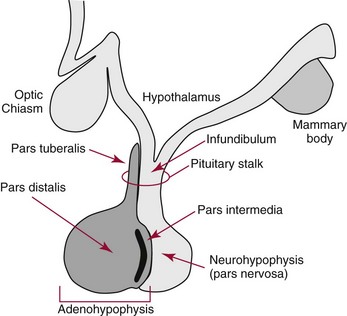
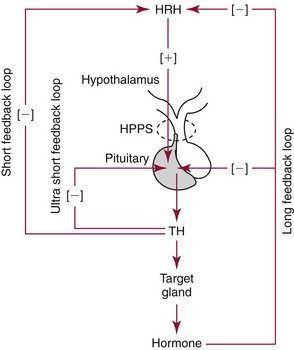
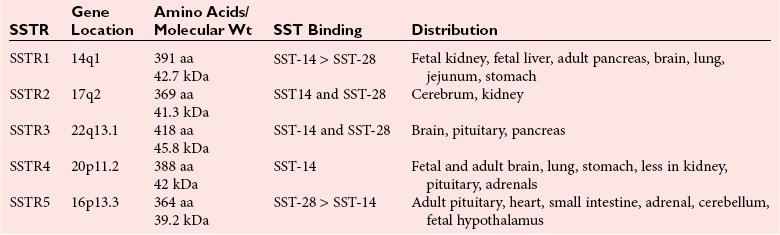
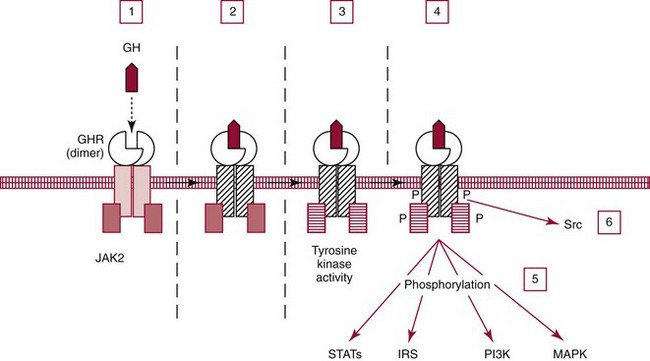
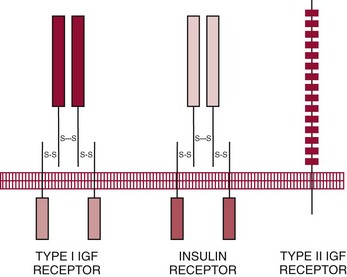
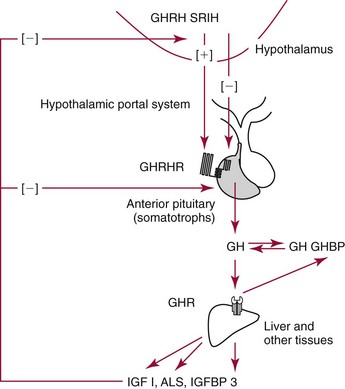
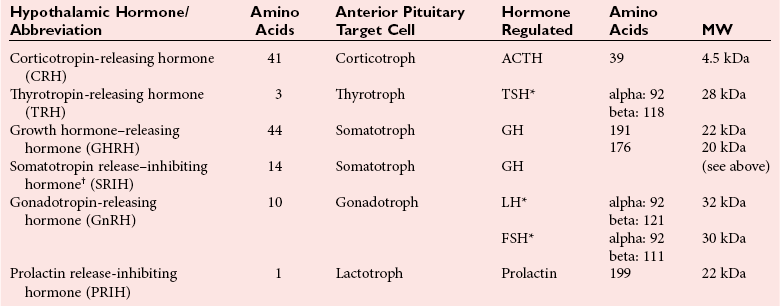
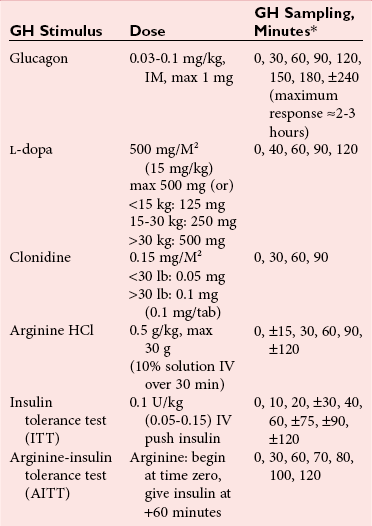
Chapter 53 William E. Winter, M.D., Ishwarlal Jialal, M.D., Ph.D., F.R.C.Path(London), D.A.B.C.C., Mary Lee Vance, M.D. and Roger L. Bertholf, Ph.D.* The pituitary gland (also called the hypophysis) regulates the endocrine system by integrating chemical signals from the brain with feedback from the concentration of circulating hormones to stimulate intermittent hormone release from target endocrine glands.115,156 The pituitary clearly serves as the master gland in maintaining homeostasis by orchestrating the many processes necessary for survival of the individual, as well as for survival of the species. The hypophysis is composed of the adenohypophysis (the anterior lobe of the pituitary; ≈75% of the mass of the pituitary) and the neurohypophysis (the posterior lobe of the pituitary, ≈25% of the mass of the pituitary; also called the pars nervosa) (Figure 53-1).10 In turn, the adenohypophysis has three parts: (1) the pars distalis, where most hormone-producing cells are located; (2) the pars tuberalis, which is part of the hypophyseal stalk; and (3) the pars intermedia. The pars intermedia may be referred to as the intermediate lobe of the pituitary, although it is really part of the adenohypophysis. The biology of the adenohypophysis is distinctly different from that of the neurohypophysis: the adenohypophysis is controlled by the hypothalamus via releasing or inhibiting hormones, whereas the cell bodies of the neurohypophysis are anatomically located in hypothalamic nuclei, with oxytocin or antidiuretic hormone (ADH) reaching the neurohypophysis through neurohypophyseal nerve axons.96 Thus the neurohypophysis is not a discrete endocrine organ, but rather functions as a reservoir for these two hormones. A newly recognized product of the pituitary gland detected in some perimenopausal and postmenopausal women is human chorionic gonadotropin (hCG).185 Usually, hCG is associated with pregnancy or gestational trophoblastic disease. In early pregnancy, hCG doubles approximately every 48 hours, whereas the concentration of hCG from pituitary or gestational trophoblastic disease origin is relatively stable and does not increase in the pattern seen in pregnancy. If an elevated hCG of 5 to 14 IU/L is detected in a postmenopausal woman, it is likely to be of pituitary origin if (1) the FSH is elevated (>45 IU/L), (2) the hCG is suppressed after 2 weeks of estrogen replacement, and (3) gestational trophoblastic disease has been excluded. The placenta produces several hormones that are similar to pituitary hormones: hCG has functional and structural similarity to LH; human placental lactogen (hPL, or somatomammotropin) has actions similar to prolactin and GH; placental GH becomes the predominant maternal GH during gestation; and placental corticotropin-releasing hormone (CRH) concentration rises in the fetus throughout gestation.131 Placental GH (GH-V) differs from pituitary GH in 13 of 191 amino acids. Furthermore, GH-V exists in glycosylated and nonglycosylated forms, whereas pituitary GH is not glycosylated. The site of glycosylation in GH-V is asparagine at residue 140. If the pituitary is greatly reduced in size or is apparently absent on magnetic resonance imaging (MRI) studies, the sella is said to be “empty.”125 In the empty sella syndrome, the sella may be normal in size or enlarged. An incompetent diaphragma sella with compression of the pituitary gland by herniating arachnoid can cause an empty sella, or the pituitary may be reduced in size as the result of some form of injury, radiotherapy, or surgery. Via the lateral hypophyseal veins, pituitary venous drainage is moved to the cavernous and intercavernous sinuses. The cavernous sinus drains to the superior and inferior petrosal sinuses, which join the transverse sinus to form the jugular vein. This anatomic relationship is clinically important because access to pituitary secretions can be afforded by cannulation of the inferior petrosal venous sinuses.193 Usually, the neuroradiologist places a catheter into a femoral vein that traverses an iliac vein, inferior vena cava, or superior vena cava to the jugular vein to enter the inferior petrosal venous sinus. Inferior petrosal venous sinus sampling can be helpful in the evaluation of patients with Cushing syndrome, to differentiate between Cushing disease (an anterior pituitary corticotropinoma) and ectopic ACTH syndrome. The internal carotid arteries are located laterally to the pituitary. Above the pituitary is the diaphragma sella, which comprises circular (intercavernous) sinuses containing venous blood. Anterior and superior to the pituitary is the optic chiasm. These relations are clinically important because pituitary neoplasms can invade or compress these structures, as well as the bony sella turcica. For example, expanding anterior pituitary adenomas can compress the optic chiasm, producing bilateral hemianopsia.202 In this condition, both lateral visual fields are lost. While walking, affected patients will bump into lateral structures in the environment because of loss of their lateral vision. The adenohypophysis develops in utero from a dorsal evagination of the roof of the stomodeum, which becomes the Rathke pouch.204 The superior portion of the Rathke pouch constitutes the pars tuberalis (see earlier), whereas the posterior portion of the Rathke pouch develops into the pars intermedia (or intermediate lobe). Transcription factors that regulate the development of the anterior pituitary gland include HESX, LHX3, LHX4, SOX3, Pit-1, PROP1, RIEG, and GLI2.54,55,117 The pars intermedia, which is active only late in pregnancy and in utero, secretes alpha-, beta-, and gamma-melanocyte stimulating hormone (MSH), corticotropin-like intermediate lobe peptide (CLIP), gamma-lipotropin, and beta-endorphin. MSH is believed to promote melanin synthesis. Lipotropins mobilize fat from adipose tissue, and endorphins are endogenous opioids.128 The clinical significance of these products as causes of disease is not well understood. The synthesis and release of the following anterior pituitary hormones are stimulated by hypothalamic releasing hormones: ACTH, TSH, GH, LH, and FSH.96 Prolactin is the sole anterior pituitary hormone whose release is predominantly regulated through suppression. Corticotrophs secrete ACTH, thyrotrophs secrete TSH, somatotrophs secrete GH, gonadotrophs secrete both LH and FSH, and lactotrophs (or mammotrophs) secrete