Picornaviridae: The Viruses and their Replication
Vincent R. Racaniello
Viruses in the family Picornaviridae have nonenveloped particles with a single-stranded RNA (ssRNA) genome of positive polarity. Among its many members are numerous important human and animal pathogens, such as poliovirus, hepatitis A virus, foot-and-mouth disease virus (FMDV), enterovirus 71,
and rhinovirus. The name of the virus family was intended to convey the small size of the viruses (pico, a small unit of measurement [10−12]) and the type of nucleic acid that constitutes the viral genome (RNA).
and rhinovirus. The name of the virus family was intended to convey the small size of the viruses (pico, a small unit of measurement [10−12]) and the type of nucleic acid that constitutes the viral genome (RNA).
Picornaviruses have played important roles in the development of modern virology. Foot-and-mouth disease virus was the first animal virus to be discovered, by Loeffler and Frosch in 1898.305 Poliovirus was isolated 10 years later,289 a discovery spurred by the emergence of epidemic poliomyelitis at the turn of the 20th century. The discovery in 1949 that poliovirus could be propagated in cultured cells led to studies of viral replication.135 The plaque assay, an essential method for quantification of viral infectivity, was developed with poliovirus.130 The first RNA-dependent RNA polymerase identified was that of mengovirus,29 a picornavirus, and the synthesis of a precursor polyprotein from which viral proteins are derived by proteolytic processing was first identified in poliovirus-infected cells.477 The first infectious DNA clone of an animal RNA virus was that of poliovirus,416 and the first three-dimensional structures of animal viruses determined by x-ray crystallography were those of poliovirus223 and rhinovirus.435 Poliovirus RNA was the first messenger RNA (mRNA) shown to lack a 5′ cap structure.215,361 This observation was subsequently explained by the finding that the genome RNA of poliovirus and other picornaviruses is translated by internal ribosome binding,243,385 a process now known to occur on cellular mRNA.248,313
Because they cause serious diseases, poliovirus and FMDV have been the best-studied picornaviruses. Research on poliovirus has produced two effective vaccines; it is likely that poliomyelitis will be eradicated from the globe in the near future. The World Health Organization (WHO) has established a goal of cessation of vaccination, at which time all poliovirus stocks must be destroyed. When this historic event takes place, all research on this virus will cease. Poliovirus is truly a virus with a brilliant past, but with no future.
Classification
The family Picornaviridae belongs to the order Picornavirales and comprises 12 genera (Table 16.1), which all contain viruses that infect vertebrates.280 Information on selected viruses is summarized below.
The genus Aphthovirus consists of four species: Foot-and-mouth disease virus (FMDV), Bovine rhinitis A virus, Bovine rhinitis B virus, and Equine rhinitis A virus. FMDV infects cloven-footed animals (e.g., cattle, goats, pigs, and sheep), primarily via the upper respiratory tract, and has been isolated from at least 70 species of mammals. Seven serotypes of FMDV have been identified; within each serotype are many subtypes. These viruses are highly labile and rapidly lose infectivity at pH values of less than 6.8.
There are two species within the genus Cardiovirus: Encephalomyocarditis virus and Theilovirus. The encephalomyocarditis viruses (which include strains known as encephalomyocarditis virus, Columbia SK virus, Maus Elberfeld virus, and Mengovirus) are murine viruses, which can also infect many other hosts, including humans, monkeys, pigs, elephants, and squirrels. The second species includes the Theiler’s murine encephalomyelitis viruses and human Saffold viruses.
Table 16.1 Members of the Family Picornaviridae | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
There are 10 species in the Enterovirus genus: Human enterovirus A, B, C, D; Simian enterovirus A, Bovine enterovirus, Porcine enterovirus B, and Human rhinovirus A, B, C. Members of this genus include poliovirus (3 serotypes), Coxsackievirus (25 serotypes), echovirus (28 serotypes), human enterovirus (43 serotypes), and many nonhuman enteric viruses. Enteroviruses such as poliovirus (3 serotypes) and Coxsackievirus (25 serotypes) replicate in the alimentary tract and are resistant to low pH. The acid-labile rhinoviruses (so named because they replicate in the nasopharynx) are important agents of the common cold. There are 100 serotypes of Human rhinovirus A and B. Rhinoviruses may also replicate in the lower respiratory tract; the newly discovered Human rhinovirus C (49 types) have been associated with severe lower respiratory tract disease.327
The genus Hepatovirus contains a single species, the human pathogen Hepatitis A virus (one serotype). Virions are highly stable and resistant to acid pH and high temperatures (60°C for 10 min). The virus infects epithelial cells of the small intestine and hepatocytes.
The Parechovirus genus contains two species, Human parechovirus and Ljungan virus, a virus of rodents. Parechoviruses are etiologic agents of respiratory and gastrointestinal disease.
Proteins of parechoviruses are substantially diverged from those of other picornaviruses, with no greater than 30% amino acid identity.
Proteins of parechoviruses are substantially diverged from those of other picornaviruses, with no greater than 30% amino acid identity.
The Erbovirus genus contains three types of Equine rhinitis B virus. These viruses cause upper respiratory tract disease in horses that is associated with viremia and fecal shedding of virus particles. The Kobuvirus genus contains two species: Aichi virus, which causes gastroenteritis in humans, and Bovine kobuvirus. The Teschovirus genus consists of 12 serotypes of Porcine teschovirus. Some strains can cause polioencephalitis in pigs, also called Teschen/Talfan disease or teschovirus encephalomyelitis. There are three species in the Sapelovirus genus: Porcine sapelovirus, Simian sapelovirus, and Avian sapelovirus. The sole species in the genus Senecavirus is Seneca Valley virus, found in pigs throughout the United States, but there is no known associated disease. It is currently in clinical trials to assess its value for treating human tumors.440 Avian encephalomyelitis virus, which causes the eponymous disease in young chickens, pheasants, quail, and turkeys, is the sole species in the genus Tremovirus.
A number of other related viruses may be members of the Picornaviridae but have not been approved as species by the International Committee on Taxonomy of Viruses (ICTV). These include human cosaviruses, identified in the stools of south Asian children,224,262 human klassevirus/salivirus,183,225,297 and many others.280
Virion Structure
Physical Properties
Picornavirus virions are spherical, with a diameter of about 30 nm (Fig. 16.1). The particles are simple, consisting of a protein shell surrounding the naked RNA genome. The virus particles lack a lipid envelope, and their infectivity is insensitive to organic solvents. Cardioviruses, enteroviruses (except rhinoviruses), hepatoviruses, and parechoviruses are acid stable and retain infectivity at pH values of 3.0 and lower. In contrast, rhinoviruses and aphthoviruses are labile at pH values of less than 6.0. Differences in pH stability influence the sites of replication of the virus. For example, rhinoviruses and aphthoviruses replicate in the respiratory tract and need not be acid stable. Because they are acid labile, they cannot replicate in the alimentary tract. Cardioviruses, enteroviruses, hepatoviruses, and parechoviruses pass through the stomach to gain access to the intestine and, therefore, must be resistant to low pH. The structural basis for the acid lability of FMDV is partly understood (see Entry into Cells).
