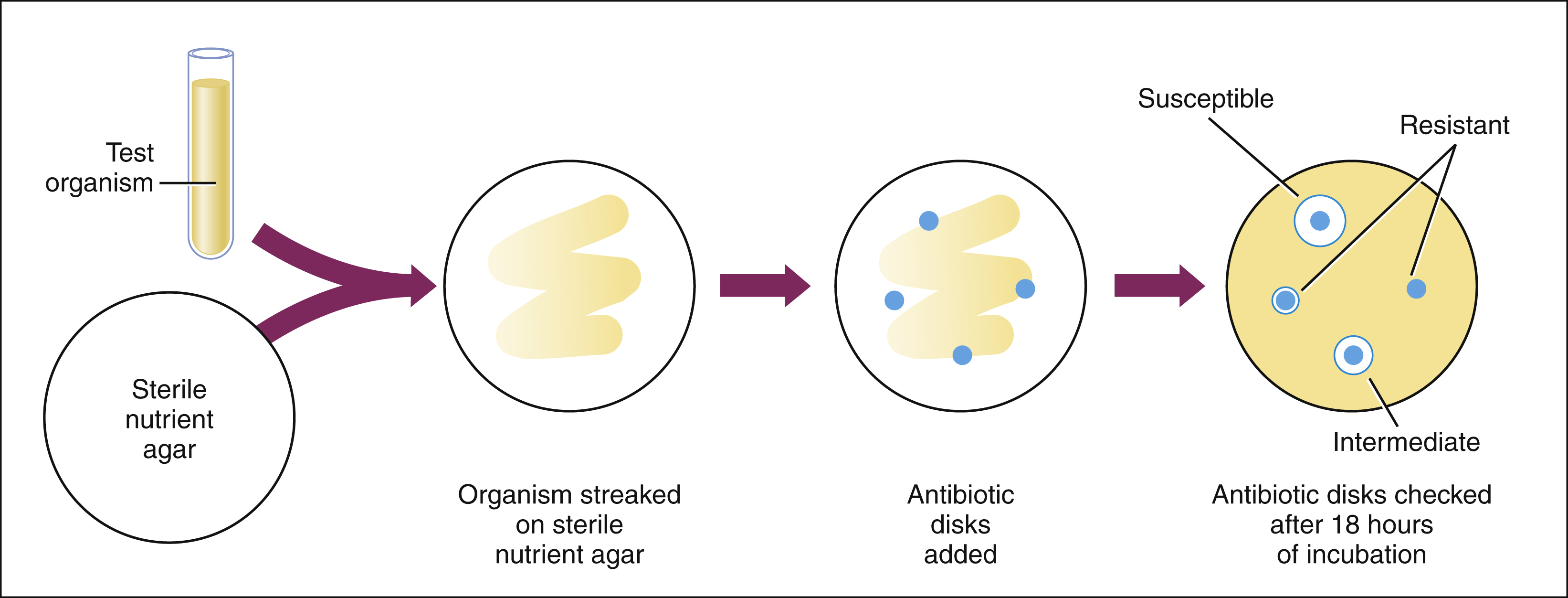
Disk Diffusion Method (Kirby-Bauer Test)
Each disk used in the disk diffusion method is impregnated with a different antibiotic. The disks are placed on agar plates seeded with the test organism. During the incubation period, the antibiotic diffuses from the disk and inhibits bacterial growth. After incubation, the zone inhibited by each antibiotic is measured. The zone diameter for each antibiotic is compared with standard values for that particular antibiotic. The organism is thereby determined to be susceptible, intermediate, or resistant to the various antibiotics tested.
Etest Method
The Etest strip is a proprietary device that uses a diffusion method to determine the MIC of an organism. The device is a plastic strip that is impregnated with a gradient of antibiotic concentrations. After the strip is placed on an agar culture of the organism, the culture is incubated. During incubation, a tear-shaped zone of inhibition is formed. The point of intersection between the zone of inhibition and the scale displayed on the strip is the MIC. In the following example, the MIC is 0.125 μg/mL.