prolactin. Except for LH and FSH, each hormone is normally produced by a unique cell type. However in pathologic circumstances, both GH and prolactin may be secreted by an anterior pituitary adenoma. The molecular composition of the anterior pituitary hormones is summarized in Table 53-1. TABLE 53-1 Hypothalamic Releasing or Inhibiting Hormones, Their Target Cells, and the Hormone That Is Regulated *All alpha glycoprotein chains are identical, including the alpha chain of human chorionic gonadotropin (hCG). Multiple levels of control of the hypothalamic-pituitary-end organ-hormone axis are known (Figure 53-2).42 Except for prolactin and LH at the midpoint of the menstrual cycle, negative feedback controls secretion of the adenohypophyseal hormones. The long feedback loop involves suppression of the hypothalamic releasing hormone and the anterior pituitary trophic hormone by the hormonal product of the target tissue. The major site of negative feedback for cortisol (regulated by ACTH), insulin-like growth factor-I (IGF-I; regulated by GH), and sex steroids and inhibins (regulated by LH and FSH) is the hypothalamus. In contrast, for thyroid hormone (regulated by TSH), the major site of negative feedback is the anterior pituitary. Retrograde flow from the pituitary to the hypothalamus via the portal system permits the existence of short negative feedback loops where pituitary hormones suppress the secretion of hypothalamic releasing hormones. Ultra-short feedback loops also exist in which pituitary hormones inhibit their own secretion. Hormones released by the hypothalmus that regulate the anterior pituitary hormones are listed in Table 53-1. With the exception of ACTH, hypothalamic hormones are structurally smaller than all of the anterior pituitary hormones. CRH has wide distribution throughout the brain and brainstem.86 In the hypothalamus, it is released by the paraventricular nucleus (PVN). CRH secretion is stimulated by systemic physiologic stress via (1) neurons of subfornical origin, (2) neurons of the nucleus tractus solitarius (NTS), (3) hypothalamic glutamatergic neurons, and (4) 5-hydroxytryptamine–secreting neurons of the raphe nucleus. Neurogenic stress to release CRH also acts via hypothalamic glutamatergic neurons. Stress inhibits hypothalamic GABAergic neurons of the PVN that otherwise would suppress CRH release. GABA (gamma-aminobutyric acid) serves as an inhibitory neurotransmitter. GABAergic neurons that innervate CRH-secreting neurons also originate from the lateral septum and the bed nucleus of the stria terminalis (BST). ACTH release is stimulated by serotonin, endorphins, and acetylcholine, and suppressed by GABA. Physiologically, stress, inflammation, and hypoglycemia stimulate ACTH release. Thyrotropin-releasing hormone (TRH) is a tripeptide the product of the PVN of the hypothalamus.155 TRH-secreting neurons in the PVN are innervated by axons that release (1) norepinephrine, (2) leptin, (3) neuropeptide Y, (4) agouti-related protein (AgRP), (5) MSH, (6) CRH, or (7) somatostatin. Leptin is produced by adipose tissue and acts to reduce appetite and raise energy expenditure as body fat stores rise. Leptin receptors are expressed in the ventromedial nucleus (VMN) of the hypothalamus. Neuropeptide Y and AgRP promote food intake. Growth hormone–releasing hormone (GHRH) is produced by neurons in the arcuate nucleus of the medial basal hypothalamus. Simulators of GHRH include dopamine- and galanin-secreting neurons, and brainstem neurons with catecholaminergic inputs.143 Galanin is a neuropeptide that is widely expressed in the endocrine, central nervous, and peripheral nervous systems. Hypothalamic somatostatin release–inhibiting hormone (SRIH) suppresses both GHRH release and anterior pituitary GH release. Leptin from adipose tissue and ghrelin from the stomach overall have the effect of increasing GHRH secretion and directly increasing GH concentrations. However, the clinical relevance of these influences and of other growth hormone–releasing peptides (e.g., GHRP-6) is not well understood. Ghrelin binds to GH secretogogue receptors (GHS-R) that increases food intake. Hormones that affect GH secretion include estrogen, testosterone, and glucocorticoids. Physiologically, amino acids and hypoglycemia stimulate GH release. In turn, the secretion of IGF-I in response to GH is influenced by nutrition, sex steroids, thyroid hormone, and the presence of chronic disease. Dopamine, endorphins, serotonin, and norepinephrine stimulate GH secretion. Gonadotropin-releasing hormone (GnRH) regulation is complicated by the fact that GnRH must differentially control LH and FSH secretion, which vary greatly during the menstrual cycle in women.129 GnRH-secreting neurons are not located in a discrete nucleus, but instead are diffusely distributed in the hypothalamus. Embryologically, these neurons are unusual in that they originate outside the central nervous system. GnRH secretion is stimulated by neurons that secrete (1) galanin-like peptide (GALP), (2) kisspeptin, (3) glutamate, (4) neuropeptide Y, and (5) norepinephrine. Kisspeptin is a neuropeptide that regulates puberty and reproduction. Neurons secreting GABA, beta-endorphins, and CRH inhibit GnRH. Gonadotropin release is stimulated by norepinephrine, GABA, and acetylcholine, and is suppressed by endorphins, dopamine, and serotonin. Corticotrophs are stimulated by high concentrations of proinflammatory cytokines such as interleukin (IL)-1, IL-6, and tumor necrosis factor (TNF)-alpha.209 This emphasizes the interrelationship of the endocrine and immune systems in a hypothalamic-pituitary-adrenal-immune system axis. Through vasopressin type 3 receptors (V1b receptors), high concentrations of ADH stimulate corticotrophs to release ACTH. The hormonal products of each anterior pituitary target cell (if applicable) are listed in Table 53-2 along with a summary of each system. Many of these hormones display circadian (daily), ultradian (>daily) or infradian (<daily) variation that reflects changes in hypothalamic control. Deficiency of an individual pituitary hormone is typically called hypopituitarism,13,179,194 whereas deficiency of all anterior pituitary hormones is termed panhypopituitarism. Linear growth is the consequence of (1) genetic potential, (2) nutrition, (3) the presence or absence of disease, and (4) hormonal effects.212 Many hormones influence growth, but the most important are GH, thyroid hormone, and sex steroids. Excess glucocorticoids can impair growth in children. GH deficiency produces disease in adults, thus GH is essential for health throughout life. Somatotropin release–inhibiting hormone (SRIH) also is known as somatostatin.195 It is widely distributed throughout the body (CNS, gut, and delta cells