The buoyant densities of picornaviruses are quite different (Table 16.2). Cardioviruses and enteroviruses have a buoyant density of 1.34 g/mL, that of FMDV is 1.45 g/mL, and rhinoviruses have an intermediate value (1.40 g/mL). The reason for the difference lies in the permeability of the viral capsid to cesium. The capsid of poliovirus does not allow cesium to reach the RNA interior; thus, the virus bands at an abnormally light buoyant density.146 In contrast, aphthovirus capsids contain pores that allow cesium to enter.1 The rhinovirus capsid is permeable to cesium, but the presence of polyamines in the capsid interior limits the amount of cesium that can enter, which provides an explanation for the intermediate buoyant density value of these viruses.146
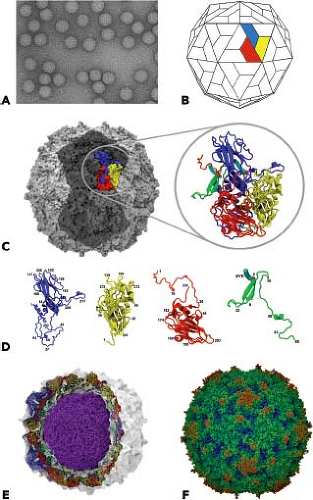 Figure 16.1. Structural features of picornaviruses. A: Electron micrograph of negatively stained poliovirus (×270,000 magnification). B: Schematic of the picornavirus capsid, showing the pseudoequivalent packing arrangement of VP1 (blue), VP2 (yellow), and VP3 (red). VP4 (green) is on the interior of the capsid. A single biologic protomer is colored. C: Model of poliovirus type 1, Mahoney, based on x-ray crystallographic structure determined at 2.9 Å. Two adjacent pentamers aligned at the twofold axis of symmetry are shaded dark gray. A single protomer is colored and also expanded at right as a cartoon of the alpha carbon backbone; capsid proteins are color coded according to B. D: Individual capsid proteins VP1, VP2, VP3, and VP4 are shown as cartoon representations of the alpha carbon backbone. E: Virion after 10 nanoseconds of atomistic molecular dynamics simulation. Volumetric (density) map representation is white with protein backbone shown as tubes and colored as in B. Lipids are represented as Van der Waals models with coloring by element. RNA is colored purple and associated magnesium ions are colored blue. F: All-atom Van der Waals representation of the poliovirus capsid with radial coloring depicting the relative distance from the center of the particle; blue = 133 Å to red = 166 Å. (A courtesy of N. Cheng and D. M. Belnap; C to F courtesy of Jason A. Roberts, WHO Poliomyelitis Regional Reference Laboratory, Victorian Infectious Diseases Reference Laboroatory, North Melbourne, Australia.) |
Table 16.2 Physical Properties of Some Picornaviruses | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
Ratio of Particles to Infectious Viruses
The ratio of particles to infectious virus is determined by dividing the number of virus particles in a sample (determined by electron microscopy [EM] or spectrophotometric measurements) by the plaque titer, yielding the particle-to-plaque forming unit (pfu) ratio. This ratio is a measurement of the fraction of virus particles that can complete an infectious cycle. The particle-to-pfu ratio of poliovirus ranges from 30 to 1,000, and that of other picornaviruses is in the same range. The high particle-to-pfu ratio may be caused by the presence of lethal mutations in the viral genome. This explanation, however, probably does not apply to all picornaviruses; it has been shown that the infectivity of aphthovirus RNA approaches one infectious unit per molecule. An alternative explanation is that all viruses do not successfully complete an infectious cycle because they fail at one of several steps that must be completed, including attachment, entry, replication, and assembly.
High-Resolution Structure of the Virion
The capsids of picornaviruses are composed of four structural proteins: VP1, VP2, VP3, and VP4. The results of x-ray diffraction studies, EM observations, and biochemical studies of virus particles and their dissociation products led to the hypothesis that the picornavirus capsid contains 60 structural proteins arranged into an icosahedral lattice.441 Our understanding of the structure of picornaviruses was substantially advanced in 1985 when the atomic structures of poliovirus type 1223 and human rhinovirus type 14435 were determined by x-ray crystallography. Since then, the high-resolution structures of many other picornaviruses have been determined.
The basic building block of the picornavirus capsid is the protomer, which contains one copy each of VP1, VP2, VP3, and VP4. The shell is formed by VP1 to VP3, and VP4 lies on its inner surface (Fig. 16.1). VP1, VP2, and VP3 have no sequence homology, yet all three proteins have the same topology: they form an eight-stranded, antiparallel β-barrel (also called a β-barrel jelly roll or a Swiss-roll β-barrel). This domain is a wedge-shaped structure made up of two antiparallel β-sheets. One β-sheet forms the wall of the wedge, and the second, which has a bend in the center, forms both a wall and the floor. The wedge shape facilitates the packing of structural units to form a dense, rigid protein shell. Packing of the β-barrel domains is strengthened by a network of protein–protein contacts on the interior of the capsid, particularly around the fivefold axes. This network, which is formed by the N-terminal extensions of VP1 to VP3 as well as VP4, is essential for the stability of the virion. VP4 differs significantly from the other three proteins in that it has an extended conformation. This protein is similar in position and conformation to the NH2-terminal sequences of VP1 and VP3 and functions as a detached NH2-terminal extension of VP2 rather than an independent capsid protein.
The main structural differences among VP1, VP2, and VP3 lie in the loops that connect the β-strands and the N- and C-terminal sequences that extend from the β-barrel domain. These amino acid sequences give each picornavirus its distinct morphology and antigenicity. The C-termini are located on the surface of the virion, and the N-termini are on the interior, indicating that significant rearrangement of the P1 precursor occurs on proteolytic cleavage.
Resolution of the structures of poliovirus and rhinovirus revealed that the β-barrel domains are strikingly similar in structure to those of plant viruses such as southern bean mosaic virus and tomato bushy stunt virus. The capsid proteins of these viruses bear no sequence homology with those of the picornaviruses. It has since become apparent that similar protein topology is found in the capsid proteins of many plant, insect, and vertebrate positive-stranded RNA viruses as well as in the DNA-containing papovaviruses and adenoviruses. These findings suggest that either the polypeptides evolved from a common ancestor or that the β-barrel domain is one of the few ways to allow proteins to pack to form a sphere.
In some viruses, such as the parechoviruses, VP0, the precursor to VP2 + VP4, remains uncleaved, while in hepatitis A virus, VP4 is very small.
Surface of the Virion
Resolution of the structures of poliovirus and human rhinovirus revealed that the surfaces of these viruses have a corrugated topography; a prominent, star-shaped plateau (mesa) is found at the fivefold axis of symmetry, surrounded by a deep depression (canyon) and another protrusion at the threefold axis (Fig. 16.1). It was originally proposed that the canyon is the receptor-binding site—this hypothesis has been proved for a number of enteroviruses. Not all picornaviruses have canyons. The surface of cardioviruses bears a series of depressions, or “pits,” which are involved in receptor binding, while that of FMDV is much smoother. As will be discussed later, a flexible loop that projects from the capsid surface binds to the cellular receptor for FMDV.
Interior of the Virion
A network formed by the N-termini on the interior of the capsid contributes significantly to the stability of the virion.
At the fivefold axis of symmetry, the N-termini of five VP3 molecules form a cylindrically parallel β-sheet. This structure is surrounded by five three-stranded β-sheets formed by the N-termini of VP4 and VP1. The myristate group attached to the N-terminus of VP4 mediates the interaction between these two structures.93 Interactions among pentamers are stabilized by a seven-stranded β-sheet, composed of four β-strands of the VP3 β-barrel and one strand from the N-terminus of VP1 that surround a two-stranded β-sheet made from the N-terminus of VP2 from a neighboring pentamer.138
At the fivefold axis of symmetry, the N-termini of five VP3 molecules form a cylindrically parallel β-sheet. This structure is surrounded by five three-stranded β-sheets formed by the N-termini of VP4 and VP1. The myristate group attached to the N-terminus of VP4 mediates the interaction between these two structures.93 Interactions among pentamers are stabilized by a seven-stranded β-sheet, composed of four β-strands of the VP3 β-barrel and one strand from the N-terminus of VP1 that surround a two-stranded β-sheet made from the N-terminus of VP2 from a neighboring pentamer.138
Poliovirus genomes containing an extra 1,500 nucleotides can be successfully packaged, indicating that the interior of the capsid is not fully occupied.9 It has been suggested that picornaviral capsids are stabilized by interactions with the RNA genome, based on findings with bean pod mottle virus, which is related to picornaviruses. In this virus, ordered RNA can be observed at the threefold axis, and packaging of viral RNA stabilizes subunit interactions.91,299 Several nucleotides have been tentatively identified in a similar location in the structures of P3/Sabin and rhinovirus type 14.25,138 In the atomic structure of poliovirus P2/Lansing, RNA bases have been observed stacking with conserved aromatic residues of VP4.294 The structure of Seneca Valley virus revealed the arrangement of the RNA within the capsid.511 Much of the nucleic acid contacts the inner surface of the capsid, particularly near the twofold axes under VP2. The RNA forms a shell that contacts both VP2 and VP4 and which could serve as a scaffold for capsid assembly or might contribute to virion stability.