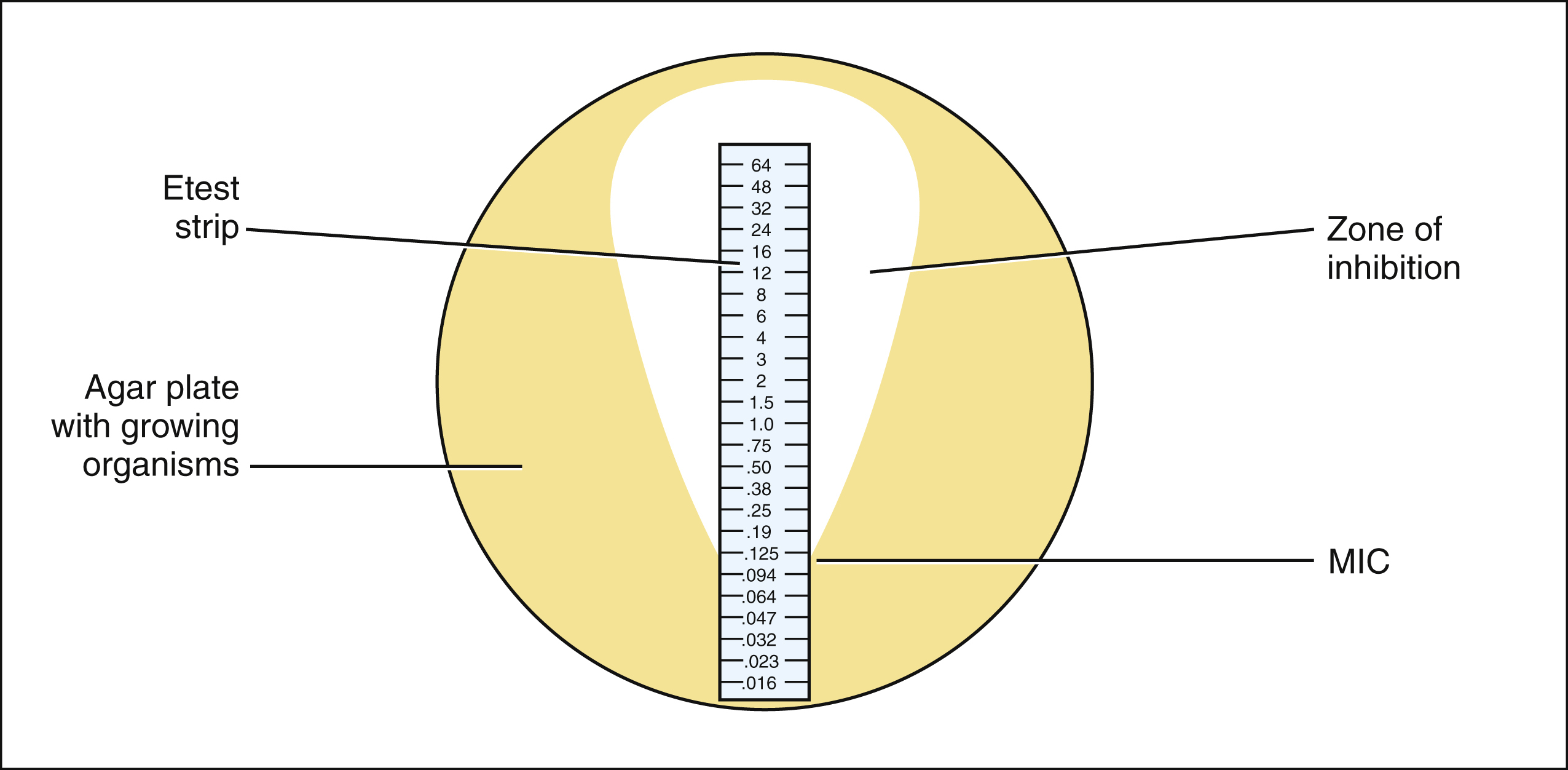
Either the broth dilution test or the Etest method can be used to determine the MIC of a drug, which is the lowest drug concentration that prevents visible growth of bacteria. On the basis of the MIC, the organism is classified as having susceptibility, intermediate sensitivity, or resistance to the drug tested. These categories are based on the relationship between the MIC and the peak serum concentration of the drug after administration of typical doses. In general, the peak serum concentration of a drug should be 4 to 10 times greater than the MIC in order for a pathogen to be susceptible to a drug (see below). Pathogens with intermediate sensitivity may respond to treatment with maximal doses of an antimicrobial drug.
Microbial Resistance to Drugs
Origin of Resistance
Resistance can be innate or acquired. Acquired drug resistance arises from mutation and selection or from the transfer of plasmids that confer drug resistance.
MUTATION AND SELECTION
Microbes can spontaneously mutate to a form that is resistant to a particular antimicrobial drug. These mutations occur at a relatively constant rate, such as 1 in 1012 organisms per unit of time. If the organisms are exposed to an antimicrobial drug during this time period, the sensitive organisms may be eradicated, enabling the resistant mutant to multiply and become the dominant strain (Figure 37–4A).