of the islets of Langerhan) and produces multiple physiologic effects. Somatostatin receptors are widely distributed (see later). Somatostatin that functions as SRIH is produced by the paraventricular nucleus of the hypothalamus. In the islets, somatostatin suppresses glucagon and insulin secretion, whereas somatostatin release is stimulated by both of these hormones. In this way, delta cell somatostatin modulates islet function by smoothing out extremes in the secretion of glucagon and insulin to maintain a stable blood glucose concentration. Somatostatin in the gut is found in highest concentration in the duodenum and jejunum. Expression of the somatostatin gene produces the 116 amino acid polypeptide preprosomatostatin. Cleavage of the signal sequence (24 amino acids) produces prosomatostatin (92 amino acids), and subsequent cleavage of the N-terminal pro-sequence (64 amino acids) yields a 28 amino acid form of somatostatin (SST-28). SST-28 has an intrachain disulfide bond between amino acids 17 and 28. In many tissues, SST-28 undergoes cleavage to a 14 amino acid form (SST-14) through removal of the N-terminal 14 amino acid sequence by the enzymes prohormone convertase 1/prohormone convertase 2 (PC1/PC2) and carboxypeptidase E (CPE). SST-14 is the major form of somtatostatin in the CNS and delta cells, whereas SST-28 is the major form in the gastrointestinal tract. SST-28 is also the major circulating form of somatostatin. Therefore, somatostatin measurements in peripheral blood do not reflect SRIH secretion. Somatostatin is highly conserved in nature: all vertebrates have the identical sequence for SST-14.195 In addition to GH suppression, somatostatin also suppresses TRH, TSH, CRH, and ACTH. However, the effect of somatostatin on regulation of the adrenal cortical and thyroid axes ordinarily is minor. In the gastrointestinal tract, somatostatin reduces the secretion of multiple hormones including (1) gastrin, (2) secretin, (3) cholecystokinin, (4) vasoactive intestinal polypeptide, (5) motilin, (6) neurotensin, and (7) pepsin and reduces gastric pH, intestinal motility, ion and nutrient absorption, and proliferation of the mucosa (see Chapter 51). Calcitonin, catecholamines, renin, and pancreatic exocrine function are suppressed by somatostatin. Somatostatin analogs (e.g., octreotide) are used pharmacologically to suppress a variety of hormone overproduction conditions.28 The anterior pituitary somatotroph GHRH receptor (GHRHR) is a member of family B-III of the G-coupled receptor superfamily (the “secretin” family).7 Receptors for (1) secretin, (2) vasoactive intestinal polypeptide (VIP), (3) parathyroid hormone (PTH), and (4) calcitonin share partial sequence identity with GHRHR. Throughout the body, there are five receptors for somatostatin (SSTR1 through SSTR5; Table 53-3). Each receptor is encoded by a gene located on a separate chromosome. SSTR2 has two alternatively spliced isoforms. All of the SSTR receptors have seven transmembrane domains, and are coupled with a pertussis toxin–sensitive G protein. SSTR3, SSTR4, and SSTR5 are expressed in the pituitary. GH has two disulfide bridges (amino acids 54 and 165, and amino acids 182 and 189).59 Structurally, GH has four main alpha-helices, and within the connecting loops three mini-helices. Two circulating forms of GH are present: a 22 kDa form that is a 191 amino acid chain (full-length GH) representing 85 to 90% of circulating GH, and a 20 kDa GH that lacks amino acids 32 through 46.184 The 20 kDa form of GH results from alternative splicing of the GH mRNA transcript. In addition to the 22 kDa and 20 kDa forms, circulating GH exists as aggregates and oligomers. “Big GH” is a dimer of GH monomers, and “big-big GH” is GH associated with its binding protein (GHBP). GHBP is the external domain of the GH receptor (GHR), which binds GH with high affinity and is produced by cleavage of the GHR. Approximately 55% of all circulating GH forms are monomeric; big GH and big-big GH represent ≈27% and ≈18% of circulating GH, respectively. Approximately 50% of GH is not bound to GHBP, ≈45% is bound to GHBP, and the remaining 5% of GH is bound to low-affinity binding proteins. Considering the multiple forms of GH, it is not surprising that significant analytical biases are observed between different immunoassays for GH. The gene for GH (chromosome 17q24.2) is a member of the GH subfamily that includes (−5′ to 3′ direction) (1) GH (the GH1 gene), (2) a chorionic somatomammotropin (human placental lactogen) pseudogene designated CSHP, (3) a chorionic somatomammotropin-A designated CSH1, (4) the placentally produced 22 kDa GH variant (GH-V; gene designation GH2), and (5) chorionic somatomammotropin-B (gene designation CSH2).40 Somatomammotropin (hPL) is a placental hormone with growth-promoting properties. Prolactin (199 amino acids) shares a homologous amino acid sequence with GH, but prolactin, encoded on chromosome 6p22, is not part of the GH complex on 17q24.2. GH has both direct and indirect activity. Its direct actions will be described in the following sections.118 The indirect activity of GH is mediated by IGF-I. To initiate its direct and indirect activity, GH binds to tissue receptors (GHR) that appear to be expressed by all tissues. The GHR is a member of the class 1 hematopoietic cytokine family.142 Other members of this family include receptors for erythropoietin, granulocyte-macrophage colony-stimulating factor, and various interferons. Structurally, the GHR is a single-chain, 620 amino acid protein (130 kDa). PreGHR includes an 18 amino acid leader sequence. The GHR structure includes an extracellular domain (246 amino acids), a transmembrane domain (24 amino acids), and a cytoplasmic domain (350 amino acids).31 When the extracellular portion of the GHR is shed, the 55 kDa GHBP moiety is released into the circulation. The GHR exists as a cell surface dimer in its inactive state (Figure 53-3). When GH binds to the GHR, the receptor recruits or activates 120 kDa Janus-associated kinase enzymes (JAK2, a type of adapter tyrosine kinase). JAK2 then exerts tyrosine kinase activity by phosphorylating itself and the GHR.132 Phosphorylation activates JAK2, which triggers several intracellular pathways involving (1) STATs (signal transducers and activators of transcription), (2) the insulin receptor substrate (IRS), (3) phosphatidylinositol 3′-kinase (PI3K), and (4) a mitogen-activated protein kinase (MAPK). STAT5b is involved, but it is unclear whether STAT5a is also involved. In its role as a transcription factor, phosphorylated, dimerized STAT5b enters the nucleus to promote gene transcription. Independent of JAK2, GHR signaling can be effected via Src, which is a tyrosine kinase. (Note: Src is the Rous sarcoma virus