Hydrophobic Pocket
Within the core of VP1, just beneath the canyon floor of many picornaviruses, is a hydrophobic tunnel or pocket (Fig. 16.2). Electron density observed in this area has been interpreted to be cell-derived lipids called “pocket factors.” In poliovirus types 1 and 3, the pocket is believed to contain sphingosine.138 An unidentified lipid has been found in human rhinoviruses types 1A and 16, a C16 fatty acid has been modeled in the pocket of coxsackievirus B3,194,271,348 coxsackievirus A21 is believed to carry myristic acid,535 and bovine enterovirus contains a mixture of palmitic and myristic acid.464 In the enterovirus 71 virion, the pocket factor is partly exposed on the floor of the canyon.403 In contrast, the pocket of human rhinovirus type 14 is empty.25 The results of introducing amino acid changes in the pocket of rhinovirus 16 suggest that the hydrophobic pocket, and not the pocket factor, is important for maintaining capsid dynamics.265
The hydrophobic pocket is also the binding site for antipicornavirus drugs such as the WIN compounds produced by Sterling-Winthrop462 as well as similar molecules produced by Janssen Pharmaceuticals (Titusville, NJ)19,104 (Fig. 16.2). Some of these drugs have been evaluated in clinical trials, such as pleconaril for treatment of common colds caused by rhinoviruses392 and for enteroviral sepsis syndrome.356 These hydrophobic, sausage-shaped compounds bind tightly in the hydrophobic tunnel, displacing any lipid that is present, inhibiting either binding or uncoating. Drug-dependent mutants of poliovirus spontaneously lose infectivity at 37°C, probably because they do not contain lipid in the pocket.347
Myristate
Myristic acid (n-tetradecanoic acid) is covalently linked to glycine at the amino terminus of VP4 of most picornaviruses.93 This fatty acid is an integral part of the viral capsid. The N-termini of VP3 intertwine around the fivefold axis to form a twisted tube of parallel β-structure.223 The five myristyl groups extend from the N-termini of VP4 and cradle the twisted tube formed by VP3.93 The myristyl groups interact with amino acid side chains of VP4 and VP3. Mutagenesis of VP4 has revealed a role for myristic acid modification in virus assembly and in the stability of the capsid.20,314,315,316,344
Neutralizing Antigenic Sites
Viral serotype is determined by the connecting loops and C-termini of the capsid proteins that decorate the outer surface of the virion. These contain the major neutralization antigenic sites of the virus, the amino acid sequences that are recognized by antibodies that block viral infectivity. Such sites are defined by mutations that confer resistance to neutralization with monoclonal antibodies directed against the viral capsid.334,335,458 Human sera contain antibodies directed against poliovirus antigenic sites identified in mice.423
The large area of contact of the poliovirus receptor (PVR) CD155 on the virion surface, compared with other picornavirus–receptor interactions, might provide a clue to why there are many rhinovirus serotypes but only three serotypes of poliovirus.209 This large interaction area could limit the viability of antibody-escape mutants because they would also compromise their ability to bind CD155.
Genome Structure and Organization
General Features
The genome of picornaviruses is a single positive-stranded RNA molecule (Fig. 16.3). The viral RNA is infectious because it is translated on entry into the cell to produce all the viral proteins required for viral replication. Picornavirus genomic RNA is unique because it is covalently linked at the 5′ end to a protein called VPg (virion protein, genome linked).142,293 VPg is covalently joined to the 5′-uridylylate moiety of the viral RNA by an O4-(5′-uridylyl)-tyrosine linkage. The tyrosine that is linked to the viral RNA is always the third amino acid from the N-terminus. VPg of different picornaviruses varies in length from 22 to 24 amino acid residues and is encoded by a single viral gene, except in the genome of FMDV, which encodes three VPg genes.145 VPg is not required for infectivity of poliovirus RNA; if it is removed from viral RNA by treatment with proteinase, the specific infectivity of the viral RNA is not reduced. VPg is not found on viral mRNA that is associated with cellular ribosomes and undergoing translation; these mRNA contain only uridine 5′-phosphate (pU) at their 5′ ends. Poliovirus mRNA differs from virion RNA only by the lack of VPg.363,389 VPg is removed from virion RNA by a host protein called unlinking enzyme.12 It is not known whether removal of VPg is a prerequisite for association with ribosomes or is a result of that association. Although VPg-linked RNA can be translated in cell-free extracts, it is possible that VPg is rapidly cleaved from the RNA such that only RNA lacking VPg are translated.169,338,514 VPg is present on nascent RNA
chains of the replicative intermediate RNA and on negative-stranded RNA, which has led to the suggestion that VPg is a primer for poliovirus RNA synthesis.362,389 The role of VPg in viral RNA synthesis is discussed in subsequent sections.
chains of the replicative intermediate RNA and on negative-stranded RNA, which has led to the suggestion that VPg is a primer for poliovirus RNA synthesis.362,389 The role of VPg in viral RNA synthesis is discussed in subsequent sections.
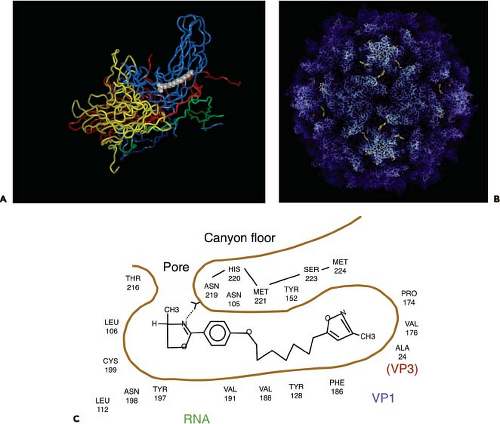 Figure 16.2. The hydrophobic pocket in the picornavirus capsid. A: Cellular lipid bound in the hydrophobic pocket of poliovirus type 1. Side view of a protomer, consisting of one copy each of VP1, VP2, VP3, and VP4. RNA is below, and the fivefold axis of symmetry is at the upper right. Gray spheres represent what is believed to be a molecule of sphingosine bound in the hydrophobic pocket. The lipid is just below the canyon floor. B: Drug bound in the poliovirus pocket. R78206, a WIN-like compound, bound in the poliovirus capsid. The bound drug is the small yellow molecule at the base of the canyon. The drug appears to be exposed on the surface but is actually in the hydrophobic pocket. The capsid model is shown in a radial depth-cued representation, in which atoms are colored according to whether they are near the center of the virus (blue) or far from the center (white). Residues at the bottom of the canyon are dark because they are at a position of low radius. The bound drug is not depth cued. C: WIN52084 bound in the hydrophobic pocket. These drugs displace the lipid from the pocket, thereby blocking infectivity. |
Nucleotide sequence analysis of many picornavirus RNAs has revealed a common organizational pattern (Fig. 16.3). The genomes vary in length from 7 to 8.8 kb. The 5′-noncoding regions of picornaviruses are long (0.5 to 1.5 kb) and highly structured. This region of the genome contains sequences that control genome replication and translation. The 5′-noncoding region contains the internal ribosome entry site (IRES) that directs translation of the mRNA by internal ribosome binding. The 5′-untranslated regions of aphthoviruses, cardioviruses, and erboviruses contain a poly(C) tract that varies in length among different virus strains (80 to 250 nucleotides in cardioviruses, 40 to 400 nucleotides in aphthoviruses). Among cardioviruses, longer poly(C) length is associated with higher virulence in animals.129,198
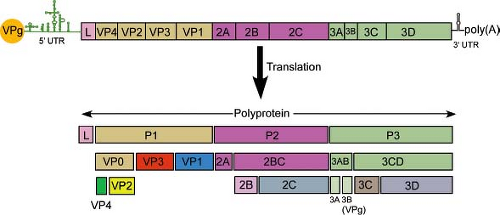 Figure 16.3. Organization of a picornavirus genome. Top: Schematic of the viral RNA genome, with the genome-linked protein VPg at the 5′ end, the 5′ untranslated region containing the IRES, the protein coding region, the 3′ untranslated region containing a pseudoknot, and the poly(A) tail. L is a leader protein encoded in the genomes of erboviruses, cardioviruses, and aphthoviruses but not other picornaviruses. Coding regions for the viral proteins are indicated. Bottom: Processing pattern of picornavirus polyprotein. Some genomes encode multiple copies of protein coding regions, e.g., there are three VPgs in the FMDV genome, two 2A motifs in Ljungan virus, and three 2A motifs in the duck hepatitis A virus genome. |
The 3′-noncoding region of picornaviruses is short, ranging in length from 40 to 330 nucleotides. This region may also contain a secondary structure, a pseudoknot, that has been implicated in controlling viral RNA synthesis.241 The entire 3′-noncoding region of poliovirus and rhinovirus is not required for infectivity, however.77,489 Both virion RNA and mRNA contain a 3′ stretch of poly(A).541 Negative-stranded RNA contains a 5′ stretch of poly(U), which is copied to form poly(A) of the positive strand.542 The average length of the poly(A) tail varies from 35 nucleotides in encephalomyocarditis virus to 100 nucleotides in aphthoviruses.5 Viral RNA from which the poly(A) tract is removed is noninfectious.469
The results of biochemical studies of poliovirus-infected cells had predicted the presence of a single, long, open reading frame (ORF) on the viral RNA that is processed to form individual viral proteins.477 This hypothesis was proved when the nucleotide sequence of the poliovirus genome was determined, which revealed that the viral RNA encodes a single ORF.276,415 A similar strategy for viral gene expression occurs during the replication of all picornaviruses. The polyprotein is cleaved during translation, so that the full-length product is not observed. Cleavage is carried out by virus-encoded proteinases to yield 11 to 15 final cleavage products. Some of the uncleaved precursors also have functions during replication.