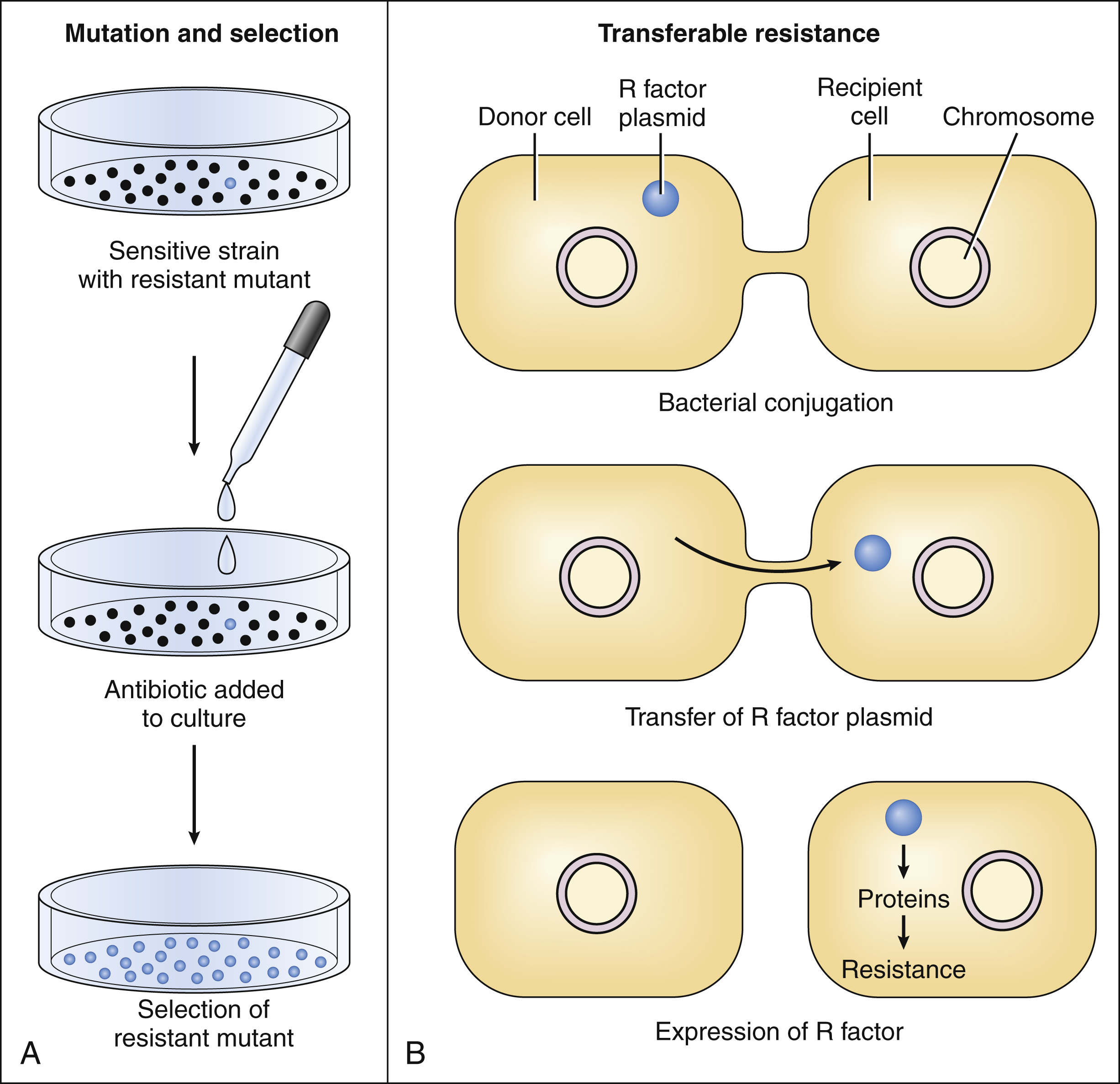
Figure 37–4 Acquired resistance to antimicrobial drugs can arise by mutation and selection or by transferable resistance. (A) Exposure of an organism to an antibiotic can result in the selection of a resistant mutant. (B) The most common mechanism of transferable resistance is bacterial conjugation and exchange of plasmids containing resistance factors.