proto-oncogene.) Most of the circulating IGF-I is produced by hepatocytes.139 However, IGF-I is also produced locally throughout the body and thus acts as a paracrine and an autocrine hormone. The possible endocrine (systemic) influence of IGF-I on growth is discussed later. Similar to IGF-I, IGF-II does not normally produce hypoglycemia. However, reports have described tumors that secrete a larger than normal form of IGF-II. This big IGF-II did not bind normally to IGFBP-3, and therefore the free IGF-II concentration was greatly elevated. As a result of IGF-II binding to the insulin receptor, the clinical syndrome was similar to hypoglycemia caused by hyperinsulinism (i.e., free fatty acids were not elevated, ketones were absent, and lactate and alanine were not elevated), yet insulin itself was suppressed because the beta cells were normal. Removal of the tumor resolved the patient’s hypoglycemia.44 Because of their very high affinity (Kd 10−10 to 10−11 M) for IGFs, IGFBPs are regarded as inhibitors of IGF action.136 IGFBPs have higher affinity for IGFs than the IGF receptors. Currently under study is the possibility that IGFBPs may directly influence growth. Receptors for IGFBPs have been described. Proteolysis of IGFBPs releases IGF-I; therefore IGFBP proteases can influence free IGF-I concentrations. Table 53-4 summarizes the features of the seven known IGFBPs. Two types of receptors for IGFs have been identified: the type I IGF receptor and the type II IGF receptor (Figure 53-4).134 Structurally, the type I IGF receptor is similar to the insulin receptor. Because this receptor does not exclusively bind IGF-I, the terminology “IGF-I receptor” is not recommended. The type I receptor is derived from a single precursor protein of 1367 amino acids that includes the 30 amino acid signal peptide. The 706 amino acid (130 kDa) alpha chain is extracellular and is bound by a disulfide bond to the transmembrane 90 kDa (627 amino acids) beta chain. Cleavage of the alpha and beta chains releases a tetrapeptide (707 to 710: arginine-lysine-arginine-arginine). Beta chain amino acids 906 to 929 form a transmembrane domain. The receptor exists as a homodimer (beta-alpha-alpha-beta) with the two alpha chains bound to each other by two disulfide bonds. Similar to the type I IGF receptor, the insulin receptor is a homodimer of two 135 kDa alpha chains and two 95 kDa beta chains. A hybrid receptor consisting of the alpha-beta chain of the insulin receptor and the alpha-beta chain of the type I IGF receptor has been described. It has been suggested that these hybrid receptors may allow cancers to respond to insulin.23 Increasing evidence suggests that IGF-I may enhance the growth of many tumors. Thus, the casual use of IGF-I injections in children to enhance growth in short but otherwise normal children is problematic and potentially carcinogenic. GH ultimately stimulates release of IGF-I, which negatively feeds back to regulate GH release via two hypothalamic hormones: somatotropin-release inhibiting hormone (SRIH, or somatostatin; from the hypothalamic paraventricular nucleus) and growth hormone–releasing hormone (GHRH; from the hypothalamic infundibular nucleus) (Figure 53-5). These hypothalamic GH-regulating hormones are carried to the anterior pituitary via the specialized hypothalamic-pituitary portal vascular system. Somatotrophs of the anterior pituitary gland have receptors for both hormones. Somatostatin inhibits GH release, whereas GHRH promotes the release of GH. However, GH release is the predominant hypothalamic effect, because surgical interruption of the pituitary stalk with destruction of the hypothalamic-pituitary portal system leads to GH deficiency, not excess. In addition to its hypothalamic negative feedback effects, IGF-I directly suppresses pituitary release of GH. Physiologically, GH secretion is episodic.138 Consequently, random measurements cannot exclude GH deficiency; between pulses, GH concentrations can be quite low and do not distinguish GH insufficiency from normal production of the hormone. During daytime hours, the plasma concentration of GH in healthy adults remains stable and relatively low (<2 µg/L), with several secretory spikes occurring approximately 3 hours after meals (particularly meals high in protein and arginine) and after exercise. In contrast, during the evening hours, adults and children show a marked rise in GH secretory activity ≈90 minutes after the onset of sleep; GH concentrations reach a peak value during the period of deepest sleep. This pattern of GH secretion may be important for anabolic and repair processes, and for proper skeletal growth. GH is also increased by psychologic or physical stress and hypoglycemia. Normal GH secretion requires thyroxine and age-appropriate concentrations of testosterone or estrogen. GH is suppressed by elevations in blood glucose. One of the tests for GH excess measures GH following an oral glucose load; the normal response is a GH concentration of less than 1 to 2 µg/L, depending on the lower limit of detection of the GH immunoassay (Box 53-1). GH also declines with increases in free fatty acid concentrations, rapid eye movement sleep, and aging. In the presence of abnormally high concentrations of glucocorticoids, GH secretion is suppressed. In addition, circulating GH is thought to influence the release of hypothalamic hormones through the short feedback loop. Other hypothalamic hormones, such as TRH and GnRH, do not affect GH release in normal subjects but may provoke GH release in patients with acromegaly. Age-associated decline in GH production has spawned an industry of dietary supplements purported to “support” growth hormone secretion.97 These supplements are amino acid preparations that theoretically stimulate release of the subject’s own GH. Such dietary supplements have no proven medical value. Use of GH by athletes to enhance strength or promote recovery from injury is prohibited in most sports. The role of non-GHRH GH secretogogues (e.g., ghrelin, various synthetic hexapeptides) in the physiologic control of GH and growth is highly debated.6 One such secretogogue is ghrelin (28 amino acids, 3.4 kDa; gene name: GHRL; chromosome 3p2). Although ghrelin is produced in the hypothalamus, its highest concentration is found in stomach tissue. In addition, ghrelin is widely distributed in the gastrointestinal tract, heart, lung, and adipose tissue. Ghrelin appears to stimulate food intake and obesity.65 Ghrelin binds to the somatotroph GH-secretogogue receptor (gene name: GHSR; isoform 1A: 366 amino acids, 41 kDa; isoform 1B: 289 amino acids, 32 kDa; chromosome 3q26.31), which is distinct from the GHRHR. GHRL, the gene that encodes ghrelin, also encodes the 23 amino acid peptide