To unify the nomenclature of picornavirus proteins, the polyprotein has been divided into three regions: P1, P2, and P3 (Fig. 16.3). The genomes of some picornaviruses encode a leader (L) protein before the P1 region. The P1 region encodes the viral capsid proteins, whereas the P2 and P3 regions encode proteins involved in protein processing (2Apro, 3Cpro, 3CDpro) and genome replication (2B, 2C, 3AB, 3BVPg, 3CDpro, 3Dpol). The genome of FMDV encodes three VPg proteins. The genome of Ljungan virus, a parechovirus, may encode two unrelated 2A proteins249 and that of duck hepatitis A virus in the genus Avihepatovirus encodes three 2A motifs.
Infectious DNA Clones of Picornavirus Genomes
Recombinant DNA techniques allow the introduction of mutations anywhere in the genome of most animal viruses. An infectious DNA clone—a double-stranded copy of the viral genome carried on a bacterial plasmid—or RNA transcripts derived by in vitro transcription, can be introduced into cultured cells by transfection to recover infectious virus. The first infectious DNA clone of an animal RNA virus was that of poliovirus.416 The infectivity of cloned poliovirus DNA (103 pfu/μg) is much lower than that of genomic RNA (106 pfu/μg). The development of plasmid vectors incorporating promoters for bacteriophage SP6, T7, or T3 RNA polymerase for the production of RNA transcripts in vitro enabled the production of infectious picornavirus RNA from cloned DNA.503 Such RNA transcripts have an infectivity approaching that of genomic RNA. A similar approach has been adopted for the recovery of many other picornaviruses from cloned DNA copies of the viral genome.
Stages of Replication
Replication of picornaviruses occurs in the cell cytoplasm. The first step is attachment to a cell receptor (Fig. 16.4). The RNA genome is then uncoated, a process that involves structural changes in the capsid. Once the positive-stranded viral RNA enters the cytoplasm, it is translated to provide viral proteins essential for genome replication and the production of new virus particles. The viral proteins are synthesized from a polyprotein precursor, which is cleaved nascently. Cleavages are carried out mainly by two viral proteinases, 2Apro and 3Cpro or 3CDpro. Among the proteins synthesized are the viral
RNA–dependent RNA polymerase and accessory proteins required for genome replication and mRNA synthesis. The first step in genome replication is copying of the positive-stranded RNA to form a negative-stranded intermediate; this step is followed by the production of additional positive strands. These events occur on small membranous vesicles that are induced by several virus proteins. Once the pool of capsid proteins is sufficiently large, encapsidation begins. Coat protein precursor P1 is cleaved to produce an immature protomer, which then assembles into pentamers. Newly synthesized, positive-stranded RNA associates with pentamers to form the infectious virus. Empty capsids that are found in infected cells are likely to be a storage form of pentamers.
RNA–dependent RNA polymerase and accessory proteins required for genome replication and mRNA synthesis. The first step in genome replication is copying of the positive-stranded RNA to form a negative-stranded intermediate; this step is followed by the production of additional positive strands. These events occur on small membranous vesicles that are induced by several virus proteins. Once the pool of capsid proteins is sufficiently large, encapsidation begins. Coat protein precursor P1 is cleaved to produce an immature protomer, which then assembles into pentamers. Newly synthesized, positive-stranded RNA associates with pentamers to form the infectious virus. Empty capsids that are found in infected cells are likely to be a storage form of pentamers.
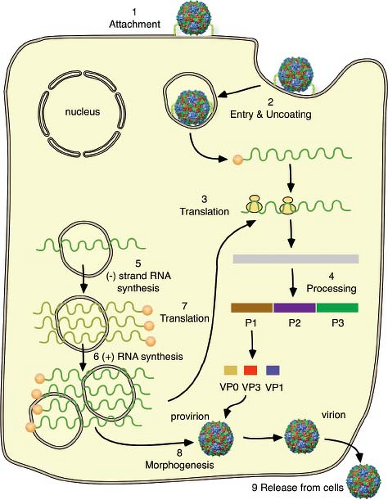 Figure 16.4. Overview of the picornavirus replication cycle. Virus binds to a cellular receptor (1) and the genome is uncoated (2). VPg (virion protein, genome linked) is removed from the viral RNA, which is then translated (3). The polyprotein is cleaved nascently to produce individual viral proteins (4). RNA synthesis occurs on membrane vesicles induced by viral proteins (not drawn to scale). Viral (+) strand RNA is copied by the viral RNA polymerase to form full-length (–) strand RNAs (5), which are then copied to produce additional (+) strand RNA (6). Early in infection, newly synthesized (+) strand RNA is translated to produce additional viral proteins (7). Later in infection, the (+) strands enter the morphogenetic pathway (8). Newly synthesized virus particles are released from the cell by lysis (9). |
The entire time required for a single replication cycle ranges from 5 to 10 hours, depending on many variables, including the particular virus, temperature, pH, host cell, and multiplicity of infection. Many picornaviruses are released as the cell loses its integrity and lyses. Other picornaviruses (e.g., hepatitis A virus) are released from cells in the absence of cytopathic effect.
Attachment
Cellular Receptors and Coreceptors
Like most viruses, picornaviruses initiate infection of cells by first attaching to a receptor on the host cell plasma membrane. The nature of picornavirus receptors remained obscure until 1989, when the receptors for poliovirus and the major group rhinoviruses were identified.184,332,471 Receptors for many other members of this virus family have since been identified (Table 16.3). Different types of cell surface molecules serve as
cellular receptors for picornaviruses; some are shared among picornaviruses and members of other virus families. For example, the cell surface protein CD55 is a receptor for certain Coxsackieviruses, echoviruses, and enterovirus 70, and the PVR CD155 is a receptor for alphaherpesviruses. For some picornaviruses (e.g., poliovirus and rhinovirus), a single type of receptor is sufficient for entry of viruses into cells. For other viruses, a second molecule, or coreceptor, is needed for virus entry into cells. For example, Coxsackievirus A21, which attaches to CD55, requires intercellular adhesion molecule 1 (ICAM-1) for entry into cells.453
cellular receptors for picornaviruses; some are shared among picornaviruses and members of other virus families. For example, the cell surface protein CD55 is a receptor for certain Coxsackieviruses, echoviruses, and enterovirus 70, and the PVR CD155 is a receptor for alphaherpesviruses. For some picornaviruses (e.g., poliovirus and rhinovirus), a single type of receptor is sufficient for entry of viruses into cells. For other viruses, a second molecule, or coreceptor, is needed for virus entry into cells. For example, Coxsackievirus A21, which attaches to CD55, requires intercellular adhesion molecule 1 (ICAM-1) for entry into cells.453
Table 16.3 Some Cell Receptors for Picornaviruses | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
One Type of Receptor Molecule for Virus Binding and Entry
For certain picornaviruses, a single type of receptor molecule is sufficient for virus binding and entry. These include the cell receptors for poliovirus (PVR/CD155), rhinoviruses (ICAM-1, low-density lipoprotein receptor LDLR, LDLR-related protein, very-low-density lipoprotein receptor), EMCV (VCAM-1), hepatitis A virus (TIM-1), and enterovirus 71 (SCBR2, PSGL-1).
The PVR is a type I transmembrane protein and a member of the immunoglobulin (Ig) superfamily of proteins, with three extracellular Ig-like domains: a membrane-distal V-type domain followed by two C2-type domains.332 Production of PVR in mice is sufficient to overcome the lack of susceptibility of this species to poliovirus infection.282,421 Because PVR transgenic mice develop poliomyelitis after inoculation with poliovirus by different routes, they have proved to be a valuable model for studying the pathogenesis of poliomyelitis.417 PVR transgenic mice are not susceptible to infection by the oral route, the natural route of infection in humans, unless the gene encoding type I interferon receptors has been deleted.370,549 PVR is synthesized in many tissues in transgenic mice, yet the main sites of poliovirus replication are the brain and spinal cord.282,422 This restricted tropism is regulated by the interferon (IFN)-α/β response, which limits viral replication in extraneural organs.232
Expression of PVR on cultured cells derived from different animal species leads to susceptibility to poliovirus infection. Therefore it is likely that PVR is the only molecule required for poliovirus binding and entry. The observation that a monoclonal antibody directed against the lymphocyte homing receptor CD44 blocks poliovirus binding to cells suggests that this protein might be a coreceptor for poliovirus entry.456,457 It was subsequently shown that CD44 is not a receptor for poliovirus and is not required for poliovirus infection of cells that produce PVR.72,149 It seems likely that PVR and CD44 are associated in the cell membrane150 and that anti-CD44 antibodies block poliovirus attachment by blocking the poliovirus-binding sites on PVR.
Orthologs of the pvr gene are present in the genomes of a number of mammals, including those not susceptible to poliovirus infection.231 The amino acid sequence of domain 1 of PVR varies extensively among the nonsusceptible mammals, especially in the regions known to contact poliovirus. The absence of a poliovirus-binding site on these PVR molecules, therefore, explains why poliovirus infection is restricted to simians.