The probability that mutation and selection of a resistant mutant will occur is increased during the exposure of an organism to suboptimal concentrations of an antibiotic, and it is also increased during prolonged exposure to an antibiotic. This observation has obvious implications for antimicrobial therapy. Laboratory tests should be used to guide the selection of an antimicrobial drug, and the dosage and duration of therapy should be appropriate for the type of infection being treated. Whenever possible, the bacteriologic response to drug therapy should be verified by culturing samples of appropriate body fluids.
TRANSFERABLE RESISTANCE
Transferable resistance usually results from bacterial conjugation and the transfer of plasmids (extrachromosomal DNA) that confer drug resistance (see Fig. 37–4B). Transferable resistance, however, can also be mediated by transformation (uptake of naked DNA) or transduction (transfer of bacterial DNA by a bacteriophage). Bacterial conjugation enables a bacterium to donate a plasmid containing genes that encode proteins responsible for resistance to an antibiotic. These genes are called resistance factors. The resistance factors can be transferred both within a particular species and between different species, so they often confer multidrug resistance. The various species need not all be present during the period in which the antibiotic is administered. Studies have shown that resident microflora of the human body can serve as reservoirs for resistance genes, allowing the transfer of these genes to organisms that later invade and colonize the host.
Several genes responsible for drug resistance have been cloned, and the factors that control their expression are being studied. In the future, drugs that block the expression of these genes may find use as adjunct therapy for infectious diseases. For example, it may be possible to develop antisense nucleotides that block the transcription or translation of genes that encode proteins responsible for drug resistance.
Mechanisms of Resistance
The three primary mechanisms of microbial resistance to an antibiotic are (1) inactivation of the drug by microbial enzymes, (2) decreased accumulation of the drug by the microbe, and (3) reduced affinity of the target macromolecule for the drug. Examples of drugs affected by these mechanisms are provided in Table 37–1.
TABLE 37–1 Mechanisms of Microbial Resistance
| Mechanism | Examples |
|---|---|
| Inactivation of the drug by microbial enzymes | Inactivation of aminoglycosides by acetylase, adenylate synthetase, and phosphorylase enzymes |
| Inactivation of penicillins and other β-lactam antibiotics by β-lactamase enzymes | |
| Decreased accumulation of the drug by the microbe | Decreased uptake of β-lactam antibiotics due to altered porins in gram-negative bacteria |
| Decreased uptake and increased efflux of fluoroquinolones | |
| Decreased uptake and increased efflux of tetracyclines | |
| Reduced affinity of the target macromolecule for the drug | Reduced affinity of DNA gyrase for fluoroquinolones |
| Reduced affinity of folate synthesis enzymes for sulfonamides and trimethoprim | |
| Reduced affinity of ribosomes for aminoglycosides, chloramphenicol, clindamycin, macrolides, or tetracyclines | |
| Reduced affinity of RNA polymerase for rifampin | |
| Reduced affinity of transpeptidase and other penicillin-binding proteins for penicillins and other β-lactam antibiotics |
Inactivation of the drug by enzymes is an important mechanism of resistance to β-lactam antibiotics, including the penicillins. This form of resistance results from bacterial elaboration of β-lactamase enzymes that destroy the β-lactam ring. Resistance to aminoglycosides (e.g., gentamicin) is partly caused by the elaboration of drug-inactivating enzymes that acetylate, adenylate, or phosphorylate these antibiotics.
Decreased accumulation of an antibiotic can result from increased efflux or decreased uptake of the drug. Both of these mechanisms contribute to the resistance of microbes to tetracyclines and fluoroquinolones. Increased drug efflux is often mediated by membrane proteins that transport antimicrobial drugs out of bacterial cells. Some of these transport proteins are similar to, or indistinguishable from, human P-glycoprotein, a glycoprotein that transports antineoplastic drugs out of human cancer cells and thereby confers resistance to the drugs (see Chapter 45). Compounds that inhibit these transport proteins are being investigated as potential therapeutic agents to reduce drug resistance. Decreased uptake of antimicrobial drugs can result from altered bacterial porins. Porins are membrane proteins containing channels through which drugs and other compounds enter bacteria. Resistance to penicillins by gram-negative bacilli is partly caused by altered porin channels that do not permit penicillin entry.
Reduced affinity of target macromolecules for antimicrobial drugs is a common mechanism of drug resistance. It is partly responsible for resistance to cell wall synthesis inhibitors, protein synthesis inhibitors, DNA gyrase inhibitors, and RNA polymerase inhibitors (see Table 37–1). This type of drug resistance often results because of bacterial mutation, followed by the selection of resistant mutants during exposure to an antimicrobial drug. Altered target affinity has been the most difficult form of drug resistance to counteract by pharmacologic agents.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