obestatin. Obestatin may decrease food intake and increase satiety, but this has been debated. Obestatin is a ligand for the orphan GPR39 receptor (453 amino acids, 51 kDa; chromosome 2q21). However, despite obestatin binding to the GPR39 receptor, receptor activation may not follow.133 GH effects can be classified as indirect or direct.173 GH directly raises blood glucose by stimulating gluconeogenesis and reducing insulin sensitivity. Also, it causes adipose tissue lipolysis and the resulting GH-induced elevations in free fatty acids provide an alternative energy source that serves to spare glucose for CNS utilization. Therefore when glucose and free fatty acid concentrations are raised at times of stress, in partnership with epinephrine, glucagon, and cortisol, fuels for the fight-or-flight response are provided. GH has other effects on intermediary metabolism: GH stimulates the uptake of nonesterified fatty acids by muscle and accelerates the mobilization and metabolism of fat from adipose tissue to the liver. In the absence of GH, IGF-I is not as effective a growth stimulant as it is when both are present. IGF-I treatment alone is not recommended as therapy for GH deficiency; IGF-I therapy is reserved for cases of GH resistance due to GHR deficiency.95 IGF-I concentrations vary widely with age and gender.83 IGF-I rises during childhood, and during puberty, IGF-I concentrations can be two to three times the adult concentration. Following adolescence, IGF-I concentrations show a gradual decline, reaching a steady state in the third decade of life. In cases of acquired GH resistance and genetic GH resistance (GHR mutation or signaling disorder), GH concentrations will rise, and high concentrations of GH produce hyperlipidemia and hyperglycemia.75 Cases of acquired GH resistance are not treated with GH or IGF-I, but instead are treated through resolution of the underlying disorder. The actions and regulation of IGF-II have been debated.160 IGF-II is believed to be important for intrauterine growth. Mice display intrauterine growth retardation when the IGF-II gene is knocked out. Although IGF-II–producing tumors are very rare, such tumors can produce hypoglycemia. The tumors produce an aberrant form of IGF- II (“big” IGF-II) that does not associate normally with IGFBPs. The production of big IGF-II, in combination with a reduction in an IGFBP concentration, results in higher concentrations of free IGF-II, which causes hypoglycemia by associating with the insulin receptor. Both IGF-I and IGF-II are of great interest to cancer researchers, but a detailed discussion of this topic is beyond the scope of this chapter. Clinically important states of GH excess or deficiency are uncommon and are often difficult to diagnose.90 GH concentrations vary widely under normal circumstances; therefore random measurements of GH, in general, are not diagnostically useful. A single GH measurement cannot distinguish between normal fluctuations and the low or high concentrations that are typical of various diseases. GH measurements are best determined as part of dynamic testing: physiologic or pharmacologic provocative stimuli are used to help diagnose GH deficiency, whereas GH suppression (or lack thereof) following glucose administration is useful in evaluating GH excess.71 IGF-I concentrations often correlate better with the clinical severity of acromegaly than with glucose-suppressed or basal GH concentrations.49 In contrast to GH, a single measurement of IGF-I is considered to be an accurate reflection of GH-IGF-I production, irrespective of the time of the day or meals. IGF-I has a much longer half-life than GH, so its concentration is more stable. The half-life of GH is slightly longer than 15 minutes, whereas the half-life of the trimeric IGF-I-IGFBP-3-ALS complex is 17 to 22 hours. The half-life of unbound IGF-I is only 10 to 20 minutes, but the unbound form of the hormone accounts for less than 1% of the total concentration. Serum concentrations of IGF-I are influenced by (1) age, (2) gender, (3) degree of sexual maturity, (4) thyroid status, and (5) nutritional status. As mentioned previously, IGF-I concentrations are low in GH deficiency and in patients with acute or chronic protein or caloric deprivation. In pediatric endocrinology, measurements of IGFBP-3 have been used in addition to IGF-I measurements to assess GH; however, the value of this approach has not been established. The diagnostic use of GH to stimulate IGF-I production is controversial and is not currently included in standard medical practice.161 Acromegaly is the rare clinical syndrome in adults that results from GH excess.57,214 Even less common is gigantism, which results from GH excess in childhood. The clinical features of acromegaly involve overgrowth of the skeleton and soft tissue, producing (1) acral enlargement (enlargement of the extremities), (2) organomegaly (enlarged heart and/or liver), (3) facial coarsening, (4) intestinal polyposis, (5) premature cardiovascular disease, (6) hyperhidrosis (increased sweating), (7) skin tags, (8) joint disease, (9) myopathy with weakness, (10) insulin resistance, and often (11) diabetes mellitus. Premature cardiovascular disease is the most common cause of death in individuals with acromegaly. Gigantism is characterized by extreme tall stature, in addition to the clinical features of acromegaly, as pathologic GH excess occurs before epiphyseal fusion is complete. Most cases of acromegaly (≈95%) result from anterior pituitary GH-secreting tumors (somatotropinomas).58 Somatotropinomas are usually large macroadenomas (>10 mm in diameter); approximately 75% of these tumors are visualized by computed tomography (CT) or magnetic resonance imaging (MRI) (the preferred method for pituitary imaging is MRI). Some anterior pituitary tumors secrete both GH and prolactin (somatomammotropinomas). Approximately 5% of acromegaly cases result from GHRH-secreting hypothalamic tumors, and less than 1% of cases result from extrapituitary somatotropinomas, GHRH-secreting islet cells, or lung or breast tumors. In severe or advanced cases of GH excess, the diagnosis may be nearly certain on the basis of physical appearance alone. However, in less severe or early cases, the physical changes may be subtle and gradual, so a high degree of clinical suspicion is needed to make an early diagnosis. The reversibility of tissue changes depends largely on the duration of the disease. In addition to soft tissue changes, acromegaly may cause severe disability or death from cardiac, pulmonary, and/or neurologic sequelae. The most important requirement for the diagnosis of acromegaly is the demonstration of inappropriate and excessive GH secretion.150 As many as 10% of patients with active acromegaly have