The PVR is an adhesion molecule that participates in the formation of adherens junctions through interaction with nectin-3, a related immunoglobulin-like protein.349 The PVR is also a recognition molecule for natural killer (NK) cells, and interacts with CD226 and CD96 on NK cells to stimulate their cytotoxic activity.71,153 It also interacts with T-cell Ig and ITIM domain (TIGIT), regulating T-cell function.308 The UL141 protein of cytomegalovirus (CMV) blocks surface expression of PVR, leading to evasion of NK cell–mediated killing.491
The cell surface receptor for the major group of human rhinoviruses (90 serotypes) was identified by using monoclonal antibodies directed against the cellular binding site to isolate the receptor protein from susceptible cells. Amino acid sequence analysis of the purified protein revealed that it is ICAM-1, a type I transmembrane protein with five immunoglobulin-like domains.184,471,492 The normal cellular functions of ICAM-1 are to bind its ligand, lymphocyte function–associated antigen 1 (LFA-1) on the surface of lymphocytes and to promote a wide range of immunologic functions.502 ICAM-1 is expressed on the surfaces of many tissues, including the nasal epithelium, which is the entry site for rhinoviruses.
Three members of the low-density lipoprotein receptor family are receptors for minor group rhinoviruses (10 serotypes). These proteins consist of 7 (LDLR), 8 (VLDLR), or 31 (LRP) ligand-binding repeats, transmembrane and cytoplasmic domains.
Many picornaviruses bind integrins, which are dimeric cell adhesion receptors with α and β subunits. Many integrin receptors recognize the tripeptide Arg-Gly-Asp (RGD) whose presence in the viral capsid suggests interaction with this type of receptor. A number of integrins can serve as entry receptors for FMDV (Table 16.3), although all may not be utilized during infection of animals. Although integrins are sufficient for FMDV infection, their interaction with the viral capsid does not lead to uncoating (see Entry Into Cells).
Receptors and Coreceptors Required for Infection
Many enteroviruses bind to decay-accelerating factor (DAF, or CD55), a member of the complement cascade (Table 16.3); it is composed of four extracellular short consensus repeat modules and is attached to the plasma membrane by a glycosylphosphatidyl inositol (GPI) anchor. For most of these viruses, however, interaction with DAF is not sufficient for infection; this molecule is an attachment receptor but does not lead to virion uncoating. For example, Coxsackievirus A21 binds to DAF, but infection does not occur unless ICAM-1 is also bound (Fig. 16.5).453 In this case, ICAM-1 inserts into the canyon where it triggers capsid uncoating.534 Coxsackievirus B3 binds DAF but virion uncoating does not occur unless Coxsackievirus-adenovirus receptor (CAR; see below) binds in the canyon.208
Echovirus 7, which normally binds DAF, can infect some DAF-negative cells; in these cases CD59 or β2-microglobulin can serve as coreceptors for entry.172,521 Some Coxsackie B viruses that bind CD55 may require αvβ6-integrin as a coreceptor.4
A specific role for coreceptors in virus entry is illustrated by Coxsackievirus B3 entry into polarized epithelial cells.105 The Coxsackievirus and adenovirus receptor, CAR, mediates cell entry of all Coxsackie B viruses.58 CAR is not present on the apical
surface of epithelial cells that line the intestinal and respiratory tracts, but is a component of the tight junction and is inaccessible to virus entry. Coxsackie B viruses first bind an attachment receptor, DAF, which is present on the apical surface of epithelial cells. Coxsackievirus B3 binding to DAF activates Abl kinase, which in turn triggers Rac-dependent actin rearrangements, leading to virus movement to the tight junction where it can bind CAR and enter cells.105
surface of epithelial cells that line the intestinal and respiratory tracts, but is a component of the tight junction and is inaccessible to virus entry. Coxsackie B viruses first bind an attachment receptor, DAF, which is present on the apical surface of epithelial cells. Coxsackievirus B3 binding to DAF activates Abl kinase, which in turn triggers Rac-dependent actin rearrangements, leading to virus movement to the tight junction where it can bind CAR and enter cells.105
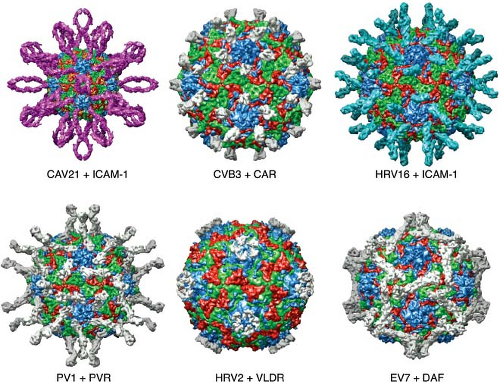 Figure 16.5. Interactions of six different picornaviruses with cellular receptors. Immunoglobulin-like cell receptors bind in the canyon, as illustrated for coxsackievirus 21 and ICAM-1, coxsackievirus B3 and CAR, rhinovirus 16 and ICAM-1, and poliovirus type 1 with PVR. A receptor for rhinovirus type 2, very-low-density lipoprotein receptor, binds on the plateau at the fivefold axis, and DAF binds echovirus 7 near the twofold icosahedral axes. Images produced with the Virus Particle Explorer at viberdb.scripps.edu.83 |
Alternative Receptors
Some viruses bind to different cell surface receptors, depending on the virus isolate or the cell line (Table 16.3). Clinical isolates of FMDV bind to integrin receptors, but passage in cell culture can select for viruses that bind to heparin sulfate, a sulfated glycan.236,320,443 Cell culture passage may also produce a virus that infects cells independent of heparin sulfate and integrins.33
How Picornaviruses Attach to Cell Receptors
Among the picornavirus members, the four capsid proteins are arranged similarly, but the surface architecture varies. These differences account for both the diverse serotypes and the varied modes of interaction with cell receptors. For example, the capsids of enteroviruses have a groove, or canyon, surrounding each fivefold axis of symmetry. In contrast, cardioviruses and aphthoviruses do not have canyons.
The canyons of enteroviruses are the sites of interaction with Ig-like cell receptors. The results of genetic and structural experiments demonstrate that the first Ig-like domain contains the site that binds poliovirus. Cells expressing the first Ig-like domain of PVR, either alone or as a hybrid with other Ig-like proteins, are susceptible to infection with poliovirus.281,342,450,451 Mutations in the first Ig-like domain of PVR interfere with poliovirus binding.21,60,343 Mutagenesis of ICAM-1 DNA has revealed that the binding site for rhinovirus is located in the first Ig-like domain.325,420,472 Models of the interaction of poliovirus, rhinovirus, and Coxsackievirus with their cellular receptors have been produced from cryo-EM and x-ray crystallographic data (Fig. 16.5).51,53,84,85,207,283,372,536 These models reveal that only domain 1 of PVR or ICAM-1 penetrates the
canyon of the respective virus. Mutations that affect receptor binding map to the virion–receptor interface as determined by these structural studies. Mutation of amino acids that line the canyons of poliovirus and rhinovirus can alter the affinity of binding to receptors.100,101,102,204,300
canyon of the respective virus. Mutations that affect receptor binding map to the virion–receptor interface as determined by these structural studies. Mutation of amino acids that line the canyons of poliovirus and rhinovirus can alter the affinity of binding to receptors.100,101,102,204,300
Although the capsids of at least two minor group rhinoviruses possess a canyon around the fivefold axes, it is not the binding site for the minor group receptor, members of the low-density lipoprotein receptor family (Fig. 16.5). Rather, the minor group receptor binds close to the fivefold axis, on the star-shaped plateau that is surrounded by the canyon.213,413,418,512 In this way multiple low-affinity interactions are combined to yield a high-avidity virus-receptor complex.
Sequence and structural comparisons have revealed why major and minor group rhinoviruses recognize different receptors. A lysine at position 224 of VP1, which is conserved in all minor group rhinoviruses, is the key amino acid that interacts with a negatively charged cluster of LDLR.512 The electrostatic attraction between Lys1224 and the acidic cluster in LDLR might initiate contact between virus and receptor. Neighboring hydrophobic and basic residues in VP1 could then lead to tight binding between virus and receptor. The conserved lysine is not present in VP1 of major group rhinoviruses, providing an explanation for failure of these viruses to bind LDLR. An exception is rhinovirus 85, a major group serotype that has the conserved lysine; presumably, it does not bind LDLR because of other amino acid differences in neighboring hydrophobic and basic VP1 residues.
It was originally believed that the picornavirus canyons were too deep and narrow to allow penetration by antibody molecules, which contain adjacent immunoglobulin domains.436 This physical barrier was believed to hide amino acids crucial for receptor binding from the immune system. Structural studies of a rhinovirus–antibody complex, however, revealed that antibody does penetrate deep into the canyon, as does ICAM-1.463 The shape of the picornavirus canyon, therefore, is not likely to play a role in concealing virus from the immune system.