random serum GH concentrations that fall within the normal reference interval. Essentially all patients with acromegaly have an abnormal GH response to oral glucose (53-1). Patients with acromegaly typically show no change in their basal concentration of GH or demonstrate a paradoxical increase in GH70; normal individuals, on the other hand, show suppression of GH concentrations to less than 1.0 µg/L after the oral glucose load. Serum IGF-I concentrations are also elevated in active acromegaly, and often correlate better with the clinical severity of acromegaly than do glucose-suppressed or basal GH concentrations. In children, short stature with a normal growth velocity (≥4 to 5 cm/y) results from (1) familial short stature, (2) primordial growth failure (prenatal-onset growth failure), or (3) constitutional delay in growth and adolescence (delayed maturation). Short stature with a low growth velocity (<4 to 5 cm/y) results from genetic short stature, chronic illness, malnutrition, deprivation (nutritional or psychologic), Turner syndrome in girls, or endocrine disorders (e.g., hypothyroidism, disorders of the GHRH-GH-IGF-I axis, poorly controlled diabetes, rickets, pseudohypoparathyroidism, pseudopseudohypoparathyroidism, and Cushing syndrome).167 GH deficiency in children is characterized by (1) short stature, (2) low growth (3) velocity, (4) immature facial appearance, (5) retarded bone age on radiologic examination, and (6) increased adiposity. In cases of congenital GH deficiency, size at birth is usually normal because in utero, IGF-I does not appear to be under GH control. Micropenis (an abnormally short penis) is evident in some boys with congenital GH deficiency and will resolve with GH replacement in childhood. Adults with GH deficiency experience (1) reduced muscle mass, (2) increased adiposity, (3) osteoporosis with decreased bone density, (4) an increase in fracture risk, (5) decreased quality of life, (6) dyslipidemia, and (7) an increased risk for cardiovascular disease.113 GH deficiency is probably the most common identifiable abnormality in adults with large pituitary adenomas33 and in patients who have undergone pituitary irradiation. Thus GH replacement therapy is an important clinical intervention in GH-deficient adults and children and is considered the standard of care. GH insufficiency can be a consequence of (1) hypothalamic disease, (2) disruption of the portal system between hypothalamic nuclei and the anterior pituitary, (3) GHRHR loss-of-function mutations, or (4) somatotroph disease. GH deficiency can occur in isolation [isolated GH deficiency] or together with other pituitary deficiencies [multiple pituitary hormone deficiencies (MPHD) or combined pituitary hormone deficiencies (CPHD)]. Patients with isolated GH deficiency should be followed clinically for the development of other pituitary hormone deficiencies because MPHD can evolve over time. Biochemical testing is necessary to establish the diagnosis of GH deficiency, growth hormone resistance, or MPHD. In most affected children, the cause of GH deficiency is unknown [idiopathic GH deficiency].159 Approximately one in four children with proven GH deficiency has an organic origin for their GH deficiency; half of these children will be diagnosed with a CNS tumor. Any type of hypothalamic disease or dysfunction can lead to GHRH deficiency, including (1) tumor, (2) inflammation, (3) previous infection, (4) trauma (including previous surgery),124 (5) bleeding, (6) irradiation, and (7) malformation [septo-optic dysplasia (SOD)].116 Low-dose irradiation of the hypothalamus/pituitary can cause idiopathic GH deficiency, whereas higher doses of irradiation can cause MPHD. SOD is defined by the triad of (1) midline brain defects such as agenesis of the septum pellucidum and/or corpus callosum, (2) hypoplasia of the optic nerve, and (3) anterior and/or posterior pituitary hormone abnormalities. A small number of cases of SOD are explained by mutations in HESX1, SOX2, and SOX3, all of which are transcription factors. HEXS1 (chromosome 3p14.3) is a paired-like homeobox gene, SOX2 (chromosome 3q26.3) is the SRY (sex-determining region on the Y chromosome) box 2 gene, and SOX3 (chromosome Xq27.1) is the SRY box 3 gene. Midline brain tumors, such as meningiomas, gliomas, germinomas, third ventricle colloid cysts, ependymomas, and optic nerve gliomas, also affect the hypothalamus. Disruptions in the hypothalamic-pituitary portal system can result from tumor, inflammation, previous infection, trauma (including previous surgery), and irradiation. Congenital GH deficiency from pituitary disease has many causes, including GHRHR gene mutations (idiopathic GH deficiency type IB), GH1 mutations (idiopathic GH deficiency types IA, IB, II, and III, and bioinactive GH), and transcription factor mutations (usually causing MPHD: LHX3, LHX4, PROP1, Pit-1, RIEG) and malformations (anencephaly and holoprosencephaly) (Table 53-5).168 Homozygous LHX3 mutations have caused panhypopituitarism, with the exception that ACTH was not affected. Heterozygous LHX4 mutations have caused deficiencies of GH, TSH, and ACTH. PROP1 (name derived from “PROphet of Pit-1”; POU1F1; Pit-1 stands for “paired-like homeodomain transcription factor”)157 mutations causes deficiencies of GH, prolactin, TSH, and gonadotropins. ACTH deficiency has also occurred in some families. PROP1 gene mutations are inherited as autosomal recessive traits. Pit-1 gene mutations cause GH, prolactin, and variable degrees of TSH deficiency. These transcription factor mutations may be inherited as autosomal recessive or dominant traits. Heterozygous RIEG gene mutations (PITX2, a paired-like homeobox gene) are the cause of Rieger syndrome, which may include GH deficiency. Features of Rieger syndrome encompass developmental abnormalities of teeth, the anterior chamber of the eye, and the umbilicus. Mutations in GLI1, GLI2, Shh, ZIC2, SIX3, tgif, PATCHED1, DGF1, and FAST1 have variously been cited as causes of holoprosencephaly.88 Children with midline facial clefts (cleft lip, cleft palate, or combined) or a single central incisor can exhibit GH deficiency. Two GH1 gene mutations have been identified to cause bioinactive GH.189 In these mutations, GH does not have full biological activity but retains normal immunoreactivity. Therefore, in contrast to other forms of pituitary or hypothalamic disease, the GH concentration is not deficient. At least one reference laboratory provides GHRHR, GH1, and GHR gene sequencing services (Athena Diagnostics, Worcester, Mass).7,62,207 TABLE 53-5 GH Deficiency of Genetic Etiology Acquired causes of pituitary disease include (1) tumor (craniopharyngioma, Rathke cleft cyst, arachnoid cyst, or anterior pituitary adenoma), (2) infiltrative