In contrast to the Ig-like receptors that bind the canyons of enteroviruses, the binding sites for DAF on the virion are diverse. For example, DAF bridges the canyon of Coxsackievirus B3.196 In contrast, the binding site of DAF on echovirus 7 (Fig. 16.5) and 12 is near the twofold axis.390,402 These interactions are not sufficient for virion uncoating. Coxsackievirus A21 must also bind ICAM-1, which inserts into the canyon, triggering capsid uncoating.453,534 Similarly, Coxsackievirus B3 binds DAF but virion uncoating does not occur unless CAR binds in the canyon.208
Integrin-binding picornaviruses attach to cell receptors through surface loops. In FMDV, an Arg-Gly-Asp sequence in the flexible, exposed βG-βH loop of the capsid protein VP1 is recognized by integrin receptors on cells.121,147,306 Arg-Gly-Asp–containing peptides block attachment of FMDV,44 and alteration of this sequence interferes with virus binding.322 In Coxsackievirus A9, the Arg-Gly-Asp sequence is present in a 17–amino acid extension of the C-terminus of VP1 and is also the site of attachment to cell receptors.87,431 Alteration of this sequence does not abolish binding to cells, suggesting that the virus can bind to another cell surface receptor.228 Echovirus 1 is unusual in that it binds the RGD-independent integrin α2β1 in the canyon.537
As discussed, FMDV binds alternative receptors, either integrin or heparan sulfate, depending on the virus isolate. The binding site for heparan sulfate on cell culture–adapted FMDV is a shallow depression on the virion surface, where the three major capsid proteins, VP1, VP2, and VP3, are located.152 Binding specificity is controlled by two preformed sulfate-binding sites on the capsid. Residue 56 of VP3 is a critical regulator of receptor recognition. In field isolates of the virus, this amino acid is histidine. Adaptation to cell culture selects for viruses with an arginine at this position, which forms the high-affinity, heparan sulfate–binding site.
The interaction of EMCV with its cellular receptor, VCAM-1, has not been studied in detail. The EMCV capsid does not have a canyon, therefore VCAM-1, an Ig-like protein, must interact with the virion in a manner different from the Ig-like receptors of poliovirus and rhinovirus.
Kinetics and Affinity of the Virus–Receptor Interaction
The affinity and kinetics of picornaviruses binding to soluble forms of their receptors have been studied by surface plasmon resonance. Two classes of receptor-binding sites, with distinct binding affinities, exist on the capsids of poliovirus and rhinovirus.84,326,536 The association rates for the two binding classes are 25 and 13 times higher for the poliovirus–sPVR interaction than for the rhinovirus–sICAM interaction at 20°C. The greater association rate of poliovirus and PVR may be caused, in part, by differences in the extent of contact between virus and receptor. In contrast, whereas two dissociation rate constants exist for the poliovirus–PVR interaction, only one has been reported for the rhinovirus 3–ICAM interaction. The dissociation rates for the poliovirus–sPVR interaction are 1.5 times and 2.0 times faster than for the rhinovirus–sICAM interaction, indicating greater instability of the former complex. The affinity constants for the poliovirus–sPVR interaction are 19 times and 6 times greater than those reported for the rhinovirus–sICAM-1 complex.
In contrast to the observations with poliovirus and rhinovirus, a single class of binding site was found on echovirus 11 for a soluble form of its receptor, CD55.292 The affinity of this interaction is at least fourfold lower than either of the binding sites on poliovirus for sPVR. The association rate for the interaction between echovirus 11 and CD55 is faster than that of poliovirus–sPVR and rhinovirus–sICAM-1. One explanation for these findings is that the contact between echovirus 11 and CD55 is more extensive than that of the other two virus–receptor complexes. The binding site for CD55 on echovirus 11 may also be more accessible than those of PVR and ICAM-1. The dissociation rate for the echovirus–CD55 interaction is at least 97 times faster than that of either the poliovirus–sPVR or the rhinovirus–sICAM-1 interaction. These findings are consistent with a more accessible binding site for CD55 on echovirus 11, compared with the receptor-binding sites on poliovirus and rhinovirus. Atomic interactions between CD55 and echovirus 11 may be weaker than between the other two viruses and their receptors. The faster dissociation rate of the echovirus 11–CD55 complex may be related to the finding that the interaction does not lead to structural changes of the virus particle,409 as occurs with poliovirus and rhinovirus. In general, there is a higher affinity for virions of receptors that can uncoat particles (“unzippers”—poliovirus/PVR, HRV/ICAM-1, CVB3/CAR) compared with attachment receptors (E6, 7, 11, 12, CVB3 with DAF). This may reflect the requirement for higher affinity to release the viral RNA.
Why do poliovirus and rhinovirus have two classes of receptor-binding sites? The receptors for both viruses make contacts at two major sites on the virus surface, one in a cleft on the south rim of the canyon, and a second on the side of the mesa on the north rim. These two contact sites may correspond to the two classes of binding sites. Two classes of binding sites may also be a consequence of the structural flexibility exhibited by both viruses, which may cause exposure of different binding sites. Normally internal parts of the poliovirus and rhinovirus capsid proteins have been shown to be transiently displayed on the virion surface, a process called breathing.296,298 As to be discussed later, the interaction of poliovirus and rhinovirus with their cellular receptors leads to irreversible and more extensive structural changes. In contrast to the findings with poliovirus and rhinovirus, binding of echovirus 11 with CD55 can be described by a simple 1:1 binding model. Such behavior, which would be expected for the interaction of two preformed binding sites, is consistent with the fact that the echovirus–CD55 interaction does not result in detectable structural changes in the capsid.409
Entry into Cells
Once picornaviruses have attached to their cellular receptor, the viral capsid is brought into the cell by the endocytic pathway, followed by genome release into the cytoplasm, the site of picornavirus replication. For some picornaviruses, interaction with a cell receptor serves only to concentrate virus on the cell surface; release of the genome is a consequence of low pH or perhaps the activity of a coreceptor. For other picornaviruses, the cell receptor is also an unzipper and initiates conformational changes in the virus that lead to release of the genome.
Entry by Clathrin-Mediated Endocytosis
Several lines of evidence indicate that FMDVs enter cells by clathrin-mediated endocytosis. Infection is inhibited by sucrose, which eliminates clathrin-coated pits and induces clathrin to polymerize into empty cages, and by expression of a dominant negative form of the clathrin coat assembly protein AP180 that is needed for assembly of clathrin cages.61 Confocal microscopy also revealed that FMDV enters cells via a clathrin-dependent mechanism.366 There is also some evidence that entry is dependent on cholesterol in the plasma membrane, a requirement generally observed for lipid rafts319; however, cholesterol might be required for clathrin-induced membrane curving.428 Another aphthovirus, equine rhinitis A virus, binds sialic acid–containing receptors but also enters into cells via clathrin-mediated endocytosis.185
LDLR family members that are receptors for minor group HRVs possess C-terminal cytoplasmic domains with tyrosine- and di-leucine–based internalization signals that lead to clustering of the receptors in clathrin-coated pits.384 Evidence for HRV entry via this pathway includes the inhibition of infection in cells producing dominant negative inhibitors of the clathrin pathway.47,465 There is some evidence that ICAM-1 binding major group HRVs also enter cells via clathrin-mediated endocytosis, including transmission EM, which shows virions in clathrin-coated pits and vesicles 5 minutes after infection187 and the fact that dominant negative dynamin inhibits infection.118 HRV infection also activates signaling pathways with links to the endocytic machinery. The cytoplasmic domain of ICAM-1 binds the adaptor protein ezrin, which links the receptor to Syk, a tyrosine kinase.519a When HRV binds ICAM-1, Syk is recruited from the cytoplasm to the plasma membrane together with clathrin. Functional Syk is required for HRV entry via ICAM-1.291
When virions enter cells by clathrin-dependent endocytosis they encounter low pH, which triggers release of the viral RNA from the capsid. A role for low pH in infection can be demonstrated by determining the effect on entry of compounds that block acidification, such as weak bases (ammonium chloride, chloroquine, methylamine) ionophores (monensin, nigericin, X537A) or inhibitors of the vacuolar proton ATPase (concanamycin A, bafilomycin A). The pH of early endosomes is 6.5; as these vesicles mature to late endosomes the pH drops to 5.5. Endosomal maturation is dependent not only on vacuolar ATPases but also on microtubules and membrane GTPases of the Rab family. Early to late endosome maturation can be inhibited by drugs that depolymerize microtubules (nocodazole), dominant negative Rabs, or inhibitors of PI3K signaling (wortmannin). In this way it is possible to determine if viral entry occurs from early or late endosomes.