disease—amyloidosis, (3) inflammation (autoimmune hypophysitis and granulomatous inflammation), (4) infection, (5) trauma (including surgery), (6) bleeding, (7) irradiation, (8) infarction (pituitary apoplexy from Sheehan syndrome), and (9) metabolic derangement (hemochromatosis, iron overload from long-term transfusion therapy, or certain anemias, such as sideroblastic anemia).5 Hemochromatosis does not cause hypopituitarism until many decades have passed.35 Some investigators believe that all patients with “idiopathic” hypopituitarism should be screened for hemochromatosis.141 A staged approach to the evaluation of GH secretion is advised.166 Initial screening can involve one of the following tests: (1) measurement of IGF-I (with or without IGFBP-3), (2) GH measurement following exercise, or (3) pharmacologic GH screening. All forms of GH testing should be performed after the subject has fasted overnight. If a GH screening test is abnormal, definitive testing should be pursued.201 If GH adequacy is demonstrated by GH screening tests, definitive GH testing need not be performed unless there is a very high index of suspicion for GH deficiency. In children, the likelihood of a falsely abnormal GH screening test can be reduced by pretreatment of both boys and girls with a short course of sex steroids [ethinylestradiol: for body weights <50 lb, 20 µg/dose 18 hours, 12 hours, and 1 hour before testing; for body weights ≥50 lb: 50 µg/dose 18, 12, and 1 hour before testing, or 100 µg/d for 3 days; Premarin: for body weights <30 lb: 2.5 mg twice daily for 3 days; for body weights ≥30 lb, 5 mg twice daily for 3 days; diethylstilbestrol (DES): 5 mg/d for 3 days; or, exclusively in boys, testosterone enanthate 50 mg injected intramuscularly 2 or 3 weeks before testing]. The mechanism is unclear; however, sex steroids appear to play a major role in increasing the response of IGF-I to GH at the time of puberty. From clinical experience, GH deficiency is overdiagnosed in some peripubertal children who are tested without the benefit of sex hormone priming. Exercise physiologically enhances GH release.72 Typically, in the fasting state, the subject exercises vigorously for ≈20 minutes (e.g., running up and down stairs, running on a treadmill). At the completion of the exercise, when the subject is tachycardic and sweating, a sample is collected for GH measurement (Box 53-2). A baseline GH measurement is not required. A GH concentration may also be obtained 40 minutes after exercise, in case of delayed GH release. Finally, screening for GH deficiency can be performed by measuring GH 60 to 90 minutes following clonidine, glucagon, or L-dopa administration. (Note: L-dopa is not currently available in the United States.) Doses of clonidine, glucagon, and L-dopa are identical to doses used in formal GH testing scenarios (see later). Reference intervals for IGF-I and IGFBP-3 are age and gender dependent.73 If IGF-I is within its reference interval for age and gender in children, GH deficiency is excluded. If IGF-I is low, definitive GH testing is required. Because IGF-I concentrations can be depressed in states of (1) malnutrition, (2) malabsorption, (3) chronic disease, (4) hypothyroidism, and (5) sex hormone deficiency, a low IGF-I concentration does not confirm GH deficiency. IGFBP-3 is less dependent on good nutrition to achieve normal concentrations, so it may be a superior marker of GH deficiency compared with IGF-I. However, one study failed to demonstrate that measuring IGFBP-3 alone or together with free IGF-I was superior to measuring IGF-I alone as a screening test for GH deficiency.48 In another study, only ≈50% of GH-deficient children had a low IGFBP-3 concentration; this finding calls into question its value as a sensitive screening test.198 Analytical advantages of IGFBP-3 over IGF-I include the following: (1) IGF-I must be separated from its binding proteins to be measured, but IGFBP-3 does not require a dissociation step; (2) IGFBP-3 is present in higher concentration than IGF-I; and (3) less age dependency is seen for IGFBP-3 compared with IGF-I.47 Because of their stability over the course of a day, IGF-I and IGFBP-3 measurements may be obtained randomly. However, some researchers have concluded that IGFBP-3 measurements are too nonspecific to be used for the evaluation of GH deficiency.45 Furthermore, reports indicate that IGF-I and IGFBP-3 exhibit imperfect sensitivity and specificity for the diagnosis of GH deficiency.169 IGF-I measurements in adults often are not diagnostically helpful.130 For reasons that are unclear, IGF-I concentrations can be normal in GH-deficient adults. Therefore, a normal IGF-I does not rule out adult GH deficiency. If the IGF-I concentration is very low and suspicion for GH deficiency is high (MPHD or childhood-onset severe GH deficiency), some experts would diagnose GH deficiency in the absence of GH testing.87 GH responses to insulin-induced hypoglycemia [insulin tolerance test (ITT); Box 53-3] and GH responses to centrally acting pharmacologic or biological agents (Box 53-4) are considered definitive tests. The stimuli can be sequential or administered on different days. The classical diagnosis of pediatric GH deficiency requires that GH responses to two different stimuli (Table 53-6) be deficient. In research settings, GHRH and GHRP-6 have been used to stimulate GH release. However, because these agents are not available for clinical use, they are not included in Table 53-6. Note that many variations of these protocols are available because endocrinologists often customize these tests. Diazepam and pentagastrin have been studied as GH secretogogues; however, experience with these agents is limited, and they are not included in Table 53-6. TABLE 53-6 *Experts may differ on the best interval of GH measurements; ± indicates an optional time point. Of children with appropriate stature for age, approximately 80% will have normal GH responses to one stimulus, and at least 95% will have normal GH responses to at least one of two stimuli. This is why two stimuli are generally recommended—to avoid overdiagnosis of GH deficiency. However, the GH Research Society advises that a single definitive abnormal test is adequate to diagnose GH deficiency if the child has (1) confirmed CNS pathology, (2) a history of CNS irradiation, (3) multiple pituitary hormone deficiencies, or (4) a genetic defect.1 A history of childhood GH deficiency, CNS disease, trauma, or irradiation is an indication to test adults for GH deficiency.39,190 Retesting of adults with the diagnosis of childhood GH deficiency is necessary because not all adults with childhood GH deficiency remain deficient as adults. In adults, a single abnormal GH response to a stimulus