Uncoating by FMDV clearly requires low pH because concanamycin A, monensin, and ammonium chloride all inhibit infection.43,61,366,517 Entry occurs from the early endosome as determined by experiments with dominant negative Rab proteins.250 FMDV that bind heparan sulfate enter cells via a caveolae-dependent route, but low pH is still required for infection.367 Consistent with this mechanism of uncoating, FMDV that has been coated with antibody can bind to, and infect, cells that express Fc receptors, in contrast to poliovirus, which cannot productively infect cells via this pathway.321 Cell receptors for FMDV are, therefore, hooks: they do not induce uncoating-related changes in the virus particle, but rather serve only to tether the virus to the cell and bring it into the endocytic pathway.
Entry of minor group HRVs requires low pH of the late endosome, as infection is inhibited by monensin, bafilomycin A1, nocodazole, and wortmannin.46,59,74,353,410 ICAM-binding HRVs are also sensitive to bafilomycin and monensin, suggesting that endosome acidification is required for entry.187,387,483
Echovirus 7, which binds DAF, enters cells by clathrin-mediated endocytosis, and trafficking into late endosomes is required.270 However, infection does not appear to require low pH, and therefore the trigger for uncoating and its intracellular location remains in question.
Entry by Caveolin-Mediated Endocytosis
As discussed above, CVB3 binds DAF, a GPI-linked protein localized within lipid rafts on the apical surface of polarized cells. The virus/DAF complex then moves to tight junctions (TJ) where it engages CAR. The virus is then internalized along with the TJ protein occludin by caveolin-1–dependent endocytosis.107 The role of occludin is not known but it could provide a scaffold for recruiting other molecules. Within 60 minutes, the virus is within caveolin-1–containing vesicles (caveolae and caveosomes). Phosphorylation of caveolin-1 by tyrosine kinases is required for CVB3 entry, but the role of this modification is not known. Dynamin is not required for uptake of this virus, suggesting that other routes of entry are involved. Inhibitors of micropinocytosis (rottlerin and dominant negative Rab34) block infection, indicating a role for this type of uptake.
Echovirus 1, which binds the integrin α2β1, is taken into the cell by the caveolin-mediated endocytic pathway. The receptor is present in raft-like membrane domains that do not contain caveolin. Internalization of the virus does not depend on dynamin, but components of the macropinocytosis pathway, such as PKC, Pak1, and Rac1, are involved. By 30 minutes after infection, the virus appears in vesicular structures that appear to be caveolae.318 These fuse with caveosomes, delivering the virus and its receptor to the perinuclear region.397 These may be novel multivesicular bodies; virus transport to them appears to depend on ESCRT proteins.263
Caveolin- and Clathrin-Independent Endocytosis
In HeLa cells, poliovirus is taken up into cells by an endocytic pathway that is dependent on actin, ATP, and a tyrosine kinase, but independent of clathrin, caveolin, flotillin, microtubules, and pinocytosis.76 RNA release from the particle occurs rapidly and within 100 to 200 nm of the cell surface. Entry was different in a highly polarized human brain microvascular endothelial cell line.106 Poliovirus enters these cells very slowly via dynamin-dependent caveolar endocytosis. Virus binding to PVR induces tyrosine phosphorylation of the receptor cytoplasmic domain, which in turn recruits and activates SHP-2, which is required for infection. These observations emphasize that virus entry pathways are likely to differ substantially according to cell type.
Uncoating
The interaction of enteroviruses and major group rhinoviruses with susceptible cells leads to the conversion of virions to a more slowly sedimenting form (135S versus 160S for native particles).112,252 The resulting particles, called altered (or A) particles, contain the viral RNA but have lost the internal capsid protein VP4. In addition, the N-terminus of VP1, which is normally on the interior of the capsid, is on the surface of the A particle.151 This sequence of VP1 is hydrophobic and, as a result, the A particles have an increased affinity for membranes compared with the native virus particle. It is believed that A particles represent a stable intermediate structure in the entry process that terminates with exit of RNA and the production of empty (80S) capsids. In one hypothesis for poliovirus entry, receptor binding leads to these conformational changes; the exposed lipophilic N-terminus of VP1 then inserts into the cell membrane, tethering the A particle to the membrane. A membrane pore is then formed, possibly by both VP4 and VP1, through which the viral RNA can travel to the cytoplasm (Fig. 16.6). The finding that A particles, when added to lipid bilayers, induce the formation of ion channels supports this hypothesis.493 The trigger for conversion of minor group HRVs is not receptor binding but low pH.410
Native poliovirus and rhinovirus particles have been shown to transiently and reversibly expose VP4 and the N-terminus of VP1, a process called “breathing”.296,298 Receptor binding (poliovirus) or low pH (rhinoviruses, cardioviruses, aphthoviruses) lowers the energy barrier for conversion to the A particle, a process that enables genome delivery to the cell. The structure of poliovirus bound to a monoclonal antibody that recognizes the N-terminus of VP1 reveals that this viral protein exits the capsid near the twofold axes, instead of near the propeller tip in 135S particles (see below).
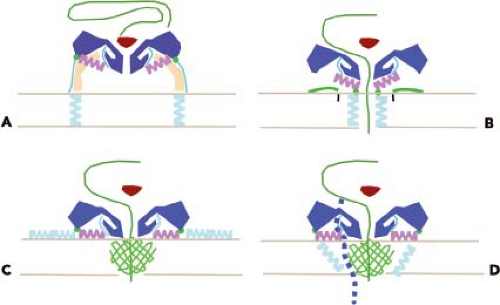 Figure 16.6. Hypothetical mechanism for translocation of poliovirus RNA across the cell membrane. A: Cross-section of a virus particle that has just bound poliovirus receptor (PVR) at the cell surface. PVR docks on the capsid in the canyon, above the hydrophobic pocket. The path of VP1 egress would not preclude continued binding to PVR. The viral RNA is in the capsid, and lipid has exited from the hydrophobic pocket. The capsid is colored blue, VP4 is green, and the N-terminus of VP1 is cyan and magenta, PVR is tan, RNA is a green line. At this stage the VP3 β-cylinder (red) blocks a channel at the fivefold axis. B–D illustrate alternative models for anchoring of virus particle to the cell membrane by the N-terminus of VP1 and formation of a pore for passage of viral RNA. Upon binding, PVR structural changes occur that move the VP3 β-cylinder out of the way like a float valve and open a channel at the fivefold axis that is contiguous with a pore in the membrane. (B) Amphipathic helices at the N-terminus of VP1 form a pore through the membrane. In C and D, VP4 (green mesh) is playing a central role in pore formation. In this case, VP1 may anchor the particle to the membrane. An alternative pathway for release of the genome from the base of the canyon is shown. |
Low-resolution, cryo-EM reconstructions of the poliovirus 135S and 80S particles did not reveal openings that could allow for release of the viral genome.52 Subsequently, higher-resolution structures of the poliovirus A particle and a derivative in which the N-terminal 31 amino acids of VP1 were removed revealed that this sequence of VP1 is likely to exit the virion at the base of the canyon, between the tips of the “stars” at the fivefold axis and the propeller-shaped feature around the threefold axis.78 It has also been suggested that VP4 exits near the twofold axis, based on the observation that insertion of a cysteine in the N-terminus of VP4 leads to disulfide linking upon breathing.266
Cryo-EM reconstructions of 80S particles of HRV2,213 HRV14, and poliovirus295 reveal two populations of particles, one with more internal density (presumably RNA) than the other. The poliovirus structures reveal different particle states: some in the process of releasing RNA, others with nucleic acid in the particle, crossing the particle walls, and outside the particle. Analysis of these particles indicates that the viral RNA exits the capsid from openings at the base of the canyon, near where the release of the VP1 N-terminus occurs.70 The trigger for release of RNA from the capsid is unknown, but may require unfolding of secondary structure.76
VP4 is released from the virion during conversion to A particles, but a small amount of this protein might remain and participate with VP1 in membrane channel formation. A virus containing an amino acid change at position 28 of VP4 can bind to cells and be converted to A particles, but these are blocked at a subsequent step in virus entry.345 Amino acid changes at this position of VP4 reduce the conductance of ion channels and the translocation of viral RNA.115 These findings suggest that VP4 might play a central role in pore formation. VP4 and VP2 are produced, during virus assembly, from the precursor VP0, which remains uncleaved until RNA encapsidation. Cleavage of VP0, therefore, can be viewed as a way of priming the capsid for uncoating because cleavage separates VP4 from VP2.