is diagnostic of GH deficiency if the deficiency is congenital or genetic, or if multiple pituitary hormone deficiencies are due to organic disease. Guidelines published in 2009 recommended that a low IGF-I concentration in a patient with three or more pituitary hormone deficiencies is sufficient to diagnose GH deficiency.8 Unfortunately GH testing is not very reproducible.103 Insulin-induced hypoglycemia (ITT) is often considered to be the “gold standard” stimulus when hypoglycemia is indeed achieved (glucose <40 to 45 mg/dL).112 The risk associated with this type of test is that untreated severe hypoglycemia can be life threatening. Venous access for infusion of glucose is very important during the ITT. Should vascular access be lost during the ITT, glucagon should be readily available for IM injection (the dose of glucagon is 1 mg). If intravenous access cannot be ensured, stimuli other than insulin should be considered. In general, GH stimulation tests are not conducted by laboratory personnel because of the risks and complexities of testing. Arginine infusion presents the danger of acidosis and even death.183 Stimulated GH concentration less than 7 to 10 µg/L defines GH deficiency in children.26 In adults, GH deficiency is present when stimulated GH is less than 5 µg/L. GH deficiency in adults can be parsed according to the stimulus. For ITTs, a deficient peak GH response is less than 3 µg/L. For GHRH plus arginine, a deficient peak GH response is less than 11 µg/L when the body mass index (BMI) is less than 25 kg/m2. However if the BMI is 25 to 30 kg/m2, deficient is defined as less than 8 µg/L, and for BMI greater than 30 kg/m2, deficient is identified at less than 4 µg/L. Because GHRH is not commercially available, and clonidine and arginine alone are not helpful in defining GH deficiency in adults, the ITT remains the best test of GH secretion in adults. Controversy continues as to what constitutes a “normal” GH response to stimuli, because insulin-induced hypoglycemia is considered by some to be a nonphysiologic stimulus. Discordance between normal stimulated GH concentration and a deficient spontaneous rise in GH concentration has been described as neurosecretory GH deficiency.53 Neurosecretory GH deficiency can result from CNS or hypothalamic disease. The diagnosis of neurosecretory GH deficiency requires overnight, every 20 minute blood sampling with GH measurements—a protocol that ordinarily requires hospitalization. The combined costs of GH assays, physician fees, and inpatient services would exceed several thousand dollars. The definition of partial GH deficiency is especially problematic because the definition of simple GH deficiency is itself controversial.91,110 Eliminating GH stimulation testing has been proposed, with the diagnosis of GH deficiency based on growth parameter, IGF-I and IGFBP-3 measurements, neuroradiologic investigation, and genetic considerations.14,137,211 In children with short stature and low growth velocity, if IGF-I is (1) below the reference interval for the child’s bone age and gender, (2) if the GH concentration is normal or elevated, and (3) if non–GH-dependent causes of IGF-I deficiency (malnutrition, malabsorption, chronic disease, hypothyroidism, and sex hormone deficiency compared with the patient’s bone age) have been excluded, GH resistance should be considered.171 As uncommon as GH deficiency is in the general pediatric population (1 in 10,000 children), GH resistance as a primary problem is far less frequent. GH resistance can be congenital, resulting from loss-of-function GHR mutations or GHR signaling defects (STAT5b mutations),123,215 or from defects in the production of IGF-I itself. Most GHR mutations involve the extracellular domain that involves GH binding to the GHR. Some GHR mutations affect homodimerization. Loss of the intracellular GHR domain can result from splice-site mutations. GHR is not expressed on the cell surface if the transmembrane domain is defective. Recall that circulating GHBP is derived from the extracellular domain of the GHR. Most cases of GHR deficiency display low or absent concentrations of GHBP. STAT5b is necessary for normal GH-GHR signaling to the cell nucleus. These are autosomal recessive disorders. Size at birth is normal because in utero IGF-I production is independent of GH. IGF-I gene inactivating mutations or deletions are rare, and only two such mutations have been described. In contrast to GH receptor and signaling defects, when IGF-I itself cannot be produced because of intrinsic IGF-I gene mutations, intrauterine growth retardation will result, in addition to extrauterine growth failure. Other consequences of IGF-I gene mutations include severe mental retardation, deafness, and micrognathia (mandibular hypoplasia).206,216 IGF-I and IGFBP-3 concentrations are very low in rare cases of ALS deficiency, while baseline and poststimulation GH concentrations are normal.67 Although adolescence was delayed, adult stature was nearly normal in persons with ALS deficiency. A number of criteria for GH resistance have been proposed, including (1) height more than 3 standard deviations below the mean for age; (2) basal GH greater than 2.5 ng/mL; (3) basal IGF-I less than 50 ng/mL; (4) basal IGFBP-3 more than 2 standard deviations below the mean for age; (5) an increase in IGF-I of less than 15 ng/mL after 4 days of GH treatment (0.05 mg/kg/d); and (6) increase in IGFBP-3 of less than 0.4 µg/mL after GH treatment. The largest concentrations of subjects with GH resistance are found in Israel and southern Ecuador.170
Pituitary Function and Pathophysiology
Anatomy
Pituitary Embryology
Regulation of Function of the Adenohypophysis
Hypothalamic Regulation
Anterior Pituitary
Growth Hormone and Insulin-like Growth Factors
Somatotropin Release–Inhibiting Hormone
Growth Hormone–Releasing Hormone Receptor (GHRHR)
Somatostatin (SRIH) Receptor
Growth Hormone (GH)
Growth Hormone Receptor (GHR)
Insulin-like Growth Factors
Insulin-like Growth Factor Binding Proteins
Receptors for Insulin-like Growth Factors
Regulation of Growth Hormone (GH) Secretion
Physiologic Actions
Clinical Significance
Growth Hormone Excess
Growth Hormone Deficiency and Growth Retardation
Gene/Classification/Inheritance
Mutation
Phenotype
GH1; IA; AR
Deletion, FS, NS
Absent GH expression; immune resistance to GH treatment is common
GH1; IB; AR
Splicing?
Reduced GH; responds to GH treatment*
GHRHR; IB; AR
Possible MS
Reduced GH; responds to GH treatment*
GH1; II; AD
DN
Reduced GH; responds to GH treatment; MPHD is possible
Unknown; III; XLR
—
Reduced GH; responds to GH treatment; agammaglobulinemia is possible
Growth Hormone Resistance
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Pituitary Function and Pathophysiology