While the enterovirus capsid maintains its icosahedral form during uncoating, the acid-labile aphthoviruses dissociate into pentamers at low pH, a process that releases the viral RNA.510 The mechanism by which low pH causes disassembly of the FMDV capsid has been illuminated by structural and genetic studies. Examination of the atomic structure of the virus revealed a high density of histidine residues lining the pentamer interfaces, which are stabilized by β-sheet interactions.1 These residues confer stability to the capsid; because the pKa of histidine is 6.8, close to the pH at which the virus dissociates, protonation of the side chains of the histidines might cause electrostatic repulsion, leading to disassembly.113 To test this hypothesis, a histidine residue at position 142 of VP3 was changed to arginine by mutagenesis. The resulting capsids were more stable at low pH than wild-type capsids,134 supporting the proposed role of the histidine residue in acid-catalyzed disassembly. Enteroviruses are stable at low pH in part because there are extra β-sheet interactions in VP1.138 Equine rhinitis A virus is also acid labile; it dissociates into pentamers due to rearrangement of loops in VP2 that disrupt the pentamer interfaces.501 How aphthoviruses breach the endosome membrane is not known; these viruses do not have a hydrophobic VP1 N-terminus, and A particles are not produced.
If aphthoviruses dissociate into pentamers in the endosome, how is the integrity of viral RNA preserved before it exits to the cytoplasm? A clue is provided by the finding that equine rhinitis A virus capsid dissociation is preceded by the formation of a transient empty particle.501 It is possible that this protects the viral RNA until it leaves the endosome.
Low pH causes particle expansion of minor group rhinoviruses and the opening of a 10 Å diameter pore at the fivefold axis through which the RNA is presumed to exit.214
Regulation of Uncoating by Cellular Molecules
Beneath the canyon floor is a hydrophobic pocket that opens at the base of the canyon and extends toward the fivefold axis of symmetry. The pocket appears to be occupied in many picornaviruses with a fatty acid or related compound (Fig. 16.2). In some picornaviruses (e.g., rhinovirus types 3 and 14), the pockets are apparently empty.25,550 The hydrophobic pocket appears to be a critical regulator of the receptor-induced structural transitions of enteroviruses. The icosahedral symmetry of the capsid would allow each virion to contain up to 60 lipid molecules. Certain antiviral drugs (e.g., the WIN compounds first identified by Sterling-Winthrop, Inc.) displace the lipid and bind tightly within the pocket.462 Binding of such drugs to rhinovirus 14 causes conformational changes in the canyon that prevent attachment to cells.391 In contrast, drug binding to rhinoviruses 1A, 3, and 16 and to poliovirus causes smaller structural changes in the capsid.182,195,218 Inhibition of rhinovirus 16 binding by these compounds is probably not a consequence of altering the receptor-binding site, but rather the result of preventing conformational changes required for receptor binding. Such compounds do not inhibit binding of poliovirus, but rather uncoating.330
The lipids that occupy the hydrophobic pocket were originally believed to contribute to the stability of the native virus particle by locking the capsid in a stable configuration and preventing conformational changes. Removal of the lipid was therefore necessary to provide the capsid with sufficient flexibility to undergo the changes that permit the RNA to leave the shell. This hypothesis comes from the study of antiviral drugs, such as the WIN compounds, that displace the lipid and bind tightly in the hydrophobic pocket. These antiviral compounds block breathing of the rhinovirus capsid, the process by which normally internal parts of the capsid proteins are transiently displayed on the virion surface.296 Polioviruses containing bound WIN compounds can bind to cells, but the interaction with PVR does not result in the production of A particles.148,548 WIN compounds appear to inhibit poliovirus infectivity by preventing PVR-mediated conformational alterations that are required for uncoating. Additional support for the role of lipids in uncoating comes from the analysis of poliovirus mutants that cannot replicate unless WIN compounds are present.347 Such WIN-dependent mutants spontaneously convert to altered particles at 37°C, in the absence of the cell receptor, probably because of the absence of lipid in the hydrophobic pocket. The lipids can be considered to be switches that determine whether the virus is stable (lipid present) or will uncoat (lipid absent). It is not known what causes lipid release from the capsid. PVR docks onto the poliovirus capsid just above the hydrophobic pocket (Fig. 16.6), which suggests that the interaction of the virus with receptor may initiate structural changes in the virion that lead to the release of the lipid.
Incubation of poliovirus with PVR for short periods at low temperatures appears to result in loss of the lipid.53 The results of computational modeling and kinetic studies suggest that stabilization of virus particles by drugs that replace the lipid is a consequence of increased compressibility rather than increased rigidity.395,470,500
Incubation of poliovirus with PVR for short periods at low temperatures appears to result in loss of the lipid.53 The results of computational modeling and kinetic studies suggest that stabilization of virus particles by drugs that replace the lipid is a consequence of increased compressibility rather than increased rigidity.395,470,500
Translation of the Viral RNA
Internal Ribosome Binding: The Internal Ribosome Entry Site
Once the picornavirus positive-stranded genomic RNA is released into the cell cytoplasm, it must be translated because it cannot be copied by any cellular RNA polymerase and no viral enzymes are brought into the cell within the viral capsid. Several experimental findings led to the belief that translation of the picornavirus genome was accomplished by an unusual mechanism. The positive-stranded RNA genomes lack 5′-terminal cap structures; although virion RNA is linked to the viral protein VPg, this protein is removed by a cellular unlinking enzyme on entry of the RNA into the cell.13 Furthermore, picornavirus genomes are efficiently translated in infected cells despite inhibition of cellular mRNA translation. Determination of the nucleotide sequence of poliovirus positive-stranded RNA revealed a 741-nucleotide 5′-untranslated region that contains seven AUG codons.276,415 Similar 5′-noncoding regions were subsequently found in other picornaviruses and shown to contain highly ordered RNA structures.426,461 These findings led to the suggestion that ribosomes do not scan through picornaviral 5′-untranslated regions, but rather bind to an internal sequence. The 5′-untranslated region of poliovirus positive-stranded RNA was subsequently shown to contain a sequence that promotes internal binding of the 40S ribosomal subunit; it was called the internal ribosome entry site (IRES) (Fig. 16.7).
All picornavirus RNA contain an IRES, as do other viral and some cellular mRNAs.313 Viral IRES have been placed in four groups based on a variety of criteria, including primary
sequence, secondary structure, location of the initiation codon, and activity in different cell types (Fig. 16.8). In the type I IRES (found in the genomes of enteroviruses and rhinoviruses), and the type III IRES (hepatitis A virus), the initiation codon is located 50 to 100 nucleotides beyond the 3′-end of the IRES, whereas it is located at the 3′-end of a type II IRES (cardioviruses and aphthoviruses). The IRES of porcine teschovirus, avian encephalomyelitis virus, and hepatitis C virus (a member of the Flaviviridae) are classified as a type IV IRES.
sequence, secondary structure, location of the initiation codon, and activity in different cell types (Fig. 16.8). In the type I IRES (found in the genomes of enteroviruses and rhinoviruses), and the type III IRES (hepatitis A virus), the initiation codon is located 50 to 100 nucleotides beyond the 3′-end of the IRES, whereas it is located at the 3′-end of a type II IRES (cardioviruses and aphthoviruses). The IRES of porcine teschovirus, avian encephalomyelitis virus, and hepatitis C virus (a member of the Flaviviridae) are classified as a type IV IRES.
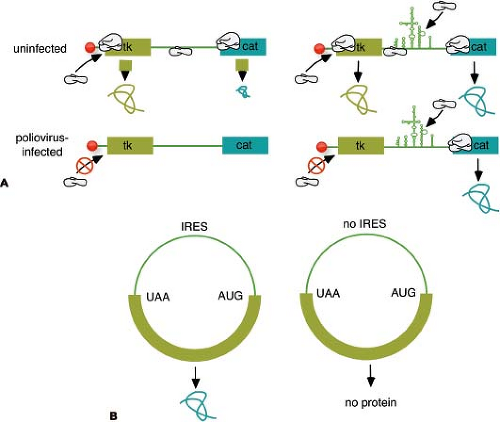 Figure 16.7. Discovery of the internal ribosome entry site (IRES). A: Bicistronic messenger RNA (mRNA) assay used to discover the IRES. Plasmids were constructed that encode two reporter molecules, thymidine kinase (tk) and chloramphenicol acetyl transferase (cat), separated by an IRES or a spacer. After introduction into mammalian cells, the plasmids give rise to mRNA of the structure shown in the figure. In uninfected cells (top line), both reporter molecules can be detected, although cat synthesis is inefficient without an IRES and is probably caused by reinitiation. In poliovirus-infected cells, 5′ end–dependent initiation is inhibited, and no proteins are detected without an IRES, demonstrating internal ribosome binding. B: Circular mRNA assay for an IRES. Circular mRNA were constructed and translated in vitro in cell extracts. In the absence of an IRES, no protein is observed because 5′-end initiation requires a free 5′ end. Inclusion of an IRES allows protein translation from the circular mRNA, demonstrating internal ribosome binding.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|